
Polygenic Risk Scores Contribute to Personalized Medicine of Parkinson’s Disease


Abstract
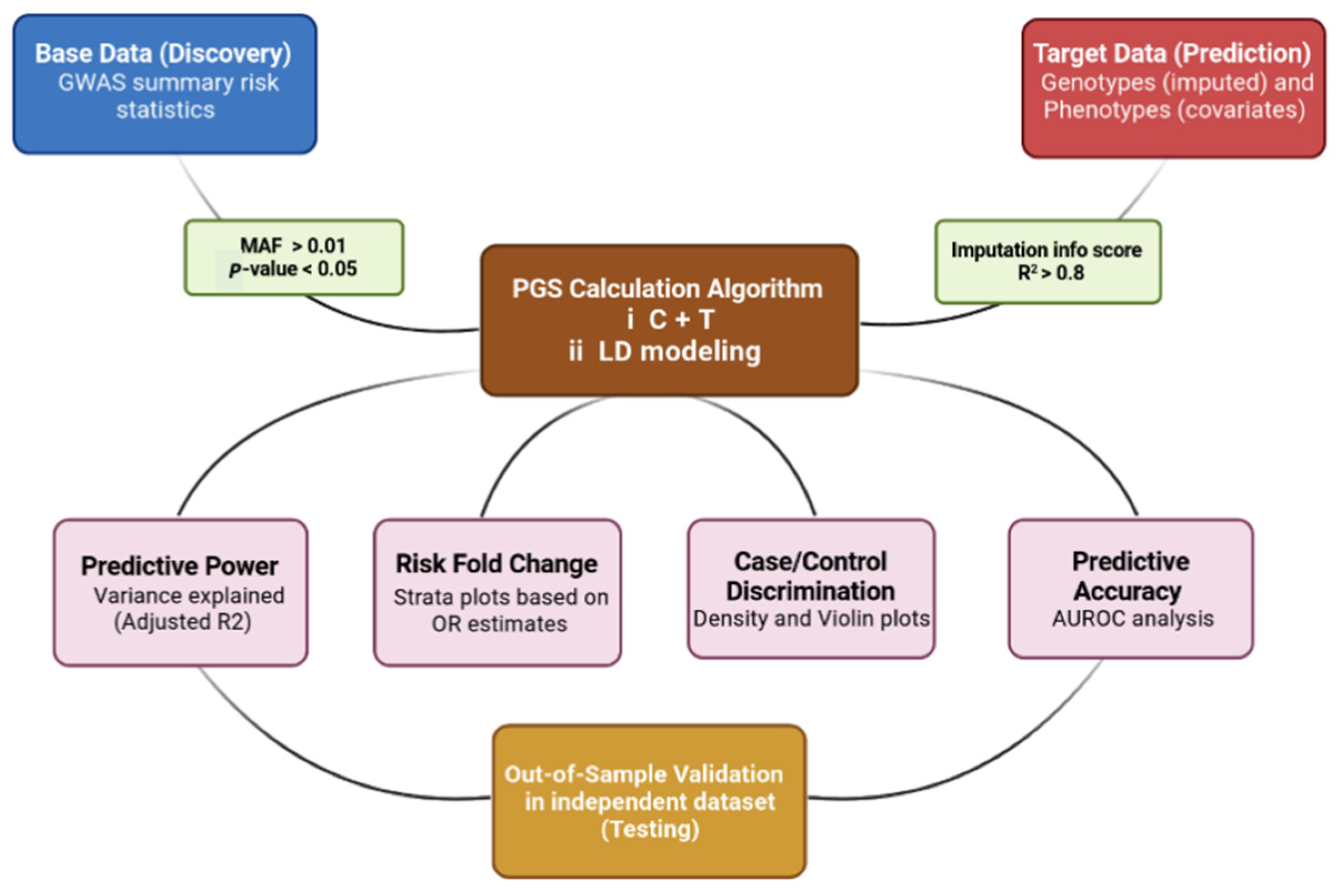
:1. Introduction
2. PRS Calculation and Data Interpretation
2.1. PRS in PD Status Prediction
2.2. PRS and PD Clinical Outcomes
2.3. PRS and Penetrance of LRRK2 and GBA
2.4. PRS and Biomarkers
2.5. PRS in the Identification of Biological Pathways
2.6. PRS for the Establishment of Stratified PD Trials
2.7. Translating PRS onto the Absolute Scale
3. Future Directions and Limitations
Author Contributions
Funding
Conflicts of Interest
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 1–21. [Google Scholar] [CrossRef]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef] [PubMed]
- Lohmueller, K.E.; Pearce, C.L.; Pike, M.; Lander, E.S.; Hirschhorn, J.N. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat. Genet. 2003, 33, 177–182. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Bloem, B.R. The Parkinson pandemic—A call to action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef]
- Von Linstow, C.U.; Gan-Or, Z.; Brundin, P. Precision medicine in Parkinson’s disease patients with LRRK2 and GBA risk variants—Let’s get even more personal. Transl. Neurodegener. 2020, 9, 39. [Google Scholar] [CrossRef]
- Ryden, L.E.; Lewis, S.J. Parkinson’s disease in the era of personalised medicine: One size does not fit all. Drugs Aging 2019, 36, 103–113. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, K.C.; Schulz, J.; Bronstein, J.M.; Lill, C.M.; Ritz, B.R. Association of polygenic risk score with cognitive decline and motor progression in Parkinson disease. JAMA Neurol. 2018, 75, 360–366. [Google Scholar] [CrossRef]
- Han, Y.; Teeple, E.; Shankara, S.; Sadeghi, M.; Zhu, C.; Liu, D.; Wang, C.; Frau, F.; Klinger, K.; Madden, S. Genome-Wide Polygenic Risk Score Identifies Individuals at Elevated Parkinson’s Disease Risk (4286). Neurology 2021, 96, 13. [Google Scholar]
- Nica, A.C.; Dermitzakis, E.T. Expression quantitative trait loci: Present and future. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120362. [Google Scholar] [CrossRef]
- Guo, J.; Wu, Y.; Zhu, Z.; Zheng, Z.; Trzaskowski, M.; Zeng, J.; Robinson, M.R.; Visscher, P.M.; Yang, J. Global genetic differentiation of complex traits shaped by natural selection in humans. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Euesden, J.; Lewis, C.M.; O’Reilly, P.F. PRSice: Polygenic risk score software. Bioinformatics 2015, 31, 1466–1468. [Google Scholar] [CrossRef] [Green Version]
- Vilhjálmsson, B.J.; Yang, J.; Finucane, H.K.; Gusev, A.; Lindström, S.; Ripke, S.; Genovese, G.; Loh, P.-R.; Bhatia, G.; Do, R. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet. 2015, 97, 576–592. [Google Scholar] [CrossRef] [Green Version]
- Ge, T.; Chen, C.-Y.; Ni, Y.; Feng, Y.-C.A.; Smoller, J.W. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 2019, 10, 1–10. [Google Scholar]
- Newcombe, P.J.; Nelson, C.P.; Samani, N.J.; Dudbridge, F. A flexible and parallelizable approach to genome-wide polygenic risk scores. Genet. Epidemiol. 2019, 43, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Mak, T.S.H.; Porsch, R.M.; Choi, S.W.; Zhou, X.; Sham, P.C. Polygenic scores via penalized regression on summary statistics. Genet. Epidemiol. 2017, 41, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Meisner, A.; Kundu, P.; Zhang, Y.D.; Lan, L.V.; Kim, S.; Ghandwani, D.; Choudhury, P.P.; Berndt, S.I.; Freedman, N.D.; Garcia-Closas, M. Combined utility of 25 disease and risk factor polygenic risk scores for stratifying risk of all-cause mortality. Am. J. Hum. Genet. 2020, 107, 418–431. [Google Scholar] [CrossRef]
- Viechtbauer, W.; Cheung, M.W.L. Outlier and influence diagnostics for meta-analysis. Res. Synth. Methods 2010, 1, 112–125. [Google Scholar] [CrossRef]
- Choi, S.W.; Mak, T.S.-H.; O’Reilly, P.F. Tutorial: A guide to performing polygenic risk score analyses. Nat. Protoc. 2020, 15, 2759–2772. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Saez-Atienzar, S.; Kim, J.; Makarious, M.; Faghri, F.; Diez-Fairen, M.; Iwaki, H.; Leonard, H.; Botia, J.; Ryten, M. Large-scale pathway specific polygenic risk and transcriptomic community network analysis identifies novel functional pathways in Parkinson disease. Acta. Neuropathol. 2020, 140, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ling, C.X. Using AUC and accuracy in evaluating learning algorithms. IEEE Trans. Knowl. Data Eng. 2005, 17, 299–310. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.W.; O’Reilly, P.F. PRSice-2: Polygenic Risk Score software for biobank-scale data. Gigascience 2019, 8, giz082. [Google Scholar] [CrossRef]
- Ibanez, L.; Dube, U.; Saef, B.; Budde, J.; Black, K.; Medvedeva, A.; Del-Aguila, J.L.; Davis, A.A.; Perlmutter, J.S.; Harari, O. Parkinson disease polygenic risk score is associated with Parkinson disease status and age at onset but not with alpha-synuclein cerebrospinal fluid levels. BMC Neurol. 2017, 17, 1–9. [Google Scholar] [CrossRef]
- Li, W.-W.; Fan, D.-Y.; Shen, Y.-Y.; Zhou, F.-Y.; Chen, Y.; Wang, Y.-R.; Yang, H.; Mei, J.; Li, L.; Xu, Z.-Q. Association of the Polygenic Risk Score with the Incidence Risk of Parkinson’s Disease and Cerebrospinal Fluid α-Synuclein in a Chinese Cohort. Neurotox. Res. 2019, 36, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.N.; Chew, E.G.Y.; Chung, S.J.; Peng, R.; Blauwendraat, C.; Nalls, M.A.; Mok, K.Y.; Satake, W.; Toda, T.; Chao, Y. Identification of risk loci for Parkinson disease in Asians and comparison of risk between Asians and Europeans: A genome-wide association study. JAMA Neurol. 2020, 77, 746–754. [Google Scholar] [CrossRef]
- Nalls, M.A.; Escott-Price, V.; Williams, N.M.; Lubbe, S.; Keller, M.F.; Morris, H.R.; Singleton, A.B.; International Parkinson’s Disease Genomics Consortium. Genetic risk and age in Parkinson’s disease: Continuum not stratum. Mov. Disord. 2015, 30, 850–854. [Google Scholar] [CrossRef] [Green Version]
- Lill, C.M.; Hansen, J.; Olsen, J.H.; Binder, H.; Ritz, B.; Bertram, L. Impact of Parkinson’s disease risk loci on age at onset. Mov. Disord. 2015, 30, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Escott-Price, V.; International Parkinson’s Disease Genomics Consortium; Nalls, M.A.; Morris, H.R.; Lubbe, S.; Brice, A.; Gasser, T.; Heutink, P.; Wood, N.W.; Hardy, J. Polygenic risk of P arkinson disease is correlated with disease age at onset. Ann. Neurol. 2015, 77, 582–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.; Mercado-Ayón, E.; Mercado-Ayón, Y.; Dong, Y.N.; Halawani, S.; Ngaba, L.; Lynch, D.R. Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch. Biochem. Biophys. 2020, 702, 108698. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, K.J.; Barbosa, I.A.; Bandrés-Ciga, S.; Quinn, J.P.; Bubb, V.J.; Deshpande, C.; Botia, J.A.; Reynolds, R.H.; Zhang, D.; Simpson, M.A. Mitochondria function associated genes contribute to Parkinson’s Disease risk and later age at onset. Npj Parkinson’s Dis. 2019, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Blauwendraat, C.; Heilbron, K.; Vallerga, C.L.; Bandres-Ciga, S.; Von Coelln, R.; Pihlstrøm, L.; Simón-Sánchez, J.; Schulte, C.; Sharma, M.; Krohn, L. Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and α-synuclein mechanisms. Mov. Disord. 2019, 34, 866–875. [Google Scholar] [CrossRef] [Green Version]
- Pihlstrøm, L.; Morset, K.R.; Grimstad, E.; Vitelli, V.; Toft, M. A cumulative genetic risk score predicts progression in P arkinson’s disease. Mov. Disord. 2016, 31, 487–490. [Google Scholar] [CrossRef]
- Aradi, S.D.; Hauser, R.A. Medical management and prevention of motor complications in Parkinson’s disease. Neurotherapeutics 2020, 17, 1339–1365. [Google Scholar] [CrossRef]
- Eusebi, P.; Romoli, M.; Paoletti, F.P.; Tambasco, N.; Calabresi, P.; Parnetti, L. Risk factors of levodopa-induced dyskinesia in Parkinson’s disease: Results from the PPMI cohort. Npj Parkinson’s Dis. 2018, 4, 1–6. [Google Scholar] [CrossRef]
- Liu, G.; Peng, J.; Liao, Z.; Locascio, J.J.; Corvol, J.-C.; Zhu, F.; Dong, X.; Maple-Grødem, J.; Campbell, M.C.; Elbaz, A. Genome-wide survival study identifies a novel synaptic locus and polygenic score for cognitive progression in Parkinson’s disease. Nat. Genet. 2021, 53, 787–793. [Google Scholar] [CrossRef]
- Zhang, Q.; Aldridge, G.M.; Narayanan, N.S.; Anderson, S.W.; Uc, E.Y. Approach to cognitive impairment in Parkinson’s disease. Neurotherapeutics 2020, 17, 1495–1510. [Google Scholar] [CrossRef]
- Adams, H.H.; de Bruijn, R.F.; Hofman, A.; Uitterlinden, A.G.; van Duijn, C.M.; Vernooij, M.W.; Koudstaal, P.J.; Ikram, M.A. Genetic risk of neurodegenerative diseases is associated with mild cognitive impairment and conversion to dementia. Alzheimer’s Dement. 2015, 11, 1277–1285. [Google Scholar] [CrossRef] [Green Version]
- Kusters, C.D.; Paul, K.C.; Folle, A.D.; Keener, A.M.; Bronstein, J.M.; Dobricic, V.; Tysnes, O.-B.; Bertram, L.; Alves, G.; Sinsheimer, J.S. Genetic risk scores and hallucinations in patients with Parkinson disease. Neurol. Genet. 2020, 6. [Google Scholar] [CrossRef]
- Ihle, J.; Artaud, F.; Bekadar, S.; Mangone, G.; Sambin, S.; Mariani, L.; Bertrand, H.; Rascol, O.; Durif, F.; Derkinderen, P. Parkinson’s disease polygenic risk score is not associated with impulse control disorders: A longitudinal study. Parkinsonism Relat. Disord. 2020, 75, 30–33. [Google Scholar] [CrossRef]
- Faouzi, J.; Couvy-Duchesne, B.; Bekadar, S.; Colliot, O.; Corvol, J.-C. Exploratory analysis of the genetics of impulse control disorders in Parkinson’s disease using genetic risk scores. Parkinsonism Relat. Disord. 2021, 86, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Goveas, L.; Mutez, E.; Chartier-Harlin, M.-C.; Taymans, J.-M. Mind the Gap: LRRK2 Phenotypes in the Clinic vs. in Patient Cells. Cells 2021, 10, 981. [Google Scholar] [CrossRef] [PubMed]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Iwaki, H.; Blauwendraat, C.; Makarious, M.B.; Bandrés-Ciga, S.; Leonard, H.L.; Gibbs, J.R.; Hernandez, D.G.; Scholz, S.W.; Faghri, F.; International Parkinson’s Disease Genomics Consortium. Penetrance of Parkinson’s disease in LRRK2 p. G2019S carriers is modified by a polygenic risk score. Mov. Disord. 2020, 35, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Lai, D.; Alipanahi, B.; Fontanillas, P.; Schwantes-An, T.H.; Aasly, J.; Alcalay, R.N.; Beecham, G.W.; Berg, D.; Bressman, S.; Brice, A. Genomewide Association Studies of LRRK2 Modifiers of Parkinson’s Disease. Ann. Neurol. 2021, 90, 76–88. [Google Scholar] [CrossRef]
- Anheim, M.; Elbaz, A.; Lesage, S.; Durr, A.; Condroyer, C.; Viallet, F.; Pollak, P.; Bonaiti, B.; Bonaïti-Pellié, C.; Brice, A. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012, 78, 417–420. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Blauwendraat, C.; Reed, X.; Krohn, L.; Heilbron, K.; Bandres-Ciga, S.; Tan, M.; Gibbs, J.R.; Hernandez, D.G.; Kumaran, R.; Langston, R. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020, 143, 234–248. [Google Scholar] [CrossRef]
- Xu, L.; Pu, J. Alpha-synuclein in Parkinson’s disease: From pathogenetic dysfunction to potential clinical application. Parkinson’s Dis. 2016, 2016, 1720621. [Google Scholar] [CrossRef] [Green Version]
- Eusebi, P.; Giannandrea, D.; Biscetti, L.; Abraha, I.; Chiasserini, D.; Orso, M.; Calabresi, P.; Parnetti, L. Diagnostic utility of cerebrospinal fluid α-synuclein in Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2017, 32, 1389–1400. [Google Scholar] [CrossRef]
- Kang, J.-H.; Mollenhauer, B.; Coffey, C.S.; Toledo, J.B.; Weintraub, D.; Galasko, D.R.; Irwin, D.J.; Van Deerlin, V.; Chen-Plotkin, A.S.; Caspell-Garcia, C. CSF biomarkers associated with disease heterogeneity in early Parkinson’s disease: The Parkinson’s Progression Markers Initiative study. Acta. Neuropathol. 2016, 131, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, L.; Bahena, J.A.; Yang, C.; Dube, U.; Farias, F.H.; Budde, J.P.; Bergmann, K.; Brenner-Webster, C.; Morris, J.C.; Perrin, R.J. Functional genomic analyses uncover APOE-mediated regulation of brain and cerebrospinal fluid beta-amyloid levels in Parkinson disease. Acta. Neuropathol. Commun. 2020, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Pak, K.; Kim, J.H.; Kim, Y.J.; Yoon, J.; Lee, J.; Lyoo, C.H.; Park, H.J.; Lee, J.-H.; Jung, N.-Y. Effect of polygenic load on striatal dopaminergic deterioration in Parkinson disease. Neurology 2019, 93, e665–e674. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, H.; Klemann, C.J.; De Witte, W.; Martens, M.B.; Martens, G.J.; Poelmans, G. Shared genetic etiology between Parkinson’s disease and blood levels of specific lipids. NPJ Parkinson’s Dis. 2021, 7, 1–8. [Google Scholar]
- Tirozzi, A.; Izzi, B.; Noro, F.; Marotta, A.; Gianfagna, F.; Hoylaerts, M.F.; Cerletti, C.; Donati, M.B.; De Gaetano, G.; Iacoviello, L. Assessing genetic overlap between platelet parameters and neurodegenerative disorders. Front. Immunol. 2020, 11, 02127. [Google Scholar] [CrossRef]
- Delva, A.; Van Weehaeghe, D.; van Aalst, J.; Ceccarini, J.; Koole, M.; Baete, K.; Nuyts, J.; Vandenberghe, W.; Van Laere, K. Quantification and discriminative power of 18 F-FE-PE2I PET in patients with Parkinson’s disease. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1913–1926. [Google Scholar] [CrossRef]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Saez-Atienzar, S.; Bonet-Ponce, L.; Billingsley, K.; Vitale, D.; Blauwendraat, C.; Gibbs, J.R.; Pihlstrøm, L.; Gan-Or, Z.; International Parkinson’s Disease Genomics Consortium. The endocytic membrane trafficking pathway plays a major role in the risk of Parkinson’s disease. Mov. Disord. 2019, 34, 460–468. [Google Scholar] [CrossRef]
- Andersen, M.S.; Bandres-Ciga, S.; Reynolds, R.H.; Hardy, J.; Ryten, M.; Krohn, L.; Gan-Or, Z.; Holtman, I.R.; Pihlstrøm, L.; International Parkinson’s Disease Genomics Consortium. Heritability enrichment implicates microglia in Parkinson’s disease pathogenesis. Ann. Neurol. 2021, 89, 942–951. [Google Scholar] [CrossRef]
- Reed, X.; Schumacher-Schuh, A.; Hu, J.; Bandres-Ciga, S. Advancing Personalized Medicine in Common Forms of Parkinson’s Disease through Genetics: Current Therapeutics and the Future of Individualized Management. J. Pers. Med. 2021, 11, 169. [Google Scholar] [CrossRef]
- Winkler-Stuck, K.; Wiedemann, F.R.; Wallesch, C.-W.; Kunz, W.S. Effect of coenzyme Q10 on the mitochondrial function of skin fibroblasts from Parkinson patients. J. Neurol. Sci. 2004, 220, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Bodemer, N.; Meder, B.; Gigerenzer, G. Communicating relative risk changes with baseline risk: Presentation format and numeracy matter. Med. Decis. Mak. 2014, 34, 615–626. [Google Scholar] [CrossRef] [Green Version]
- Pain, O.; Glanville, K.P.; Hagenaars, S.P.; Selzam, S.; Fürtjes, A.E.; Gaspar, H.A.; Coleman, J.R.; Rimfeld, K.; Breen, G.; Plomin, R. Evaluation of polygenic prediction methodology within a reference-standardized framework. PLoS. Genet. 2021, 17, e1009021. [Google Scholar] [CrossRef]
- Kerminen, S.; Martin, A.R.; Koskela, J.; Ruotsalainen, S.E.; Havulinna, A.S.; Surakka, I.; Palotie, A.; Perola, M.; Salomaa, V.; Daly, M.J. Geographic variation and bias in the polygenic scores of complex diseases and traits in Finland. Am. J. Hum. Genet. 2019, 104, 1169–1181. [Google Scholar] [CrossRef] [Green Version]
- Frieser, M.J.; Wilson, S.; Vrieze, S. Behavioral impact of return of genetic test results for complex disease: Systematic review and meta-analysis. Health Psychol. 2018, 37, 1134. [Google Scholar] [CrossRef] [PubMed]
- Torkamani, A.; Wineinger, N.E.; Topol, E.J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018, 19, 581–590. [Google Scholar] [CrossRef]
- Wray, N.R.; Lin, T.; Austin, J.; McGrath, J.J.; Hickie, I.B.; Murray, G.K.; Visscher, P.M. From basic science to clinical application of polygenic risk scores: A primer. JAMA Psychiatry 2021, 78, 101–109. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Study | Base Data | Number of SNPs | Target Population Size | PRS Calculation Tool | Predictive Accuracy (AUC) |
|---|---|---|---|---|---|
| Nalls (Nalls, Blauwendraat et al. 2019) [4] | 2019 Nalls metaGWAS | 90 | 37,688 cases, 18,618 UK Biobank proxy-cases (i.e., individuals who do not have Parkinson’s disease but have a first-degree relative that does), and 1.4 million controls | PRSice2 | 65% |
| Nalls (Nalls, Blauwendraat et al. 2019) [4] | 2019 Nalls metaGWAS | 1805 | 37,688 cases, 18,618 UK Biobank proxy-cases (i.e., individuals who do not have Parkinson’s disease but have a first-degree relative that does), and 1.4 million controls | PRSice2 | 69% |
| Ibanez (Ibanez, Dube et al. 2017) [24] | 2014 Nalls metaGWAS | 26 | 829 cases and 432 controls | Plink | Not published |
| Han (Han, Teeple et al. 2021) [10] | 2019 Nalls metaGWAS | 90 | 1654 PD Cases: 79,123 controls | LDpred | 76% |
| Li (Li, Fan et al. 2019) [25] | 2014 Nalls metaGWAS 2009 Satake GWAS 2017 Redensek metaGWAS | 46 | 418 PD patients and 426 controls | Plink | 61% |
| Foo (Foo, Chew et al. 2020) [26] | Asian GWAS | 11 | 2536 PD cases and 21,840 controls | Plink | 60.20% |
| Foo (Foo, Chew et al. 2020) [26] | Asian GWAS Nalls GWAS | 11 + 90 | 2536 PD cases and 21,840 controls | Plink | 63.10% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dehestani, M.; Liu, H.; Gasser, T. Polygenic Risk Scores Contribute to Personalized Medicine of Parkinson’s Disease. J. Pers. Med. 2021, 11, 1030. https://doi.org/10.3390/jpm11101030
Dehestani M, Liu H, Gasser T. Polygenic Risk Scores Contribute to Personalized Medicine of Parkinson’s Disease. Journal of Personalized Medicine. 2021; 11(10):1030. https://doi.org/10.3390/jpm11101030
Chicago/Turabian StyleDehestani, Mohammad, Hui Liu, and Thomas Gasser. 2021. "Polygenic Risk Scores Contribute to Personalized Medicine of Parkinson’s Disease" Journal of Personalized Medicine 11, no. 10: 1030. https://doi.org/10.3390/jpm11101030