
Molecular and Genetic Immune Biomarkers of Primary and Immune-Therapy Induced Hypophysitis: From Laboratories to the Clinical Practice

 ,
,
Abstract
:1. Introduction
2. Pathogenesis of Primary Autoimmune Hypophysitis
3. The Immune Response in Primary- and Immunotherapy-Induced Hypophysitis
3.1. Antibodies in Primary Autoimmune Hypophysitis
3.2. Putative Antigens of Primary Autoimmune Hypophysitis
3.3. Cell-Mediated Immune Response in Primary Autoimmune Hypophysitis
3.4. The Genetics of Primary Autoimmune Hypophysitis
3.5. Molecular Mechanisms in Immunotherapy Induced Hypophysitis
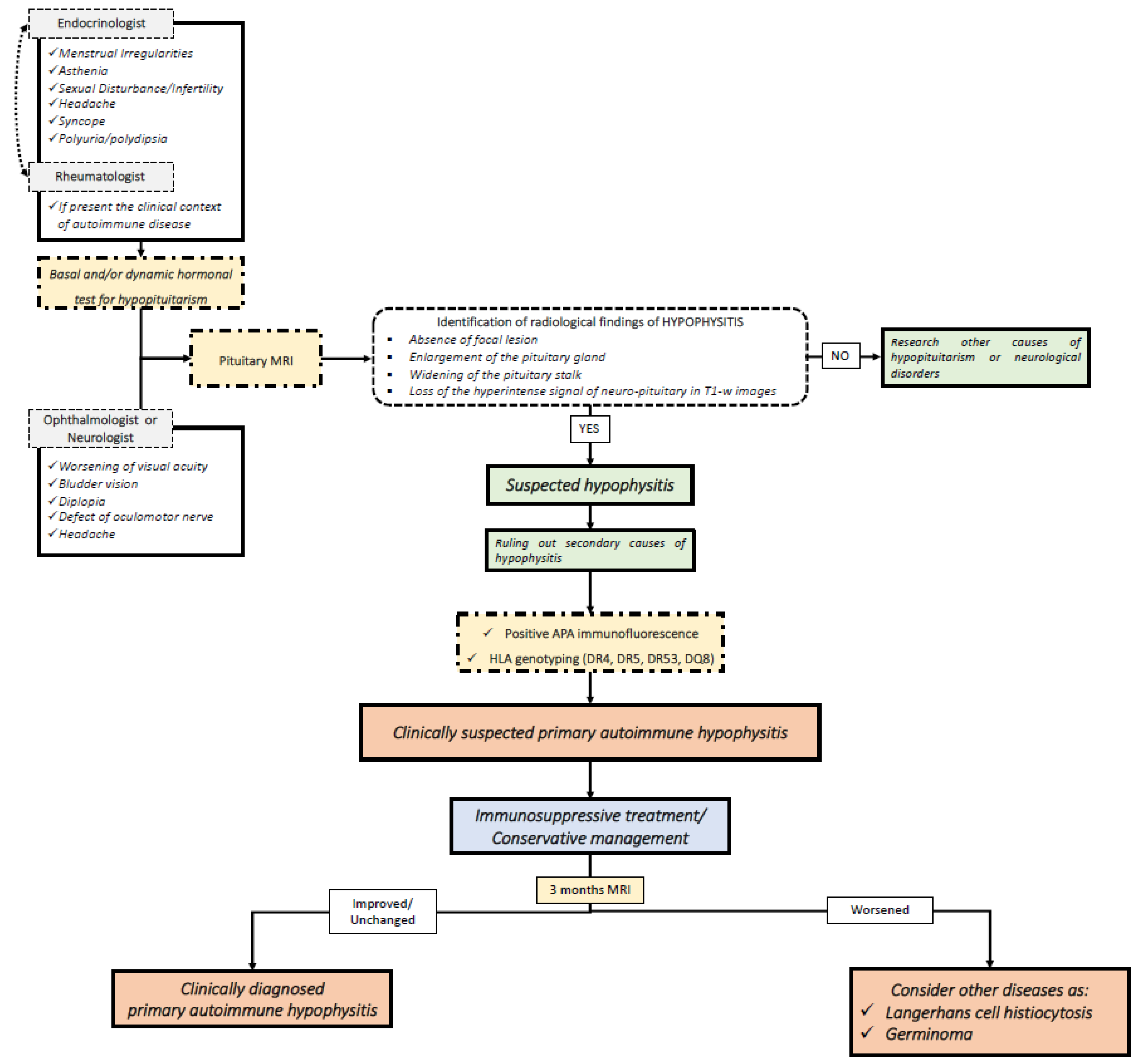
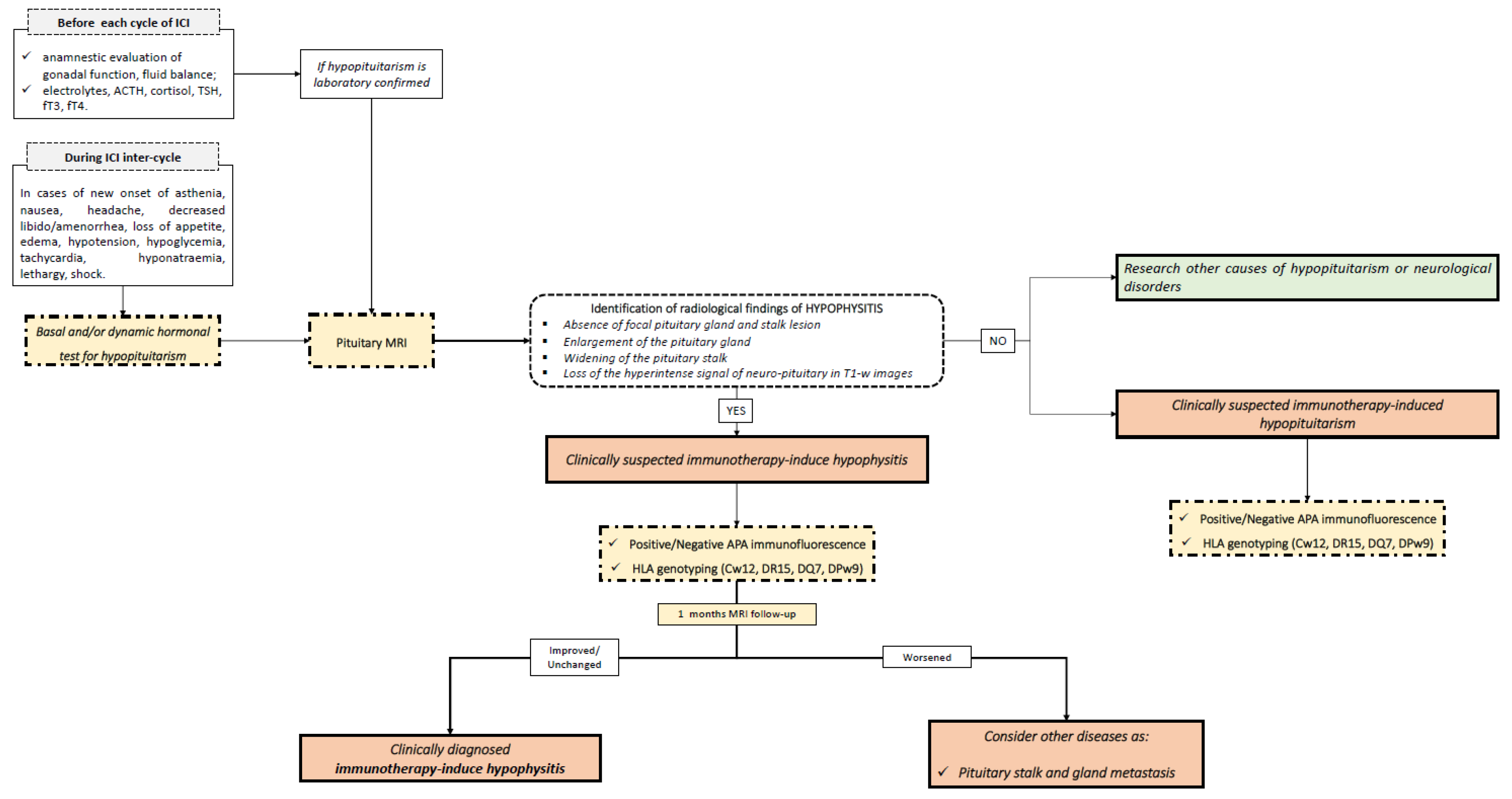
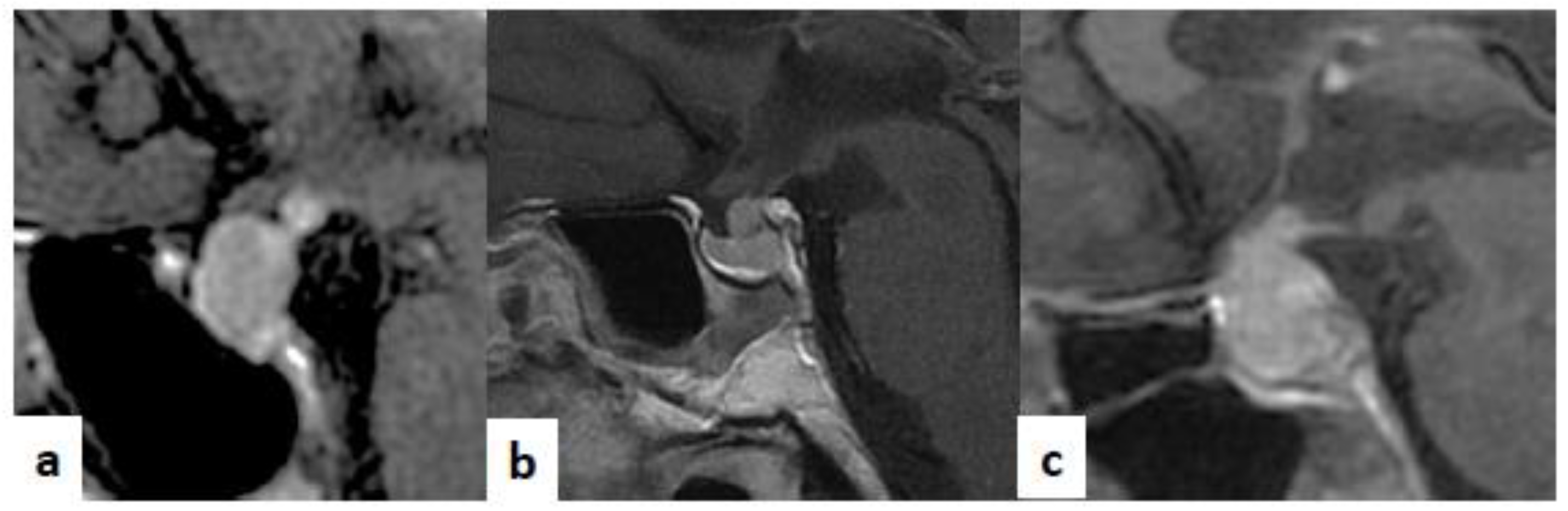
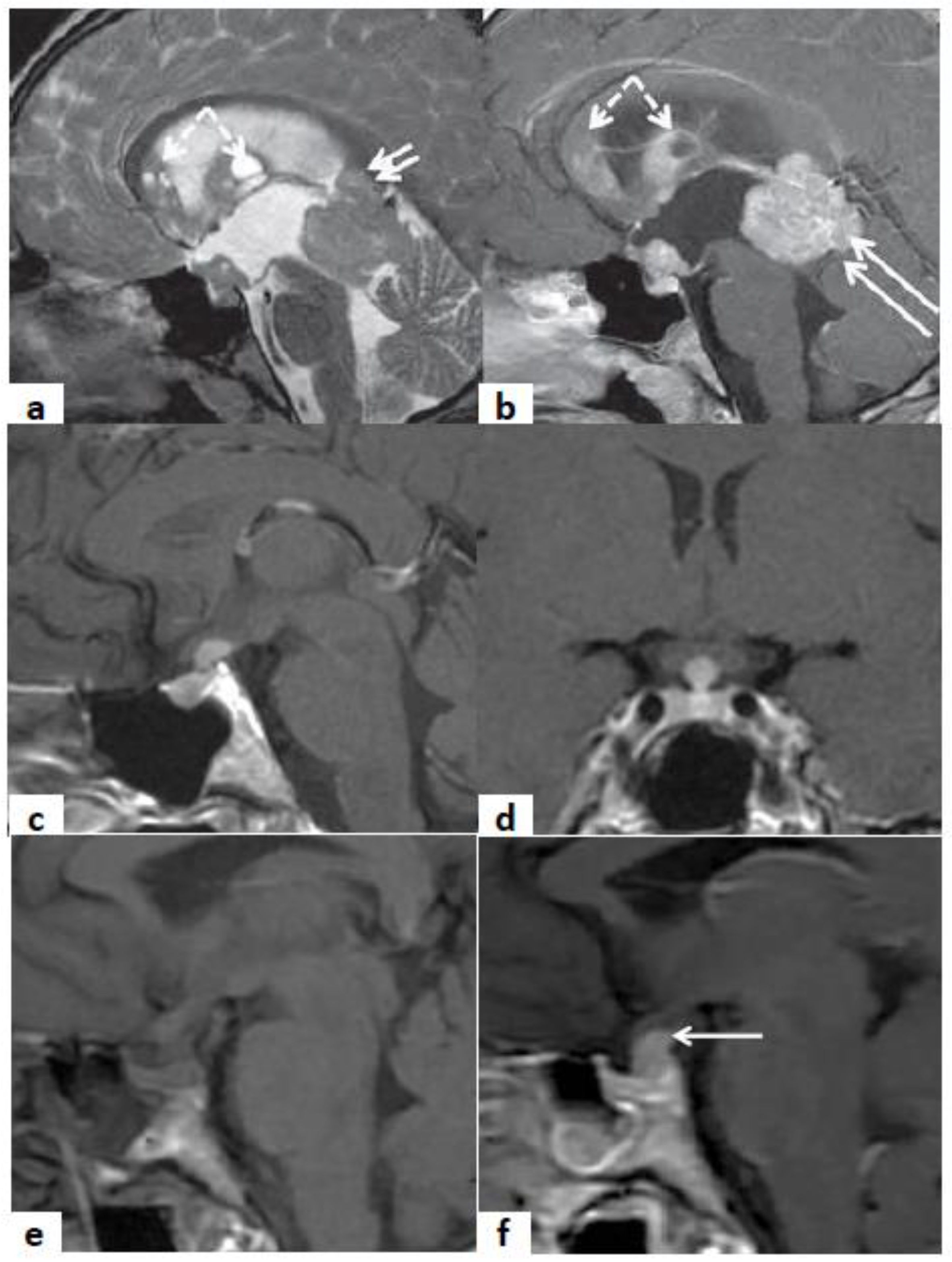
4. Clinical Application of Molecular Biomarkers in Hypophysitis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACTH | adrenocorticotropic hormone |
| APA | anti-pituitary antibodies |
| APC | antigen presenting cell |
| CTLA-4 | cytotoxic T lymphocyte antigen 4 |
| FITC | fluorescein isothiocyanate |
| GDI | rad guanine nucleotide dissociation inhibitor |
| GH | growth hormone |
| HLA | human leucocyte antigen |
| ICI | immune check-point inhibitor |
| IIH | immune-therapy induced hypophysitis |
| mAb | monoclonal antibody |
| MHC | major histocompatibility complex |
| PAH | primary autoimmune hypophysitis |
| PCNA | proliferating cell nuclear antigen |
| PD-1 | programmed cell death-1 |
| PD-L1 | programmed cell death ligand 1 |
| PD-L2 | programmed cell death ligand 2 |
| PIT-1 | pituitary-specific positive transcription factor 1 |
| PGSF1a | pituitary gland specific factor 1a |
| PGSF2 | pituitary gland specific factor 2 |
| POMC | pro-opiomelanocortin |
| TDRD | tudor domain containing protein |
| TSH | thyroid stimulating hormone |
References
- Chiloiro, S.; Capoluongo, E.D.; Tartaglione, T.; Giampietro, A.; Bianchi, A.; Giustina, A.; Pontecorvi, A.; De Marinis, L. The Changing Clinical Spectrum of Hypophysitis. Trends Endocrinol. Metab. 2019, 30, 590–602. [Google Scholar] [CrossRef]
- Tartaglione, T.; Chiloiro, S.; Laino, M.E.; Giampietro, A.; Gaudino, S.; Zoli, A.; Bianchi, A.; Pontecorvi, A.; Colosimo, C.; De Marinis, L. Neuro-Radiological Features Can Predict Hypopituitarism in Primary Autoimmune Hypophysitis. Pituitary 2018, 21, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Chiloiro, S.; Angelini, F. An Overview of Diagnosis of Primary Autoimmune Hypophysitis in a Prospective Single-Center Experience. Neuroendocrinology 2017, 104, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Freda, P.U.; Beckers, A.M.; Katznelson, L.; Molitch, M.E.; Montori, V.M.; Post, K.D.; Lee Vance, M. Pituitary Incidentaloma: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2011, 96, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Chiloiro, S.; Tartaglione, T.; Capoluongo, E.D.; Angelini, F.; Arena, V.; Giampietro, A.; Bianchi, A.; Zoli, A.; Pontecorvi, A.; Colosimo, C.; et al. Hypophysitis Outcome and Factors Predicting Responsiveness to Glucocorticoid Therapy: A Prospective and Double-Arm Study. J. Clin. Endocrinol. Metab. 2018, 103, 3877–3889. [Google Scholar] [CrossRef] [Green Version]
- Khare, S.; Jagtap, V.S.; Budyal, S.R.; Kasaliwal, R.; Kakade, H.R.; Bukan, A.; Sankhe, S.; Lila, A.R.; Bandgar, T.; Menon, P.S.; et al. Primary (Autoimmune) Hypophysitis: A Single Centre Experience. Pituitary 2015, 18, 16–22. [Google Scholar] [CrossRef]
- Caturegli, P.; Newschaffer, C.; Olivi, A.; Pomper, M.G.; Burger, P.C.; Rose, N.R. Autoimmune Hypophysitis. Endocr. Rev. 2005, 26, 599–614. [Google Scholar] [CrossRef] [Green Version]
- Cironi, K.A.; Decater, T.; Iwanaga, J.; Dumont, A.S.; Tubbs, R.S. Arterial Supply to the Pituitary Gland: A Comprehensive Review. World Neurosurg. 2020, 142, 206–211. [Google Scholar] [CrossRef]
- Tzou, S.C.; Landek-Salgado, M.A.; Kimura, H.; Caturegli, P. Preparation of Mouse Pituitary Immunogen for the Induction of Experimental Autoimmune Hypophysitis. J. Vis. Exp. 2010, 46, e2181. [Google Scholar] [CrossRef] [Green Version]
- Lupi, I.; Zhang, J.; Gutenberg, A.; Landek-Salgado, M.; Tzou, S.C.; Mori, S.; Caturegli, P. From Pituitary Expansion to Empty Sella: Disease Progression in a Mouse Model of Autoimmune Hypophysitis. Endocrinology 2011, 152, 4190–4198. [Google Scholar] [CrossRef]
- Chiloiro, S.; Giampietro, A.; Bianchi, A.; Tartaglione, T.; Capobianco, A.; Anile, C.; De Marinis, L. Diagnosis of Endocrine Disease: Primary Empty Sella: A Comprehensive Review. Eur. J. Endocrinol. 2017, 177, R275–R285. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, D.T.; Ian Smith, A.; Matthew, M.L.; Andronicos, N.M.; Ranson, M.; Robinson, P.J.; Crock, P.A. Identification of the 49-KDa Autoantigen Associated with Lymphocytic Hypophysitis as α-Enolase. J. Clin. Endocrinol. Metab. 2002, 87, 752–757. [Google Scholar] [CrossRef] [Green Version]
- De Bellis, A.; Bizzarro, A.; Bellastella, A. Pituitary Antibodies and Lymphocytic Hypophysitis. Best Pract. Res. Clin. Endocrinol. Metab. 2005, 19, 67–84. [Google Scholar] [CrossRef] [PubMed]
- De Bellis, A.; Bizzarrot, A.; Perrino, S.; Coronella, C.; Conte, M.; Pasquali, D.; Sinisi, A.A.; Betterle, C.; Bellastella, A. Characterization of Antipituitary Antibodies Targeting Pituitary Hormone-Secreting Cells in Idiopathic Growth Hormone Deficiency and Autoimmune Endocrine Diseases. Clin. Endocrinol. 2005, 63, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Lupi, I.; Manetti, L.; Raffaelli, V.; Grasso, L.; Sardella, C.; Cosottini, M.; Iannelli, A.; Gasperi, M.; Bogazzi, F.; Caturegli, P.; et al. Pituitary Autoimmunity Is Associated with Hypopituitarism in Patients with Primary Empty Sella. J. Endocrinol. Investig. 2011, 34 (Suppl. S8), 240–244. [Google Scholar] [CrossRef]
- Mele, C.; Pingue, V.; Caputo, M.; Zavattaro, M.; Pagano, L.; Prodam, F.; Nardone, A.; Aimaretti, G.; Marzullo, P. Neuroinflammation and Hypothalamo-Pituitary Dysfunction: Focus of Traumatic Brain Injury. Int. J. Mol. Sci. 2021, 22, 2686. [Google Scholar] [CrossRef]
- Chiloiro, S.; Giampietro, A.; Angelini, F.; Arena, V.; Stigliano, E.; Tartaglione, T.; Mattogno, P.P.; D’Alessandris, Q.G.; Lauretti, L.; Pontecorvi, A.; et al. Markers of Humoral and Cell-Mediated Immune Response in Primary Autoimmune Hypophysitis: A Pilot Study. Endocrine 2021, 73, 308–315. [Google Scholar] [CrossRef]
- Crock, P.A. Cytosolic Autoantigens in Lymphocytic Hypophysitis. J. Clin. Endocrinol. Metab. 1998, 83, 609–618. [Google Scholar] [CrossRef]
- Tanaka, S.; Tatsumi, K.I.; Takano, T.; Murakami, Y.; Takao, T.; Yamakita, N.; Tahara, S.; Teramoto, A.; Hashimoto, K.; Kato, Y.; et al. Anti-Alpha-Enolase Antibodies in Pituitary Disease. Endocr. J. 2003, 50, 697–702. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.J.A.; Bensing, S.; Burns, C.; Robinson, P.J.; Kasperlik-Zaluska, A.A.; Scott, R.J.; Kämpe, O.; Crock, P.A. Identification of TPIT and Other Novel Autoantigens in Lymphocytic Hypophysitis; Immunoscreening of a Pituitary CDNA Library and Development of Immunoprecipitation Assays. Eur. J. Endocrinol. 2012, 166, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Iguchi, G.; Takeno, R.; Okimura, Y.; Sano, T.; Takahashi, M.; Nishizawa, H.; Handayaningshi, A.E.; Fukuoka, H.; Tobita, M.; et al. Adult Combined GH, Prolactin, and TSH Deficiency Associated with Circulating PIT-1 Antibody in Humans. J. Clin. Investig. 2011, 121, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landek-Salgado, M.A.; Leporati, P.; Lupi, I.; Geis, A.; Caturegli, P. Growth Hormone and Proopiomelanocortin Are Targeted by Autoantibodies in a Patient with Biopsy-Proven IgG4-Related Hypophysitis. Pituitary 2012, 15, 412–419. [Google Scholar] [CrossRef]
- Kiyota, A.; Iwama, S.; Sugimura, Y.; Takeuchi, S.; Takagi, H.; Iwata, N.; Nakashima, K.; Suzuki, H.; Nishioka, T.; Kato, T.; et al. Identification of the Novel Autoantigen Candidate Rab GDP Dissociation Inhibitor Alpha in Isolated Adrenocorticotropin Deficiency. Endocr. J. 2015, 62, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupi, I.; Broman, K.W.; Tzou, S.C.; Gutenberg, A.; Martino, E.; Caturegli, P. Novel Autoantigens in Autoimmune Hypophysitis. Clin. Endocrinol. 2008, 69, 269–278. [Google Scholar] [CrossRef]
- Tatsumi, K.I.; Tanaka, S.; Takano, T.; Tahara, S.; Murakami, Y.; Takao, T.; Hashimoto, K.; Kato, Y.; Teramoto, A.; Amino, N. Frequent Appearance of Autoantibodies Against Prohormone Convertase 1/3 and Neuroendocrine Protein 7B2 in Patients with Nonfunctioning Pituitary Macroadenoma. Endocrine 2003, 22, 335–340. [Google Scholar] [CrossRef]
- Bensing, S.; Hulting, A.L.; Höög, A.; Ericson, K.; Kämpe, O. Lymphocytic Hypophysitis: Report of Two Biopsy-Proven Cases and One Suspected Case with Pituitary Autoantibodies. J. Endocrinol. Investig. 2007, 30, 153–162. [Google Scholar] [CrossRef]
- Iwama, S.; Sugimura, Y.; Kiyota, A.; Kato, T.; Enomoto, A.; Suzuki, H.; Iwata, N.; Takeuchi, S.; Nakashima, K.; Takagi, H.; et al. Rabphilin-3A as a Targeted Autoantigen in Lymphocytic Infundibulo-Neurohypophysitis. J. Clin. Endocrinol. Metab. 2015, 100, E946–E954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kume, Y.; Sakuma, H.; Sekine, H.; Sumikoshi, M.; Sugimura, Y.; Hosoya, M. Lymphocytic Infundibuloneurohypophysitis with Positive Anti-Rabphilin-3A Antibodies Nine Years Post-Onset of Central Diabetes Insipidus. Clin. Pediatr. Endocrinol. 2021, 30, 65–69. [Google Scholar] [CrossRef]
- Nielsen, C.; Hansen, D.; Husby, S.; Jacobsen, B.B.; Lillevang, S.T. Association of a Putative Regulatory Polymorphism in the PD-1 Gene with Susceptibility to Type 1 Diabetes. Tissue Antigens 2003, 62, 492–497. [Google Scholar] [CrossRef]
- Lin, H.H.; Gutenberg, A.; Chen, T.Y.; Tsai, N.M.; Lee, C.J.; Cheng, Y.C.; Cheng, W.H.; Tzou, Y.M.; Caturegli, P.; Tzou, S.C. In Situ Activation of Pituitary-Infiltrating T Lymphocytes in Autoimmune Hypophysitis. Sci. Rep. 2017, 7, 43492. [Google Scholar] [CrossRef]
- Penta, L.; Bizzarri, C.; Panichi, M.; Novelli, A.; Lepri, F.R.; Cappa, M.; Esposito, S. Identification of a Novel PROP1 Mutation in a Patient with Combined Pituitary Hormone Deficiency and Enlarged Pituitary. Int. J. Mol. Sci. 2019, 20, 1875. [Google Scholar] [CrossRef] [Green Version]
- Bertko, E.; Klammt, J.; Dusatkova, P.; Bahceci, M.; Gonc, N.; Ten Have, L.; Kandemir, N.; Mansmann, G.; Obermannova, B.; Oostdijk, W.; et al. Combined Pituitary Hormone Deficiency Due to Gross Deletions in the POU1F1 (PIT-1) and PROP1 Genes. J. Hum. Genet. 2017, 62, 755–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beressi, N.; Beresi, J.P.; Cohen, R.; Modigliani, E. Lymphocytic Hypophysitis. Ann. Med. Interne 1999, 150, 327–341. [Google Scholar] [CrossRef]
- Heaney, A.P.; Sumerel, B.; Rajalingam, R.; Bergsneider, M.; Yong, W.H.; Liau, L.M. HLA Markers DQ8 and DR53 Are Associated with Lymphocytic Hypophysitis and May Aid in Differential Diagnosis. J. Clin. Endocrinol. Metab. 2015, 100, 4092–4097. [Google Scholar] [CrossRef]
- Chiloiro, S.; Capoluongo, E.D.; Tartaglione, T.; Bianchi, A.; Giampietro, A.; Angelini, F.; Arena, V.; Pontecorvi, A.; De Marinis, L. Human Leucocyte Antigens Coeliac Haplotypes and Primary Autoimmune Hypophysitis in Caucasian Patients. Clin. Endocrinol. 2018, 88, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Megiorni, F.; Mora, B.; Bonamico, M.; Barbato, M.; Nenna, R.; Maiella, G.; Lulli, P.; Mazzilli, M.C. HLA-DQ and Risk Gradient for Celiac Disease. Hum. Immunol. 2009, 70, 55–59. [Google Scholar] [CrossRef]
- Choo, S.Y. The HLA System: Genetics, Immunology, Clinical Testing, and Clinical Implications. Yonsei Med. J. 2007, 48, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, J.D.; Zhang, X.; Fedorov, A.A. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature 2001, 410, 604–608. [Google Scholar] [CrossRef]
- Stamper, C.C.; Tobin, J.F.; Erbe, D.V.; Stahl, M.L.; Seehra, J.; Somers, W.S.; Mosyak, L. Crystal Structure of the B7-1/CTLA-4 Complex That Inhibits Human Immune Responses. Nature 2001, 410, 608–611. [Google Scholar] [CrossRef]
- Anderson, B.; Morganstein, D.L. Endocrine Toxicity of Cancer Immunotherapy: Clinical Challenges. Endocr. Connect. 2021, 10, R116–R124. [Google Scholar] [CrossRef]
- Okazaki, T.; Maeda, A.; Nishimura, H.; Kurosaki, T.; Honjo, T. PD-1 Immunoreceptor Inhibits B Cell Receptor-Mediated Signaling by Recruiting Src Homology 2-Domain-Containing Tyrosine Phosphatase 2 to Phosphotyrosine. Proc. Natl. Acad. Sci. USA 2001, 98, 13866–13871. [Google Scholar] [CrossRef] [Green Version]
- Freeman, B.G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. PD-1 Freeman JEM 2000. J. Exp. Med. 2000, 192, 1028–1034. [Google Scholar]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 Is a Second Ligand for PD-1 and Inhibits T Cell Activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-Associated B7-H1 Promotes T-Cell Apoptosis: A Potential Mechanism of Immune Evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Amarnath, S.; Mangus, C.W.; Wang, J.C.M.; Wei, F.; He, A.; Kapoor, V.; Foley, J.E.; Massey, P.R.; Felizardo, T.C.; Riley, J.L.; et al. The PDL1-PD1 Axis Converts Human T H1 Cells into Regulatory T Cells. Sci. Transl. Med. 2011, 3, 111ra120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azuma, T.; Yao, S.; Zhu, G.; Flies, A.S.; Flies, S.J.; Chen, L. B7-H1 Is a Ubiquitous Antiapoptotic Receptor on Cancer Cells. Blood 2008, 111, 3635–3643. [Google Scholar] [CrossRef]
- Queirolo, P.; Dozin, B.; Morabito, A.; Banelli, B.; Carosio, R.; Fontana, V.; Ferrucci, P.F.; Martinoli, C.; Cocorocchio, E.; Ascierto, P.A.; et al. CTLA-4 Gene Variant -1661A>G May Predict the Onset of Endocrine Adverse Events in Metastatic Melanoma Patients Treated with Ipilimumab. Eur. J. Cancer 2018, 97, 59–61. [Google Scholar] [CrossRef]
- Kula, A.; Dawidowicz, M.; Kiczmer, P.; Prawdzic Seńkowska, A.; Świętochowska, E. The Role of Genetic Polymorphism within PD-L1 Gene in Cancer. Review. Exp. Mol. Pathol. 2020, 116, 104494. [Google Scholar] [CrossRef]
- Pulichino, A.M.; Vallette-Kasic, S.; Couture, C.; Gauthier, Y.; Brue, T.; David, M.; Malpuech, G.; Deal, C.; Van Vliet, G.; De Vroede, M.; et al. Human and Mouse TPIT Gene Mutations Cause Early Onset Pituitary ACTH Deficiency. Genes Dev. 2003, 17, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Iwama, S.; Sugiyama, D.; Yasuda, Y.; Okuji, T.; Ito, M.; Ito, S.; Sugiyama, M.; Onoue, T.; Takagi, H.; et al. Anti-Pituitary Antibodies and Susceptible Human Leukocyte Antigen Alleles as Predictive Biomarkers for Pituitary Dysfunction Induced by Immune Checkpoint Inhibitors. J. Immunother. Cancer 2021, 9, e002493. [Google Scholar] [CrossRef]
- Kanie, K.; Iguchi, G.; Bando, H.; Urai, S.; Shichi, H.; Fujita, Y.; Matsumoto, R.; Suda, K.; Yamamoto, M.; Fukuoka, H.; et al. Mechanistic Insights into Immune Checkpoint Inhibitor-Related Hypophysitis: A Form of Paraneoplastic Syndrome. Cancer Immunol. Immunother. 2021. [Google Scholar] [CrossRef]
- Yano, S.; Ashida, K.; Sakamoto, R.; Sakaguchi, C.; Ogata, M.; Maruyama, K.; Sakamoto, S.; Ikeda, M.; Ohe, K.; Akasu, S.; et al. Human Leucocyte Antigen DR15, a Possible Predictive Marker for Immune Checkpoint Inhibitor–Induced Secondary Adrenal Insufficiency. Eur. J. Cancer 2020, 130, 198–203. [Google Scholar] [CrossRef]
- Dillard, T.; Yedinak, C.G.; Alumkal, J.; Fleseriu, M. Anti-CTLA-4 Antibody Therapy Associated Autoimmune Hypophysitis: Serious Immune Related Adverse Events across a Spectrum of Cancer Subtypes. Pituitary 2010, 13, 29–38. [Google Scholar] [CrossRef]
- Iwama, S.; Arima, H. Anti-Pituitary Antibodies as a Marker of Autoimmunity in Pituitary Glands. Endocr. J. 2020, 67, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Gellner, V.; Kurschel, S.; Scarpatetti, M.; Mokry, M. Lymphocytic Hypophysitis in the Pediatric Population. Child’s Nerv. Syst. 2008, 24, 785–792. [Google Scholar] [CrossRef]
- Fernandes, S.; Varlamov, E.V.; McCartney, S.; Fleseriu, M. A Novel Etiology of Hypophysitis: Immune Checkpoint Inhibitors. Endocrinol. Metab. Clin. N. Am. 2020, 49, 387–399. [Google Scholar] [CrossRef]
- Pal, R.; Rai, A.; Vaiphei, K.; Gangadhar, P.; Gupta, P.; Mukherjee, K.K.; Singh, P.; Ray, N.; Bhansali, A.; Dutta, P. Intracranial Germinoma Masquerading as Secondary Granulomatous Hypophysitis: A Case Report and Review of Literature. Neuroendocrinology 2020, 110, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Frobisher, C.; Hawkins, M.; Jenney, M.; Lancashire, E.; Reulen, R.; Taylor, A.; Winter, D. The British Childhood Cancer Survivor Study: Objectives, Methods, Population Structure, Response Rates and Initial Descriptive Information. Pediatr. Blood Cancer 2008, 50, 1018–1025. [Google Scholar] [CrossRef]
- Carl, D.; Grüllich, C.; Hering, S.; Schabet, M. Steroid Responsive Encephalopathy Associated with Autoimmune Thyroiditis Following Ipilimumab Therapy: A Case Report. BMC Res. Notes 2015, 8, 10–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Etiological classification | Primary Hypophysitis | The Inflammatory Process Involves Only the Pituitary Gland | |
| Secondary hypophysitis | Cystic/neoplastic pituitary lesions | Craniopharyngioma, adenoma, germinoma, cyst and Rathke cleft cyst | |
| Autoimmune systemic diseases | IgG4-disease, sarcoidosis, granulomatosis, vasculitis, Crohn’s disease | ||
| Histiocytic proliferative diseases | Langerhans cell histiocytosis, Erdheim–Chester, Rosai–Dorfman diseases | ||
| Infective Systemic Disease | Syphilis, tuberculosis, fungal and viral infection in immune-depressed patients | ||
| Immuno-therapy induced hypophysitis | in patients on treatment with check-point inhibitors, as monoclonal antibodies anti-CTLA-4, anti-PD-1 and anti-PD1 ligand | ||
| Biomarkers | Primary Autoimmune Hypophysitis | Immunotherapy-Induced Hypophysitis | Immunotherapy-Induced Hypopituitarism/ACTH Deficit | |
|---|---|---|---|---|
| Humoral immune response | Anti-pituitary antibodies | Yes | Yes | Yes |
| Putative antigens | Alpha-enolase Chorionic somatomammotropin hormone Growth hormone (typically in AH) Prohormone convertase PC2 regulatory protein PGSF 1a and 2 (typically in AH and INH) Pit-1 POMC Alpha-rab guanine nucleotide dissociation inhibitor Rabphilin-3A (typically in INH) Secretogranin II T-PIT Tudor Domain Containing Protein 6 | ACTH CTLA-4 Prolactin | ACTH | |
| Cytokine | Gamma-interferon 17 interleukin | No data | No data | |
| Cell-mediated immune response | Lymphocytes | B220, CD3, CD4, CD8, CD20, CD44 positive | No data | No data |
| Other | CD11 positive dendritic cells monocytes/macrophages granulocytes | No data | No data | |
| Genetic biomarkers | HLA haplotypes | DR4, DR5, DR53, DQ8 | Cw12, DR15 | Cw12, DR15, DQ7, DPw9 |
| Other | No data | CTLA-4 and PD-1 gene | No data | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiloiro, S.; Russo, F.; Tartaglione, T.; Capoluongo, E.D. Molecular and Genetic Immune Biomarkers of Primary and Immune-Therapy Induced Hypophysitis: From Laboratories to the Clinical Practice. J. Pers. Med. 2021, 11, 1026. https://doi.org/10.3390/jpm11101026
Chiloiro S, Russo F, Tartaglione T, Capoluongo ED. Molecular and Genetic Immune Biomarkers of Primary and Immune-Therapy Induced Hypophysitis: From Laboratories to the Clinical Practice. Journal of Personalized Medicine. 2021; 11(10):1026. https://doi.org/10.3390/jpm11101026
Chicago/Turabian StyleChiloiro, Sabrina, Filippo Russo, Tommaso Tartaglione, and Ettore Domenico Capoluongo. 2021. "Molecular and Genetic Immune Biomarkers of Primary and Immune-Therapy Induced Hypophysitis: From Laboratories to the Clinical Practice" Journal of Personalized Medicine 11, no. 10: 1026. https://doi.org/10.3390/jpm11101026