
Malignant Arrhythmogenic Role Associated with RBM20: A Comprehensive Interpretation Focused on a Personalized Approach

 , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Dilated Cardiomyopathy
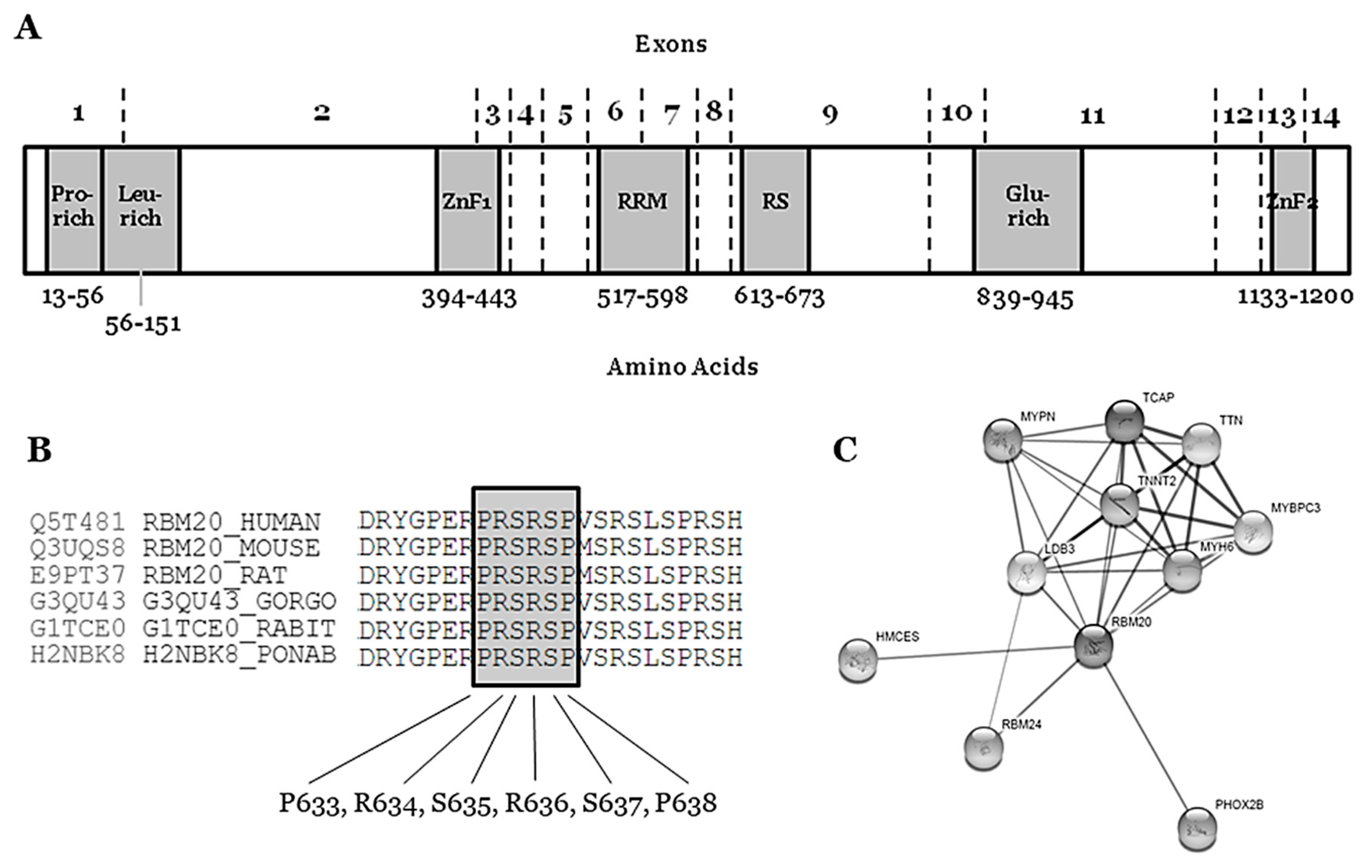
3. RBM20
4. Rare RBM20 Variants in Familial Dilated Cardiomyopathy
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics and Genomics |
| AR-DCM | Arrhythmogenic phenotype in dilated cardiomyopathy |
| CC | Cardio Classifier, ClinVar: Clinical Variation |
| CMR | Cardiac Magnetic Resonance |
| CNV | Copy Number Variation |
| DM | Disease Mutation |
| DCM | Dilated Cardiomyopathy |
| ECG | Electrocardiogram |
| FDCM | Familial DCM |
| gnomAD | Genome Aggregation Database |
| HGMD | Human Genome Mutation Database |
| hiPSC-CMs | human induced-pluripotent stem cell-derived cardiomyocytes |
| ICD | Implantable Cardiac Defibrillator |
| LB | Likely Benign |
| LP | Likely Pathogenic |
| MAF | Minor Allele Frequency |
| NGS | Next Generation Sequencing |
| P | Pathogenic |
| SCD | Sudden Cardiac Death |
| SD | Standard Deviation |
| VF | Ventricular Fibrillation |
| VT | Ventricular Tachycardia |
| VUS | Variant of Uncertain Significance |
References
- Rampersaud, E.; Siegfried, J.D.; Norton, N.; Li, D.; Martin, E.; Hershberger, R.E. Rare variant mutations identified in pediatric patients with dilated cardiomyopathy. Prog. Pediatric Cardiol. 2011, 31, 39–47. [Google Scholar] [CrossRef] [Green Version]
- McKenna, W.J.; Maron, B.J.; Thiene, G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 2017, 121, 722–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manolio, T.A.; Baughman, K.L.; Rodeheffer, R.; Pearson, T.A.; Bristow, J.D.; Michels, V.V.; Abelmann, W.H.; Harlan, W.R. Prevalence and etiology of idiopathic dilated cardiomyopathy (summary of a National Heart, Lung, and Blood Institute workshop. Am. J. Cardiol 1992, 69, 1458–1466. [Google Scholar] [CrossRef]
- Miura, K.; Nakagawa, H.; Morikawa, Y.; Sasayama, S.; Matsumori, A.; Hasegawa, K.; Ohno, Y.; Tamakoshi, A.; Kawamura, T.; Inaba, Y. Epidemiology of idiopathic cardiomyopathy in Japan: Results from a nationwide survey. Heart 2002, 87, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Amoah, A.G.; Kallen, C. Aetiology of heart failure as seen from a National Cardiac Referral Centre in Africa. Cardiology 2000, 93, 11–18. [Google Scholar] [CrossRef]
- Den Boer, S.L.; Lennie van Osch-Gevers, M.; van Ingen, G.; du Marchie Sarvaas, G.J.; van Iperen, G.G.; Tanke, R.B.; Backx, A.P.; Ten Harkel, A.D.; Helbing, W.A.; Delhaas, T.; et al. Management of children with dilated cardiomyopathy in The Netherlands: Implications of a low early transplantation rate. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2015, 34, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Kirk, R.; Naftel, D.; Hoffman, T.M.; Almond, C.; Boyle, G.; Caldwell, R.L.; Kirklin, J.K.; White, K.; Dipchand, A.I. Pediatric Heart Transplant Study I: Outcome of pediatric patients with dilated cardiomyopathy listed for transplant: A multi-institutional study. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2009, 28, 1322–1328. [Google Scholar] [CrossRef] [Green Version]
- Towbin, J.A. Inflammatory cardiomyopathy: There is a specific matrix destruction in the course of the disease. Ernst Scher. Res. Found. Workshop 2006, 55, 219–250. [Google Scholar]
- Towbin, J.A.; Bowles, N.E. Dilated cardiomyopathy: A tale of cytoskeletal proteins and beyond. J. Cardiovasc Electrophysiol 2006, 17, 919–926. [Google Scholar] [PubMed]
- Lund, L.H.; Edwards, L.B.; Kucheryavaya, A.Y.; Benden, C.; Dipchand, A.I.; Goldfarb, S.; Levvey, B.J.; Meiser, B.; Rossano, J.W.; Yusen, R.D.; et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-second Official Adult Heart Transplantation Report--2015; Focus Theme: Early Graft Failure. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2015, 34, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Bohm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlo, M.; Pivetta, A.; Pinamonti, B.; Stolfo, D.; Zecchin, M.; Barbati, G.; Di Lenarda, A.; Sinagra, G. Long-term prognostic impact of therapeutic strategies in patients with idiopathic dilated cardiomyopathy: Changing mortality over the last 30 years. Eur. J. Heart Fail. 2014, 16, 317–324. [Google Scholar] [CrossRef]
- Merlo, M.; Cannata, A.; Vitagliano, A.; Zambon, E.; Lardieri, G.; Sinagra, G. Clinical management of dilated cardiomyopathy: Current knowledge and future perspectives. Expert Rev. Cardiovasc Ther. 2016, 14, 137–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ware, J.S.; Amor-Salamanca, A.; Tayal, U.; Govind, R.; Serrano, I.; Salazar-Mendiguchia, J.; Garcia-Pinilla, J.M.; Pascual-Figal, D.A.; Nunez, J.; Guzzo-Merello, G.; et al. Genetic Etiology for Alcohol-Induced Cardiac Toxicity. J. Am. Coll Cardiol 2018, 71, 2293–2302. [Google Scholar] [PubMed]
- Ware, J.S.; Seidman, J.G.; Arany, Z. Shared Genetic Predisposition in Peripartum and Dilated Cardiomyopathies. N. Engl. J. Med. 2016, 374, 2601–2602. [Google Scholar] [CrossRef] [PubMed]
- Mathew, T.; Williams, L.; Navaratnam, G.; Rana, B.; Wheeler, R.; Collins, K.; Harkness, A.; Jones, R.; Knight, D.; O’Gallagher, K.; et al. Diagnosis and assessment of dilated cardiomyopathy: A guideline protocol from the British Society of Echocardiography. Echo Res. Pract. 2017, 4, G1–G13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toro, R.; Blasco-Turrion, S.; Morales-Ponce, F.J.; Gonzalez, P.; Martinez-Camblor, P.; Lopez-Granados, A.; Brugada, R.; Campuzano, O.; Perez-Serra, A.; Rosa Longobardo, F.; et al. Plasma microRNAs as biomarkers for Lamin A/C-related dilated cardiomyopathy. J. Mol. Med. (Berl) 2018, 96, 845–856. [Google Scholar] [CrossRef]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Muller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015, 36, 1123–1135. [Google Scholar] [PubMed]
- Mestroni, L.; Maisch, B.; McKenna, W.J.; Schwartz, K.; Charron, P.; Rocco, C.; Tesson, F.; Richter, A.; Wilke, A.; Komajda, M. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur. Heart J. 1999, 20, 93–102. [Google Scholar] [CrossRef]
- Mestroni, L.; Rocco, C.; Gregori, D.; Sinagra, G.; Di Lenarda, A.; Miocic, S.; Vatta, M.; Pinamonti, B.; Muntoni, F.; Caforio, A.L.; et al. Familial dilated cardiomyopathy: Evidence for genetic and phenotypic heterogeneity. Heart Muscle Disease Study Group. J. Am. Coll Cardiol 1999, 34, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Perez-Serra, A.; Toro, R.; Sarquella-Brugada, G.; de Gonzalo-Calvo, D.; Cesar, S.; Carro, E.; Llorente-Cortes, V.; Iglesias, A.; Brugada, J.; Brugada, R.; et al. Genetic basis of dilated cardiomyopathy. Int. J. Cardiol. 2016, 224, 461–472. [Google Scholar] [CrossRef]
- Gacita, A.M.; McNally, E.M. Genetic Spectrum of Arrhythmogenic Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005850. [Google Scholar] [CrossRef] [PubMed]
- Fatkin, D.; Huttner, I.G. Titin-truncating mutations in dilated cardiomyopathy: The long and short of it. Curr Opin Cardiol 2017, 32, 232–238. [Google Scholar] [CrossRef]
- Gigli, M.; Begay, R.L.; Morea, G.; Graw, S.L.; Sinagra, G.; Taylor, M.R.; Granzier, H.; Mestroni, L. A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; American Heart, A.; et al. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [PubMed] [Green Version]
- Ivanov, A.; Dabiesingh, D.S.; Bhumireddy, G.P.; Mohamed, A.; Asfour, A.; Briggs, W.M.; Ho, J.; Khan, S.A.; Grossman, A.; Klem, I.; et al. Prevalence and Prognostic Significance of Left Ventricular Noncompaction in Patients Referred for Cardiac Magnetic Resonance Imaging. Circ. Cardiovasc. Imaging 2017, 10, e006174. [Google Scholar] [CrossRef] [Green Version]
- Mazzarotto, F.; Hawley, M.H.; Beltrami, M.; Beekman, L.; de Marvao, A.; McGurk, K.A.; Statton, B.; Boschi, B.; Girolami, F.; Roberts, A.M.; et al. Systematic large-scale assessment of the genetic architecture of left ventricular noncompaction reveals diverse etiologies. Genet. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Dauksaite, V.; Gotthardt, M. Molecular basis of titin exon exclusion by RBM20 and the novel titin splice regulator PTB4. Nucleic Acids Res. 2018, 46, 5227–5238. [Google Scholar] [CrossRef] [PubMed]
- Parikh, V.N.; Caleshu, C.; Reuter, C.; Lazzeroni, L.C.; Ingles, J.; Garcia, J.; McCaleb, K.; Adesiyun, T.; Sedaghat-Hamedani, F.; Kumar, S.; et al. Regional Variation in RBM20 Causes a Highly Penetrant Arrhythmogenic Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005371. [Google Scholar] [CrossRef] [PubMed]
- Spezzacatene, A.; Sinagra, G.; Merlo, M.; Barbati, G.; Graw, S.L.; Brun, F.; Slavov, D.; Di Lenarda, A.; Salcedo, E.E.; Towbin, J.A.; et al. Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias. J. Am. Heart Assoc. 2015, 4, e002149. [Google Scholar] [CrossRef] [Green Version]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Dal Ferro, M.; Altinier, A.; et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J. Am. Coll Cardiol. 2019, 74, 1480–1490. [Google Scholar] [PubMed]
- Van Rijsingen, I.A.; Arbustini, E.; Elliott, P.M.; Mogensen, J.; Hermans-van Ast, J.F.; van der Kooi, A.J.; van Tintelen, J.P.; van den Berg, M.P.; Pilotto, A.; Pasotti, M.; et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J. Am. Coll Cardiol. 2012, 59, 493–500. [Google Scholar] [CrossRef]
- Helio, T.; Elliott, P.; Koskenvuo, J.W.; Gimeno, J.R.; Tavazzi, L.; Tendera, M.; Kaski, J.P.; Mansencal, N.; Bilinska, Z.; Carr-White, G.; et al. ESC EORP Cardiomyopathy Registry: Real-life practice of genetic counselling and testing in adult cardiomyopathy patients. ESC Heart Fail. 2020, 7, 3013–3021. [Google Scholar] [CrossRef]
- Kober, L.; Thune, J.J.; Nielsen, J.C.; Haarbo, J.; Videbaek, L.; Korup, E.; Jensen, G.; Hildebrandt, P.; Steffensen, F.H.; Bruun, N.E.; et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. N. Engl. J. Med. 2016, 375, 1221–1230. [Google Scholar] [CrossRef] [Green Version]
- Peters, S.; Kumar, S.; Elliott, P.; Kalman, J.M.; Fatkin, D. Arrhythmic Genotypes in Familial Dilated Cardiomyopathy: Implications for Genetic Testing and Clinical Management. Heart Lung Circ. 2019, 28, 31–38. [Google Scholar]
- Priori, S.G.; Blomstrom-Lundqvist, C. 2015 European Society of Cardiology Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death summarized by co-chairs. Eur. Heart J. 2015, 36, 2757–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zegkos, T.; Panagiotidis, T.; Parcharidou, D.; Efthimiadis, G. Emerging concepts in arrhythmogenic dilated cardiomyopathy. Heart Fail. Rev. 2020. [Google Scholar] [CrossRef]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padron-Barthe, L.; Duro-Aguado, I.; Jimenez-Jaimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Zahr, H.C.; Jaalouk, D.E. Exploring the Crosstalk Between LMNA and Splicing Machinery Gene Mutations in Dilated Cardiomyopathy. Front. Genet. 2018, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Murayama, R.; Kimura-Asami, M.; Togo-Ohno, M.; Yamasaki-Kato, Y.; Naruse, T.K.; Yamamoto, T.; Hayashi, T.; Ai, T.; Spoonamore, K.G.; Kovacs, R.J.; et al. Phosphorylation of the RSRSP stretch is critical for splicing regulation by RNA-Binding Motif Protein 20 (RBM20) through nuclear localization. Sci. Rep. 2018, 8, 8970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippello, A.; Lorenzi, P.; Bergamo, E.; Romanelli, M.G. Identification of nuclear retention domains in the RBM20 protein. FEBS Lett. 2013, 587, 2989–2995. [Google Scholar]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Giron, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Lennermann, D.; Backs, J.; van den Hoogenhof, M.M.G. New Insights in RBM20 Cardiomyopathy. Curr. Heart Fail. Rep. 2020, 17, 234–246. [Google Scholar] [CrossRef]
- Fochi, S.; Lorenzi, P.; Galasso, M.; Stefani, C.; Trabetti, E.; Zipeto, D.; Romanelli, M.G. The Emerging Role of the RBM20 and PTBP1 Ribonucleoproteins in Heart Development and Cardiovascular Diseases. Genes 2020, 11, 402. [Google Scholar] [CrossRef] [Green Version]
- Wyles, S.P.; Li, X.; Hrstka, S.C.; Reyes, S.; Oommen, S.; Beraldi, R.; Edwards, J.; Terzic, A.; Olson, T.M.; Nelson, T.J. Modeling structural and functional deficiencies of RBM20 familial dilated cardiomyopathy using human induced pluripotent stem cells. Hum. Mol. Genet. 2016, 25, 254–265. [Google Scholar] [PubMed] [Green Version]
- Wilsbacher, L.D. Clinical Implications of the Genetic Architecture of Dilated Cardiomyopathy. Curr. Cardiol. Rep. 2020, 22, 170. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Den Dunnen, J.T. Sequence Variant Descriptions: HGVS Nomenclature and Mutalyzer. Curr Protoc Hum. Genet. 2016, 90, 7–13. [Google Scholar] [CrossRef]
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, Y.; Yang, S.; Nykamp, K.; Garcia, J.; Lincoln, S.E.; Topper, S.E. Pathogenic variant burden in the ExAC database: An empirical approach to evaluating population data for clinical variant interpretation. Genome Med. 2017, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Muller, R.D.; McDonald, T.; Pope, K.; Cragun, D. Evaluation of Clinical Practices Related to Variants of Uncertain Significance Results in Inherited Cardiac Arrhythmia and Inherited Cardiomyopathy Genes. Circ. Genom. Precis. Med. 2020, 13, e002789. [Google Scholar] [CrossRef] [PubMed]
- Briganti, F.; Sun, H.; Wei, W.; Wu, J.; Zhu, C.; Liss, M.; Karakikes, I.; Rego, S.; Cipriano, A.; Snyder, M.; et al. iPSC Modeling of RBM20-Deficient DCM Identifies Upregulation of RBM20 as a Therapeutic Strategy. Cell Rep. 2020, 32, 108117. [Google Scholar] [CrossRef]
- Brauch, K.M.; Karst, M.L.; Herron, K.J.; de Andrade, M.; Pellikka, P.A.; Rodeheffer, R.J.; Michels, V.V.; Olson, T.M. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll Cardiol 2009, 54, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Morales, A.; Gonzalez-Quintana, J.; Norton, N.; Siegfried, J.D.; Hofmeyer, M.; Hershberger, R.E. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin. Transl. Sci. 2010, 3, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Wells, Q.S.; Becker, J.R.; Su, Y.R.; Mosley, J.D.; Weeke, P.; D’Aoust, L.; Ausborn, N.L.; Ramirez, A.H.; Pfotenhauer, J.P.; Naftilan, A.J.; et al. Whole exome sequencing identifies a causal RBM20 mutation in a large pedigree with familial dilated cardiomyopathy. Circ. Cardiovasc Genet. 2013, 6, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Chami, N.; Tadros, R.; Lemarbre, F.; Lo, K.S.; Beaudoin, M.; Robb, L.; Labuda, D.; Tardif, J.C.; Racine, N.; Talajic, M.; et al. Nonsense mutations in BAG3 are associated with early-onset dilated cardiomyopathy in French Canadians. Can. J. Cardiol. 2014, 30, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Bouvagnet, P.; Chevalier, P.; Sebbag, L.; Dulac, A.; Dauphin, C.; Jouk, P.S.; Delrue, M.A.; Thambo, J.B.; Le Metayer, P.; et al. Clinical and mutational spectrum in a cohort of 105 unrelated patients with dilated cardiomyopathy. Eur. J. Med Genet. 2011, 54, e570–e575. [Google Scholar] [CrossRef] [PubMed]
- Refaat, M.M.; Lubitz, S.A.; Makino, S.; Islam, Z.; Frangiskakis, J.M.; Mehdi, H.; Gutmann, R.; Zhang, M.L.; Bloom, H.L.; MacRae, C.A.; et al. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm 2012, 9, 390–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaertner, A.; Klauke, B.; Brodehl, A.; Milting, H. RBM20 mutations in left ventricular non-compaction cardiomyopathy. Pediatr Investig. 2020, 4, 61–63. [Google Scholar] [CrossRef] [Green Version]
- Gaertner, A.; Klauke, B.; Felski, E.; Kassner, A.; Brodehl, A.; Gerdes, D.; Stanasiuk, C.; Ebbinghaus, H.; Schulz, U.; Dubowy, K.O.; et al. Cardiomyopathy-associated mutations in the RS domain affect nuclear localization of RBM20. Hum. Mutat 2020, 41, 1931–1943. [Google Scholar] [CrossRef]
- Streckfuss-Bomeke, K.; Tiburcy, M.; Fomin, A.; Luo, X.; Li, W.; Fischer, C.; Ozcelik, C.; Perrot, A.; Sossalla, S.; Haas, J.; et al. Severe DCM phenotype of patient harboring RBM20 mutation S635A can be modeled by patient-specific induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell Cardiol. 2017, 113, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebs, S.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Meder, B.; Streckfuss-Bomeke, K. Generation of pluripotent stem cell lines and CRISPR/Cas9 modified isogenic controls from a patient with dilated cardiomyopathy harboring a RBM20 p.R634W mutation. Stem Cell Res. 2020, 47, 101901. [Google Scholar] [CrossRef] [PubMed]
- Norton, N.; Siegfried, J.D.; Li, D.; Hershberger, R.E. Assessment of LMNA copy number variation in 58 probands with dilated cardiomyopathy. Clin. Transl. Sci. 2011, 4, 351–352. [Google Scholar] [CrossRef]
- Norton, N.; Li, D.; Rieder, M.J.; Siegfried, J.D.; Rampersaud, E.; Zuchner, S.; Mangos, S.; Gonzalez-Quintana, J.; Wang, L.; McGee, S.; et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Mates, J.; Mademont-Soler, I.; Del Olmo, B.; Ferrer-Costa, C.; Coll, M.; Perez-Serra, A.; Pico, F.; Allegue, C.; Fernandez-Falgueras, A.; Alvarez, P.; et al. Role of copy number variants in sudden cardiac death and related diseases: Genetic analysis and translation into clinical practice. Eur. J. Hum. Genet. 2018, 26, 1014–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mates, J.; Mademont-Soler, I.; Fernandez-Falgueras, A.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Garcia-Alvarez, A.; Jorda, P.; Toro, R.; Coll, M.; et al. Sudden Cardiac Death and Copy Number Variants: What Do We Know after 10 Years of Genetic Analysis? Forensic Sci. Int. Genet. 2020, 47, 102281. [Google Scholar] [CrossRef] [PubMed]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Amr, A.; Lai, A.; Haas, J.; Holzer, D.B.; Frese, K.S.; Keller, A.; Jensen, K.; Katus, H.A.; et al. Genotype-phenotype associations in dilated cardiomyopathy: Meta-analysis on more than 8000 individuals. Clin. Res. Cardiol. 2017, 106, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Hey, T.M.; Rasmussen, T.B.; Madsen, T.; Aagaard, M.M.; Harbo, M.; Molgaard, H.; Moller, J.E.; Eiskjaer, H.; Mogensen, J. Pathogenic RBM20-Variants Are Associated With a Severe Disease Expression in Male Patients With Dilated Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 Mutations Induce an Arrhythmogenic Dilated Cardiomyopathy Related to Disturbed Calcium Handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.B.; Fritsche, L.G.; Zhou, W.; Teslovich, T.M.; Holmen, O.L.; Gustafsson, S.; Gabrielsen, M.E.; Schmidt, E.M.; Beaumont, R.; Wolford, B.N.; et al. Genome-wide Study of Atrial Fibrillation Identifies Seven Risk Loci and Highlights Biological Pathways and Regulatory Elements Involved in Cardiac Development. Am. J. Hum. Genet. 2018, 102, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roselli, C.; Chaffin, M.D.; Weng, L.C.; Aeschbacher, S.; Ahlberg, G.; Albert, C.M.; Almgren, P.; Alonso, A.; Anderson, C.D.; Aragam, K.G.; et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat. Genet. 2018, 50, 1225–1233. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Nucleotide Change | Protein Change | dbSNP | gnomAD (MAF%) | ClinVar (Disease) | HGMD (Disease) | CC | ACMG Score | RBM20 Domain | Arrhythmogenic Phenotype |
|---|---|---|---|---|---|---|---|---|---|
| c.247C > A | p.(Leu83Ile) | rs536357058 | 1/155140 (0.0006%) | VUS (DCM) | CM1111132 | VUS | VUS | Exon 2 | Yes |
| (DM; DCM) | Leucine-rich region | ||||||||
| c.680G > T | p.(Gly227Val) | rs202238753 | 225/185204 (0.12%) | LB (DCM) | CM1821953 | LB | VUS | Exon 2 | No |
| (DM; DCM) | |||||||||
| c.769A > G | p.(Thr257Ala) | rs1418674149 | 1/153900 (0.0006%) | NA | CM1815813 | VUS | VUS | Exon 2 | Yes |
| (DM; DCM) | |||||||||
| c.1175G > A | p.(Arg392Gln) | rs751788298 | 3/185862 (0.0016%) | NA | CM1815814 | VUS | VUS | Exon 2 | NA |
| (DM; DCM) | |||||||||
| c.1364C > T | p.(Ser455Leu) | rs189569984 | 862/153884 (0.56%) | LB | NA | LB | LB | Exon 4 | No |
| c.1494C > A | p.(Ser498Arg) | rs774916799 | 2/153882 (0.0013%) | VUS (DCM) | CM1815816 | VUS | VUS | Exon 4 | Yes |
| (DM; DCM) | |||||||||
| c.1528-1G > C | - | rs534513476 | NA | NA | CS183215 | VUS | P | Intron 5–6 | Yes |
| (DM; DCM) | |||||||||
| c.1603G > A | p.(Val535Ile) | rs183007628 | 6/188686 (0.0031%) | VUS (DCM) | CM107458 | VUS | VUS | Exon 6 | Yes |
| (DM; DCM) | RNA Recognition Motif | ||||||||
| c.1760T > A | p.(Leu587His) | NA | NA | NA | CM1815817 | VUS | VUS | Exon 7 | Yes |
| (DM; DCM) | RNA Recognition Motif | ||||||||
| c.1764T > G | p.(Ile588Met) | NA | NA | NA | CM183216 | VUS | VUS | Exon 7 | Yes |
| (DM; DCM) | RNA Recognition Motif | ||||||||
| c.1880 + 4_1880 + 6dupAGG | - | rs1227694990 | 200/187706 (0.1%) | LB (DCM) | CI1516347 | VUS | VUS | Intron 7−8 | No |
| (DM; DCM) | |||||||||
| c.1898C > T | p.(Pro633Leu) | rs747880281 | 1/151498 (0.0006%) | VUS (DCM) | NA | VUS | P | Exon 9 | Yes |
| Arginine-Serine Domain | |||||||||
| c.1900C > T | p.(Arg634Trp) | NA | NA | NA | CM107456 | VUS | LP | Exon 9 | Yes |
| (DM; DCM) | Arginine-Serine Domain | ||||||||
| c.1901G > A | p.(Arg634Gln) | rs267607001 | 1/152378 (0.0006%) | P (DCM) | CM095004 | VUS | LP | Exon 9 | Yes |
| c.1901G > T | p.(Arg634Leu) | (DM; DCM) | Arginine-Serine Domain | ||||||
| c.1903T > G | p.(Ser635Ala) | NA | NA | NA | CM125867 | VUS | LP | Exon 9 | Yes |
| (DM; DCM) | Arginine-Serine Domain | ||||||||
| c.1906C > A | p.(Arg636Ser) | rs267607002 | NA | NA | CM095005 | LP | LP | Exon 9 | Yes |
| (DM; DCM) | Arginine-Serine Domain | ||||||||
| c.1906C > T | p.(Arg636Cys) | rs267607002 | NA | NA | CM107457 | LP | LP | Exon 9 | Yes |
| (DM; DCM) | Arginine-Serine Domain | ||||||||
| c.1907G > A | p.(Arg636His) | rs267607004 | NA | NA | CM095006 | VUS | LP | Exon 9 | Yes |
| (DM; DCM) | Arginine-Serine Domain | ||||||||
| c.1909A > G | p.(Ser637Gly) | rs267607005 | NA | NA | CM095007 | VUS | LP | Exon 9 | Yes |
| (DM; DCM) | Arginine-Serine Domain | ||||||||
| c.1913C > T | p.(Pro638Leu) | rs267607003 | NA | NA | CM095008 | VUS | LP | Exon 9 | Yes |
| (DM; DCM) | Arginine-Serine Domain | ||||||||
| c.1997G > A | p.(Arg666Gln) | rs202011408 | 5/154830 (0.003%) | NA | CM1716804 | VUS | VUS | Exon 9 | Yes |
| (DM; DCM) | Arginine-Serine Domain | ||||||||
| c.2021A > G | p.(Asp674Gly) | rs1475557145 | 1/155286 (0.0006%) | VUS (DCM) | NA | VUS | VUS | Exon 9 | NA |
| c.2042A > G | p.(Tyr681Cys) | rs372048968 | 23/186630 (0.01%) | VUS (DCM) | CM1815818 | LB | LB | Exon 9 | No |
| (DM; DCM) | |||||||||
| c.2062C > T | p.(Arg688Ter) | rs794729150 | 1/31344 (0.003%) | VUS (DCM) | CM1516720 | VUS | P | Exon 9 | Yes |
| (DM; DCM) | |||||||||
| c.2109G > C | p.(Arg703Ser) | rs988797559 | 2/186026 (0.001%) | NA | CM1111134 | VUS | VUS | Exon 9 | Yes |
| (DM; DCM) | |||||||||
| c.2147G > A | p.(Arg716Gln) | rs375798246 | 21/155108 (0.013%) | VUS (DCM) | NA | LB | LB | Exon 9 | No |
| c.2282G > A | p.(Arg761Gln) | rs556897484 | 4/156496 (0.002%) | NA | NA | VUS | VUS | Exon 9 | NA |
| c.2662G > A | p.(Asp888Asn) | rs201370621 | 603/155726 (0.3%) | VUS (DCM) | NA | LB | LB | Exon 11 | No |
| c.2737G > A | p.(Glu913Lys) | rs397516607 | NA | LP (DCM) | NA | LP | LP | Exon 11 | Yes |
| c.2741T > C | p.(Val914Ala) | rs794729154 | NA | NA | NA | VUS | VUS | Exon 11 | Yes |
| c.2714T > A | p.(Met950Lys) | NA | NA | NA | NA | VUS | VUS | Exon 11 | NA |
| c.3091G > T | p.(Gly1031Ter) | rs794729157 | NA | NA | CM1111136 | VUS | P | Exon 11 | Yes |
| (DM; DCM) | |||||||||
| c.3115C > T | p.(Pro1039Ser) | rs727503392 | 40/188260 (0.02%) | LB (DCM) | CM1815819 | VUS | LB | Exon 11 | No |
| (DM; DCM) | |||||||||
| c.3242C > G | p.(Pro1081Arg) | rs1268330519 | NA | NA | CM1111137 | VUS | VUS | Exon 12 | Yes |
| (DM; DCM) | |||||||||
| c.3545G > A | p.(Arg1182His) | rs563762318 | 47/185298 (0.025%) | LB (DCM) | CM1510988 | VUS | VUS | Exon 12 | Yes |
| (DM; DCM) | Zinc Finger domain 2 | ||||||||
| c.3616G > A | p.(Glu1206Lys) | rs757389650 | 8/181254 (0.004%) | VUS (DCM) | CM1111138 | VUS | VUS | Exon 14 | NA |
| (DM; DCM) |
| Brauch et al., 2009 (n = 39, DCM) NC | Li et al., 2010 (n = 16, DCM) NC | Refaat et al., 2012 (n = 8, DCM) | Wells et al., 2013 (n = 19 carriers) NC | Van den Hoogenhof et al., 2018 (n = 18, DCM) | Hey et al., 2019 (n = 53, DCM) | Parikh et al., 2019 (n = 74, carriers) | |
|---|---|---|---|---|---|---|---|
| Age diagnosis | 36 ± 13.2 | 37.6 ± 9 | - | 33.8 ± 11.5 | 42 ± 14 | 37 ± 15 & | 37 ± 15 † |
| Males | 19 (49%) | 8 (50%) | 4 (50%) | 14 (82%) | 8 (44%) | 31 (58%) | - |
| Follow-up (months) | 60 (12−204) | - | 27.4 ± 15.7 | - | 71 ± 65 | 86 (24−150) | - |
| Mean LVEF | 35.3 ± 11.5 | 29.3 ± 8.6 | - | 48.8 ± 13 | 37 ± 17 | 32 ± 12 && | 40 ± 17 |
| FH SCD | 39 (100%) | - | - | - | 13 (72%) | - | 22/43 (51%) †† |
| NSVT | - | 1 (6%) | - | - | 5 (28%) | - | 21/59 (36%) |
| Sustained VT or VF | 9 (23%) | 1 (6%) | 0 | - | 8 (44%) ¶ | 11 (21%) &&& | - |
| ICD therapy | - | 0 | 1 (12.5%) | - | - | - | 9/32 (28%) ††† |
| SCD | 3 (7.7%) | 1 (6%) | 0 | - | - | 6 (11.3%) &&& | 5/60 (8%)-SCA ††† |
| AF | 3 (7.7%) | 2 (12.5%) | 3 (37.5%) * | 6 (33%) | - | 10/58 (17%) †††† | |
| HTx | 4 (mean age 28.5) | 2 (12.5%) | 1 (12.5%) | 1 (5.2%, 17 years old) + | - | 11 (21%) &&&& | 5/74 (7%) NC |
| Death | 11 (28%, mean age 45): 4 HF (mean age 54.7), 3 SCD (mean age 39) | 3 (11.5%) | 0 | 11 (57.9%) + | - | 2 (4%, end-stage HF at 54 and 73 years old) | 3/74 (4%) NC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jordà, P.; Toro, R.; Diez, C.; Salazar-Mendiguchía, J.; Fernandez-Falgueras, A.; Perez-Serra, A.; Coll, M.; Puigmulé, M.; Arbelo, E.; García-Álvarez, A.; et al. Malignant Arrhythmogenic Role Associated with RBM20: A Comprehensive Interpretation Focused on a Personalized Approach. J. Pers. Med. 2021, 11, 130. https://doi.org/10.3390/jpm11020130
Jordà P, Toro R, Diez C, Salazar-Mendiguchía J, Fernandez-Falgueras A, Perez-Serra A, Coll M, Puigmulé M, Arbelo E, García-Álvarez A, et al. Malignant Arrhythmogenic Role Associated with RBM20: A Comprehensive Interpretation Focused on a Personalized Approach. Journal of Personalized Medicine. 2021; 11(2):130. https://doi.org/10.3390/jpm11020130
Chicago/Turabian StyleJordà, Paloma, Rocío Toro, Carles Diez, Joel Salazar-Mendiguchía, Anna Fernandez-Falgueras, Alexandra Perez-Serra, Monica Coll, Marta Puigmulé, Elena Arbelo, Ana García-Álvarez, and et al. 2021. "Malignant Arrhythmogenic Role Associated with RBM20: A Comprehensive Interpretation Focused on a Personalized Approach" Journal of Personalized Medicine 11, no. 2: 130. https://doi.org/10.3390/jpm11020130