
Horses for Courses in the Era of CARs: Advancing CAR T and CAR NK Cell Therapies

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Lymphocyte Heterogeneity
3. Cytokines
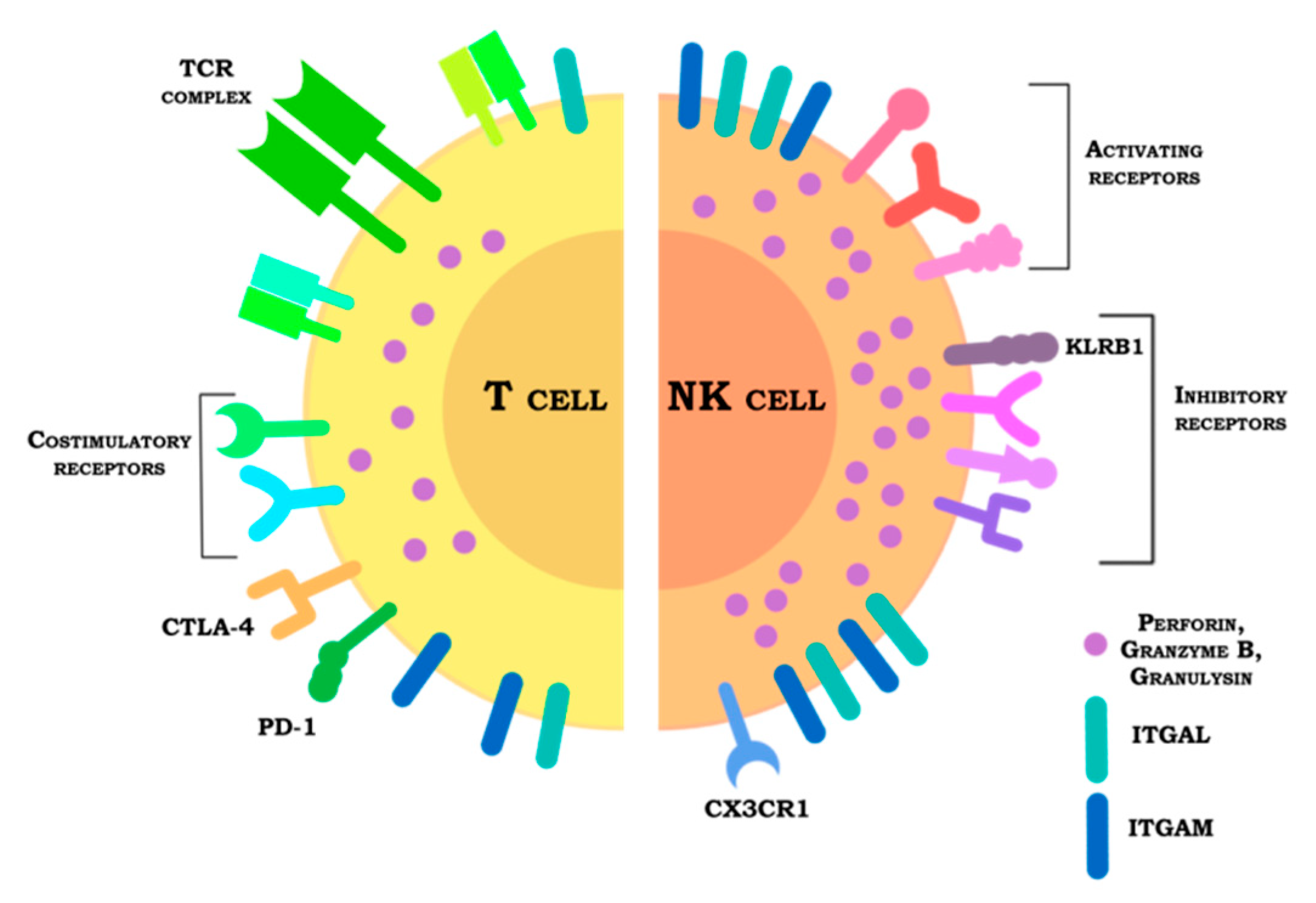
4. Receptors
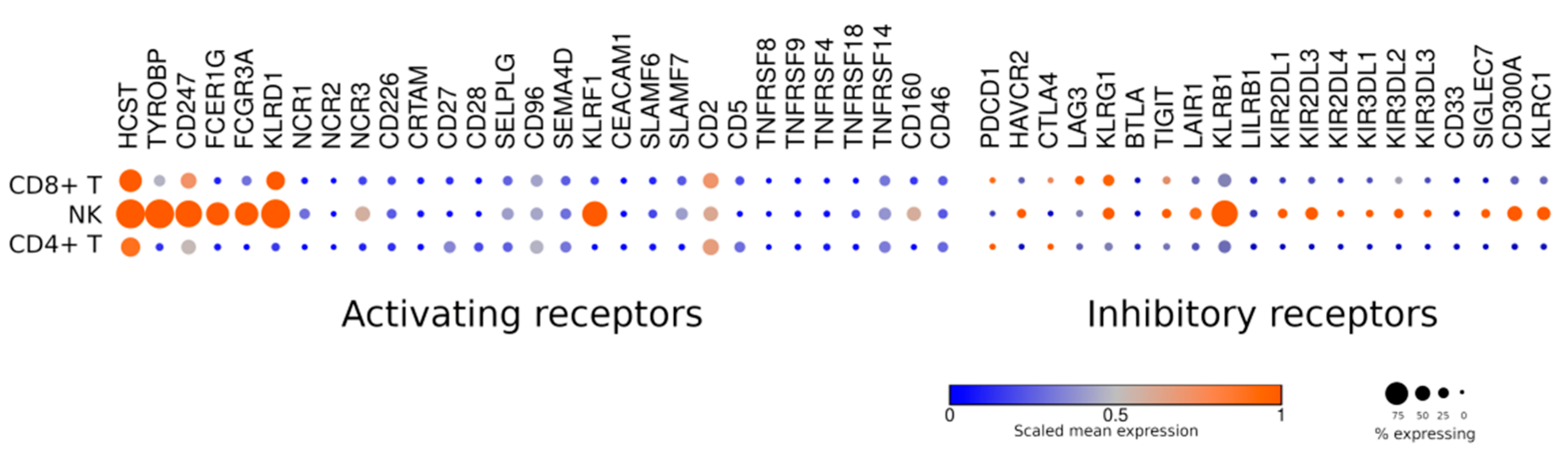
4.1. Inhibitory Receptors
4.2. Activating Receptors
5. Immune Synapse and Cytotoxicity
6. Hypoxia
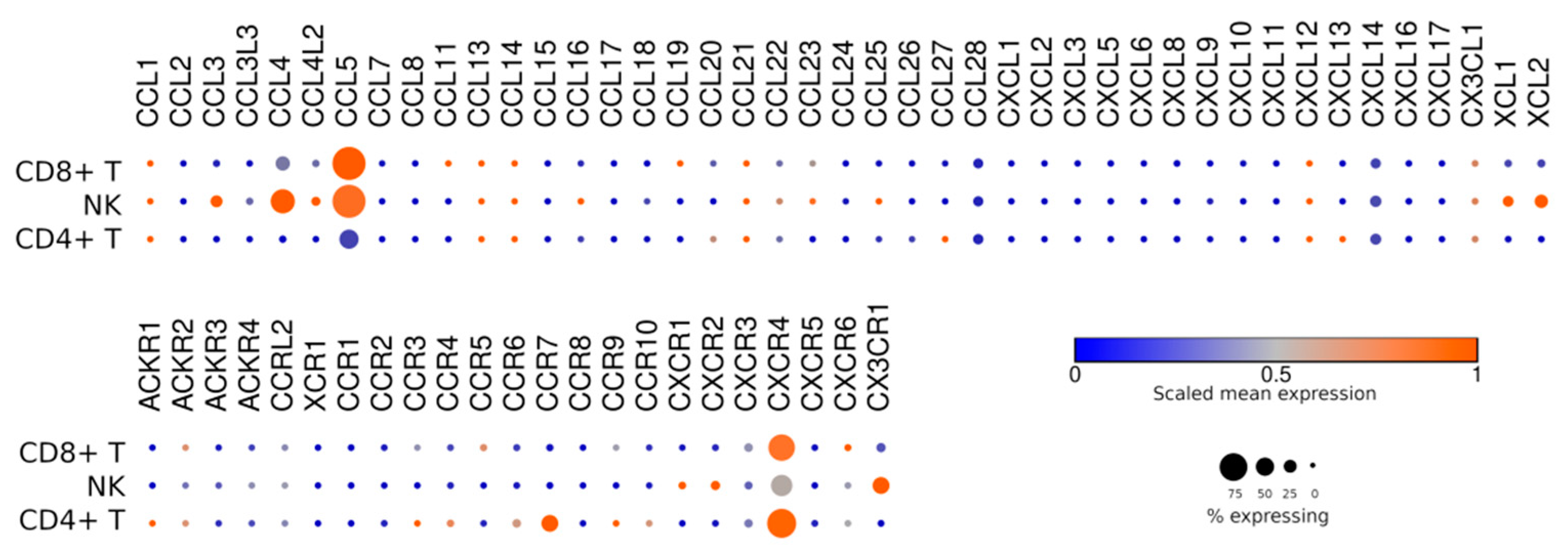
7. Homing and Taxis
8. Manufacturing of CAR T and CAR NK Products
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Adami, A.; Maher, J. An overview of CAR T-cell clinical trial activity to 2021. Immunother. Adv. 2021, 1, ltab004. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Basar, R.; Daher, M.; Rezvani, K. Next-generation cell therapies: The emerging role of CAR-NK cells. Blood Adv. 2020, 4, 5868–5876. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jiang, J.; Wu, C. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. 2020, 472, 175–180. [Google Scholar] [CrossRef]
- Moeller, M.; Haynes, N.M.; Kershaw, M.H.; Jackson, J.T.; Teng, M.W.L.; Street, S.E.; Cerutti, L.; Jane, S.M.; Trapani, J.A.; Smyth, M.J.; et al. Adoptive transfer of gene-engineered CD4+ helper T cells induces potent primary and secondary tumor rejection. Blood 2005, 106, 2995–3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeller, M.; Kershaw, M.H.; Cameron, R.; Westwood, J.A.; Trapani, J.A.; Smyth, M.J.; Darcy, P.K. Sustained antigen-specific antitumor recall response mediated by gene-modified CD4+ T helper-1 and CD8+ T cells. Cancer Res. 2007, 67, 11428–11437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudecek, M.; Lupo-Stanghellini, M.-T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [Green Version]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Sabatino, M.; Hu, J.; Sommariva, M.; Gautam, S.; Fellowes, V.; Hocker, J.D.; Dougherty, S.; Qin, H.; Klebanoff, C.A.; Fry, T.J.; et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood 2016, 128, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, A.; Strauss-Albee, D.M.; Leipold, M.; Kubo, J.; Nemat-Gorgani, N.; Dogan, O.C.; Dekker, C.L.; Mackey, S.; Maecker, H.; Swan, G.E.; et al. Genetic and environmental determinants of human NK cell diversity revealed by mass cytometry. Sci. Transl. Med. 2013, 5, 208ra145. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef]
- Singh, N.; Hofmann, T.J.; Gershenson, Z.; Levine, B.L.; Grupp, S.A.; Teachey, D.T.; Barrett, D.M. Monocyte lineage-derived IL-6 does not affect chimeric antigen receptor T-cell function. Cytotherapy 2017, 19, 867–880. [Google Scholar] [CrossRef]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M.; et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 2019, 133, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-D.; Bae, S.-Y.; Hong, J.-W.; Azam, T.; Dinarello, C.A.; Her, E.; Choi, W.-S.; Kim, B.-K.; Lee, C.-K.; Yoon, D.-Y.; et al. Identification of the most active interleukin-32 isoform. Immunology 2009, 126, 535–542. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.M.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobie, C.; Skropeta, D. Insights into the role of sialylation in cancer progression and metastasis. Br. J. Cancer 2021, 124, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Muccio, L.; Bertaina, A.; Falco, M.; Pende, D.; Meazza, R.; Lopez-Botet, M.; Moretta, L.; Locatelli, F.; Moretta, A.; Della Chiesa, M. Analysis of memory-like natural killer cells in human cytomegalovirus-infected children undergoing αβ+T and B cell-depleted hematopoietic stem cell transplantation for hematological malignancies. Haematologica 2016, 101, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, A.; Asahina, Y.; Chang, L.-Y.; Angata, T.; Tanaka, H.; Kitajima, K.; Sato, C. Identification and functional characterization of a Siglec-7 counter-receptor on K562 cells. J. Biol. Chem. 2021, 296, 100477. [Google Scholar] [CrossRef] [PubMed]
- Rygiel, T.P.; Stolte, E.H.; de Ruiter, T.; van de Weijer, M.L.; Meyaard, L. Tumor-expressed collagens can modulate immune cell function through the inhibitory collagen receptor LAIR-1. Mol. Immunol. 2011, 49, 402–406. [Google Scholar] [CrossRef]
- Lankry, D.; Rovis, T.L.; Jonjic, S.; Mandelboim, O. The interaction between CD300a and phosphatidylserine inhibits tumor cell killing by NK cells. Eur. J. Immunol. 2013, 43, 2151–2161. [Google Scholar] [CrossRef] [Green Version]
- Montaldo, E.; Vitale, C.; Cottalasso, F.; Conte, R.; Glatzer, T.; Ambrosini, P.; Moretta, L.; Mingari, M.C. Human NK cells at early stages of differentiation produce CXCL8 and express CD161 molecule that functions as an activating receptor. Blood 2012, 119, 3987–3996. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.B.; Bettadapura, J.; Alsharifi, M.; Mathew, P.A.; Warren, H.S.; Lanier, L.L. Cutting edge: Lectin-like transcript-1 is a ligand for the inhibitory human NKR-P1A receptor. J. Immunol. 2005, 175, 7796–7799. [Google Scholar] [CrossRef]
- Marrufo, A.M.; Mathew, S.O.; Chaudhary, P.; Malaer, J.D.; Vishwanatha, J.K.; Mathew, P.A. Blocking LLT1 (CLEC2D, OCIL)-NKRP1A (CD161) interaction enhances natural killer cell-mediated lysis of triple-negative breast cancer cells. Am. J. Cancer Res. 2018, 8, 1050–1063. [Google Scholar]
- Mathew, S.O.; Chaudhary, P.; Powers, S.B.; Vishwanatha, J.K.; Mathew, P.A. Overexpression of LLT1 (OCIL, CLEC2D) on prostate cancer cells inhibits NK cell-mediated killing through LLT1-NKRP1A (CD161) interaction. Oncotarget 2016, 7, 68650–68661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front. Immunol. 2019, 10, 3038. [Google Scholar] [CrossRef] [PubMed]
- Koshy, S.; Wu, D.; Hu, X.; Tajhya, R.B.; Huq, R.; Khan, F.S.; Pennington, M.W.; Wulff, H.; Yotnda, P.; Beeton, C. Blocking KCa3.1 channels increases tumor cell killing by a subpopulation of human natural killer lymphocytes. PLoS ONE 2013, 8, e76740. [Google Scholar] [CrossRef]
- Bonavita, E.; Bromley, C.P.; Jonsson, G.; Pelly, V.S.; Sahoo, S.; Walwyn-Brown, K.; Mensurado, S.; Moeini, A.; Flanagan, E.; Bell, C.R.; et al. Antagonistic inflammatory phenotypes dictate tumor fate and response to immune checkpoint blockade. Immunity 2020, 53, 1215–1229.e8. [Google Scholar] [CrossRef] [PubMed]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Masoumi, E.; Jafarzadeh, L.; Mirzaei, H.R.; Alishah, K.; Fallah-Mehrjardi, K.; Rostamian, H.; Khakpoor-Koosheh, M.; Meshkani, R.; Noorbakhsh, F.; Hadjati, J. Genetic and pharmacological targeting of A2a receptor improves function of anti-mesothelin CAR T cells. J. Exp. Clin. Cancer Res. 2020, 39, 49. [Google Scholar] [CrossRef]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.-F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef]
- Burga, R.A.; Yvon, E.; Chorvinsky, E.; Fernandes, R.; Cruz, C.R.Y.; Bollard, C.M. Engineering the tgfβ receptor to enhance the therapeutic potential of natural killer cells as an immunotherapy for neuroblastoma. Clin. Cancer Res. 2019, 25, 4400–4412. [Google Scholar] [CrossRef] [Green Version]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.-M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, N.; Michen, S.; Tietze, S.; Töpfer, K.; Schulte, A.; Lamszus, K.; Schmitz, M.; Schackert, G.; Pastan, I.; Temme, A. Engineering NK Cells Modified With an EGFRvIII-specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1α-secreting Glioblastoma. J. Immunother. 2015, 38, 197–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Töpfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Müller, N.; Lindemann, D.; Bachmann, M.; Füssel, M.; Schackert, G.; Temme, A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wulfing, C.; Purtic, B.; Klem, J.; Schatzle, J.D. Stepwise cytoskeletal polarization as a series of checkpoints in innate but not adaptive cytolytic killing. Proc. Natl. Acad. Sci. USA 2003, 100, 7767–7772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orange, J.S. Formation and function of the lytic NK-cell immunological synapse. Nat. Rev. Immunol. 2008, 8, 713–725. [Google Scholar] [CrossRef] [Green Version]
- Bengsch, B.; Ohtani, T.; Herati, R.S.; Bovenschen, N.; Chang, K.-M.; Wherry, E.J. Deep immune profiling by mass cytometry links human T and NK cell differentiation and cytotoxic molecule expression patterns. J. Immunol. Methods 2018, 453, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sutton, V.R.; Sedelies, K.; Dewson, G.; Christensen, M.E.; Bird, P.I.; Johnstone, R.W.; Kluck, R.M.; Trapani, J.A.; Waterhouse, N.J. Granzyme B triggers a prolonged pressure to die in Bcl-2 overexpressing cells, defining a window of opportunity for effective treatment with ABT-737. Cell Death Dis. 2012, 3, e344. [Google Scholar] [CrossRef]
- Purbhoo, M.A.; Irvine, D.J.; Huppa, J.B.; Davis, M.M. T cell killing does not require the formation of a stable mature immunological synapse. Nat. Immunol. 2004, 5, 524–530. [Google Scholar] [CrossRef]
- Wiedemann, A.; Depoil, D.; Faroudi, M.; Valitutti, S. Cytotoxic T lymphocytes kill multiple targets simultaneously via spatiotemporal uncoupling of lytic and stimulatory synapses. Proc. Natl. Acad. Sci. USA 2006, 103, 10985–10990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, Y.; Kawasaki-Koyanagi, A.; Sueyoshi, N.; Kanai, A.; Yagita, H.; Okumura, K. Localization of Fas ligand in cytoplasmic granules of CD8+ cytotoxic T lymphocytes and natural killer cells: Participation of Fas ligand in granule exocytosis model of cytotoxicity. Biochem. Biophys. Res. Commun. 2002, 296, 328–336. [Google Scholar] [CrossRef]
- Zamai, L.; Ahmad, M.; Bennett, I.M.; Azzoni, L.; Alnemri, E.S.; Perussia, B. Natural killer (NK) cell-mediated cytotoxicity: Differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J. Exp. Med. 1998, 188, 2375–2380. [Google Scholar] [CrossRef] [PubMed]
- Ang, S.; Silva, M.L.D.; Dawson, M.; Figliola, M.; Maiti, S.; Huls, H.; Shpall, E.J.; Champlin, R.; Lee, D.A.; Silla, L.; et al. NK cell proliferation and cytolytic function are compromised in the hypoxic tumor microenvironment. Blood 2010, 116, 4291. [Google Scholar] [CrossRef]
- Sarkar, S.; Germeraad, W.T.V.; Rouschop, K.M.A.; Steeghs, E.M.P.; van Gelder, M.; Bos, G.M.J.; Wieten, L. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef]
- Hamann, I.; Unterwalder, N.; Cardona, A.E.; Meisel, C.; Zipp, F.; Ransohoff, R.M.; Infante-Duarte, C. Analyses of phenotypic and functional characteristics of CX3CR1-expressing natural killer cells. Immunology 2011, 133, 62–73. [Google Scholar] [CrossRef]
- Hertwig, L.; Hamann, I.; Romero-Suarez, S.; Millward, J.M.; Pietrek, R.; Chanvillard, C.; Stuis, H.; Pollok, K.; Ransohoff, R.M.; Cardona, A.E.; et al. CX3CR1-dependent recruitment of mature NK cells into the central nervous system contributes to control autoimmune neuroinflammation. Eur. J. Immunol. 2016, 46, 1984–1996. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, I.; Erreni, M.; van Brakel, M.; Debets, R.; Allavena, P. Enhanced recruitment of genetically modified CX3CR1-positive human T cells into Fractalkine/CX3CL1 expressing tumors: Importance of the chemokine gradient. J. Immunother. Cancer 2016, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Bari, R.; Granzin, M.; Tsang, K.S.; Roy, A.; Krueger, W.; Orentas, R.; Schneider, D.; Pfeifer, R.; Moeker, N.; Verhoeyen, E.; et al. A Distinct Subset of Highly Proliferative and Lentiviral Vector (LV)-Transducible NK Cells Define a Readily Engineered Subset for Adoptive Cellular Therapy. Front. Immunol. 2019, 10, 2001. [Google Scholar] [CrossRef] [Green Version]
- Kremer, V.; Ligtenberg, M.A.; Zendehdel, R.; Seitz, C.; Duivenvoorden, A.; Wennerberg, E.; Colón, E.; Scherman-Plogell, A.-H.; Lundqvist, A. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J. Immunother. Cancer 2017, 5, 73. [Google Scholar] [CrossRef]
- Idorn, M.; Skadborg, S.K.; Kellermann, L.; Halldórsdóttir, H.R.; Holmen Olofsson, G.; Met, Ö.; Thor Straten, P. Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model. Oncoimmunology 2018, 7, e1450715. [Google Scholar] [CrossRef]
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin αvβ6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef] [Green Version]
- Wennerberg, E.; Pfefferle, A.; Ekblad, L.; Yoshimoto, Y.; Kremer, V.; Kaminskyy, V.O.; Juhlin, C.C.; Höög, A.; Bodin, I.; Svjatoha, V.; et al. Human anaplastic thyroid carcinoma cells are sensitive to NK cell-mediated lysis via ULBP2/5/6 and chemoattract NK cells. Clin. Cancer Res. 2014, 20, 5733–5744. [Google Scholar] [CrossRef] [Green Version]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.-C.S.; Kapoor, V.; Predina, J.; Powell, D.J.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonanni, V.; Antonangeli, F.; Santoni, A.; Bernardini, G. Targeting of CXCR3 improves anti-myeloma efficacy of adoptively transferred activated natural killer cells. J. Immunother. Cancer 2019, 7, 290. [Google Scholar] [CrossRef] [Green Version]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef] [Green Version]
- Bachier, C.; Borthakur, G.; Hosing, C.; Blum, W.; Rotta, M.; Ojeras, P.; Barnett, B.; Rajangam, K.; Majhail, N.S.; Nikiforow, S. A Phase 1 Study of NKX101, an Allogeneic CAR Natural Killer (NK) Cell Therapy, in Subjects with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML) or Higher-Risk Myelodysplastic Syndrome (MDS). Blood 2020, 136, 42–43. [Google Scholar] [CrossRef]
- Goodridge, J.P.; Mahmood, S.; Zhu, H.; Gaidarova, S.; Blum, R.; Bjordahl, R.; Cichocki, F.; Chu, H.; Bonello, G.; Lee, T.; et al. FT596: Translation of First-of-Kind Multi-Antigen Targeted Off-the-Shelf CAR-NK Cell with Engineered Persistence for the Treatment of B Cell Malignancies. Blood 2019, 134, 301. [Google Scholar] [CrossRef]
- Bjordahl, R.; Gaidarova, S.; Goodridge, J.P.; Mahmood, S.; Bonello, G.; Robinson, M.; Ruller, C.; Pribadi, M.; Lee, T.; Abujarour, R.; et al. FT576: A Novel Multiplexed Engineered Off-the-Shelf Natural Killer Cell Immunotherapy for the Dual-Targeting of CD38 and Bcma for the Treatment of Multiple Myeloma. Blood 2019, 134, 3214. [Google Scholar] [CrossRef]
- Sutlu, T.; Nyström, S.; Gilljam, M.; Stellan, B.; Applequist, S.E.; Alici, E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: Implications for gene therapy. Hum. Gene Ther. 2012, 23, 1090–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colamartino, A.B.L.; Lemieux, W.; Bifsha, P.; Nicoletti, S.; Chakravarti, N.; Sanz, J.; Roméro, H.; Selleri, S.; Béland, K.; Guiot, M.; et al. Efficient and Robust NK-Cell Transduction With Baboon Envelope Pseudotyped Lentivector. Front. Immunol. 2019, 10, 2873. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Iwamoto, S.; Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005, 106, 376–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomeroy, E.J.; Lahr, W.S.; Chang, J.W.; Krueger, J.; Wick, B.J.; Slipek, N.J.; Skeate, J.G.; Webber, B.R.; Moriarity, B.S. Non-Viral Engineering of CAR-NK and CAR-T cells using the Tc Buster Transposon SystemTM. BioRxiv 2021. [Google Scholar] [CrossRef]
- Robbins, G.M.; Wang, M.; Pomeroy, E.J.; Moriarity, B.S. Nonviral genome engineering of natural killer cells. Stem Cell Res. Ther. 2021, 12, 350. [Google Scholar] [CrossRef]
- Fujisaki, H.; Kakuda, H.; Shimasaki, N.; Imai, C.; Ma, J.; Lockey, T.; Eldridge, P.; Leung, W.H.; Campana, D. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009, 69, 4010–4017. [Google Scholar] [CrossRef] [Green Version]
- Denman, C.J.; Senyukov, V.V.; Somanchi, S.S.; Phatarpekar, P.V.; Kopp, L.M.; Johnson, J.L.; Singh, H.; Hurton, L.; Maiti, S.N.; Huls, M.H.; et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE 2012, 7, e30264. [Google Scholar] [CrossRef]
- Min, B.; Choi, H.; Her, J.H.; Jung, M.Y.; Kim, H.-J.; Jung, M.-Y.; Lee, E.-K.; Cho, S.Y.; Hwang, Y.K.; Shin, E.-C. Optimization of Large-Scale Expansion and Cryopreservation of Human Natural Killer Cells for Anti-Tumor Therapy. Immune Netw. 2018, 18, e31. [Google Scholar] [CrossRef]
- Oh, E.; Min, B.; Li, Y.; Lian, C.; Hong, J.; Park, G.-M.; Yang, B.; Cho, S.Y.; Hwang, Y.K.; Yun, C.-O. Cryopreserved Human Natural Killer Cells Exhibit Potent Antitumor Efficacy against Orthotopic Pancreatic Cancer through Efficient Tumor-Homing and Cytolytic Ability (Running Title: Cryopreserved NK Cells Exhibit Antitumor Effect). Cancers 2019, 11, 966. [Google Scholar] [CrossRef] [Green Version]
- Mark, C.; Czerwinski, T.; Roessner, S.; Mainka, A.; Hörsch, F.; Heublein, L.; Winterl, A.; Sanokowski, S.; Richter, S.; Bauer, N.; et al. Cryopreservation impairs 3-D migration and cytotoxicity of natural killer cells. Nat. Commun. 2020, 11, 5224. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Good, Z.; Belk, J.A.; Daniel, B.; Klysz, D.; et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 2021, 372, eaba1786. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wu, X.; Chan, I.H.; Trager, J.B. Abstract 4235: A combination of CAR-NK and CAR-T cells results in rapid and persistent anti-tumor efficacy while reducing CAR-T cell mediated cytokine release and T-cell proliferation. In Clinical Research (Excluding Clinical Trials); American Association for Cancer Research: Philadelphia, PA, USA, 2020; p. 4235. [Google Scholar]
- Bachiller, M.; Perez-Amill, L.; Battram, A.M.; Carné, S.C.; Najjar, A.; Verhoeyen, E.; Juan, M.; Urbano-Ispizua, A.; Martin-Antonio, B. NK cells enhance CAR-T cell antitumor efficacy by enhancing immune/tumor cells cluster formation and improving CAR-T cell fitness. J. Immunother. Cancer 2021, 9, e002866. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulemzin, S.; Evsyukov, I.; Belovezhets, T.; Taranin, A.; Gorchakov, A. Horses for Courses in the Era of CARs: Advancing CAR T and CAR NK Cell Therapies. J. Pers. Med. 2021, 11, 1182. https://doi.org/10.3390/jpm11111182
Kulemzin S, Evsyukov I, Belovezhets T, Taranin A, Gorchakov A. Horses for Courses in the Era of CARs: Advancing CAR T and CAR NK Cell Therapies. Journal of Personalized Medicine. 2021; 11(11):1182. https://doi.org/10.3390/jpm11111182
Chicago/Turabian StyleKulemzin, Sergey, Igor Evsyukov, Tatiana Belovezhets, Alexander Taranin, and Andrey Gorchakov. 2021. "Horses for Courses in the Era of CARs: Advancing CAR T and CAR NK Cell Therapies" Journal of Personalized Medicine 11, no. 11: 1182. https://doi.org/10.3390/jpm11111182