The Road so Far in Colorectal Cancer Pharmacogenomics: Are We Closer to Individualised Treatment?

 , ,
, ,
Abstract
:1. Introduction
2. Chemotherapeutic Agents in CRC Treatment
3. Pharmacogenetics: Candidate Gene Studies
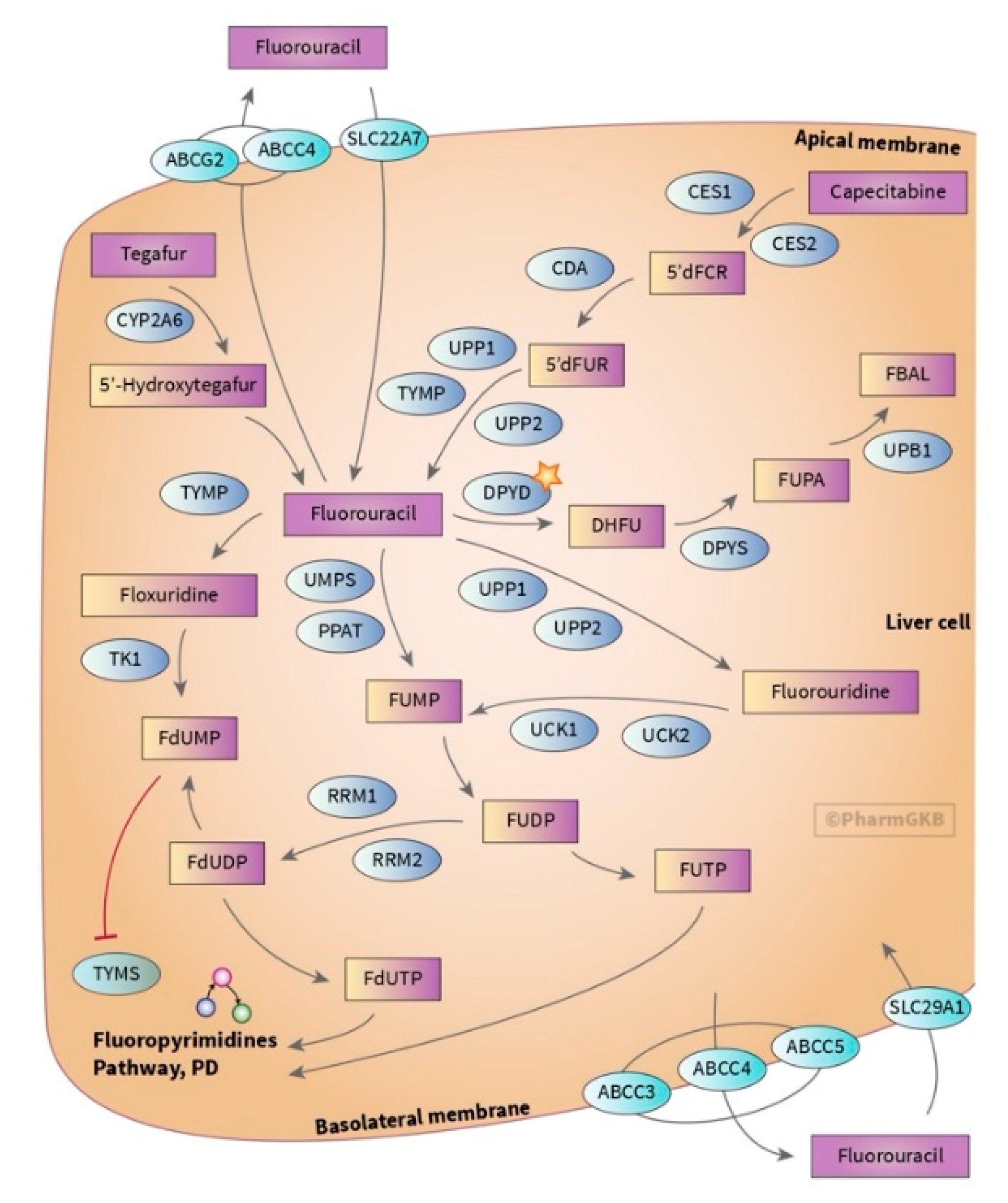
3.1. Dihydropyrimidine Dehydrogenase (DPYD)
3.2. Thymidylate Synthetase (TYMS)
3.3. Methylenetetrahydrofolate Reductase (MTHFR)
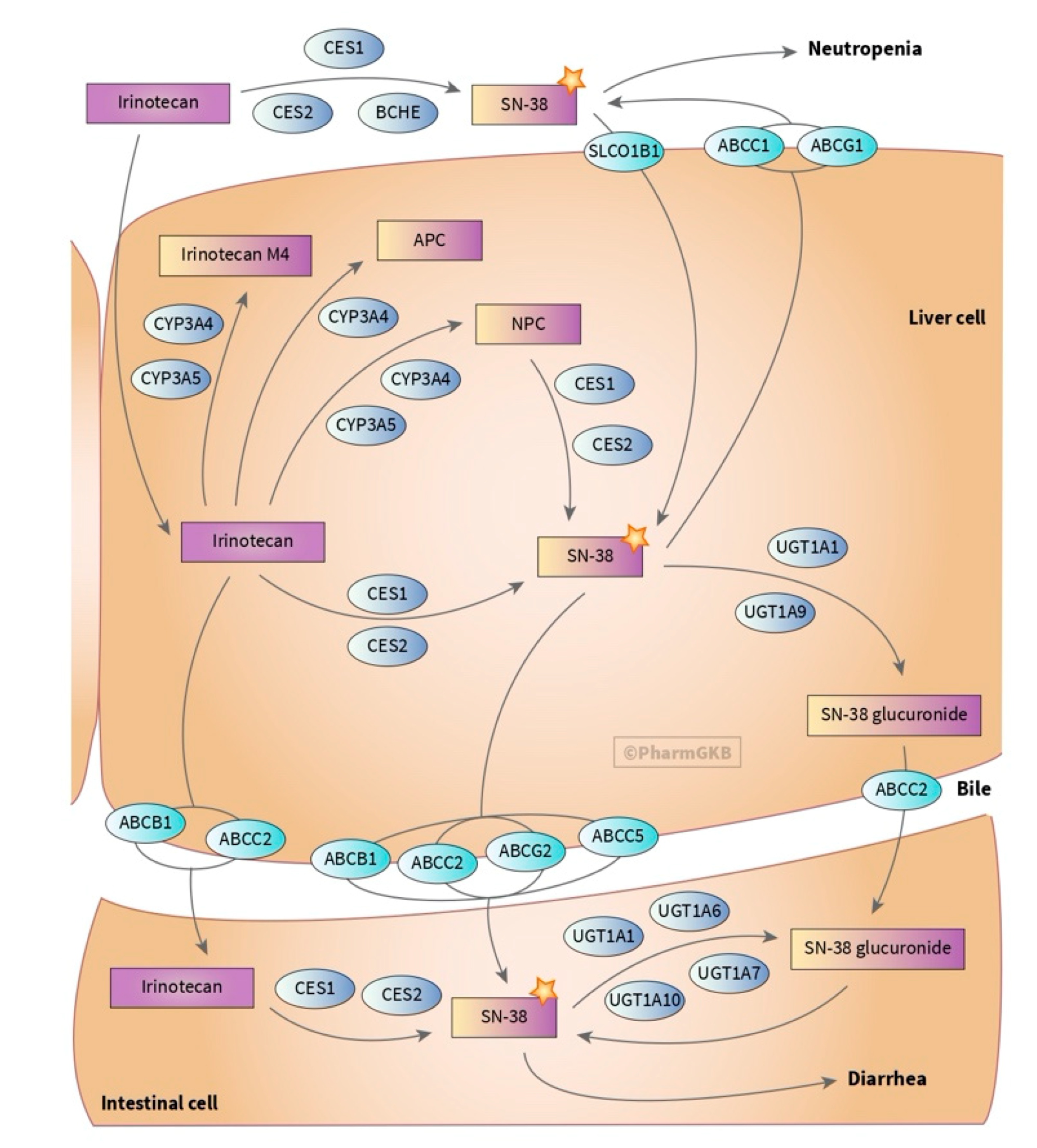
3.4. Carboxyl Esterases (CES) and Cytidine Deaminase (CDA)
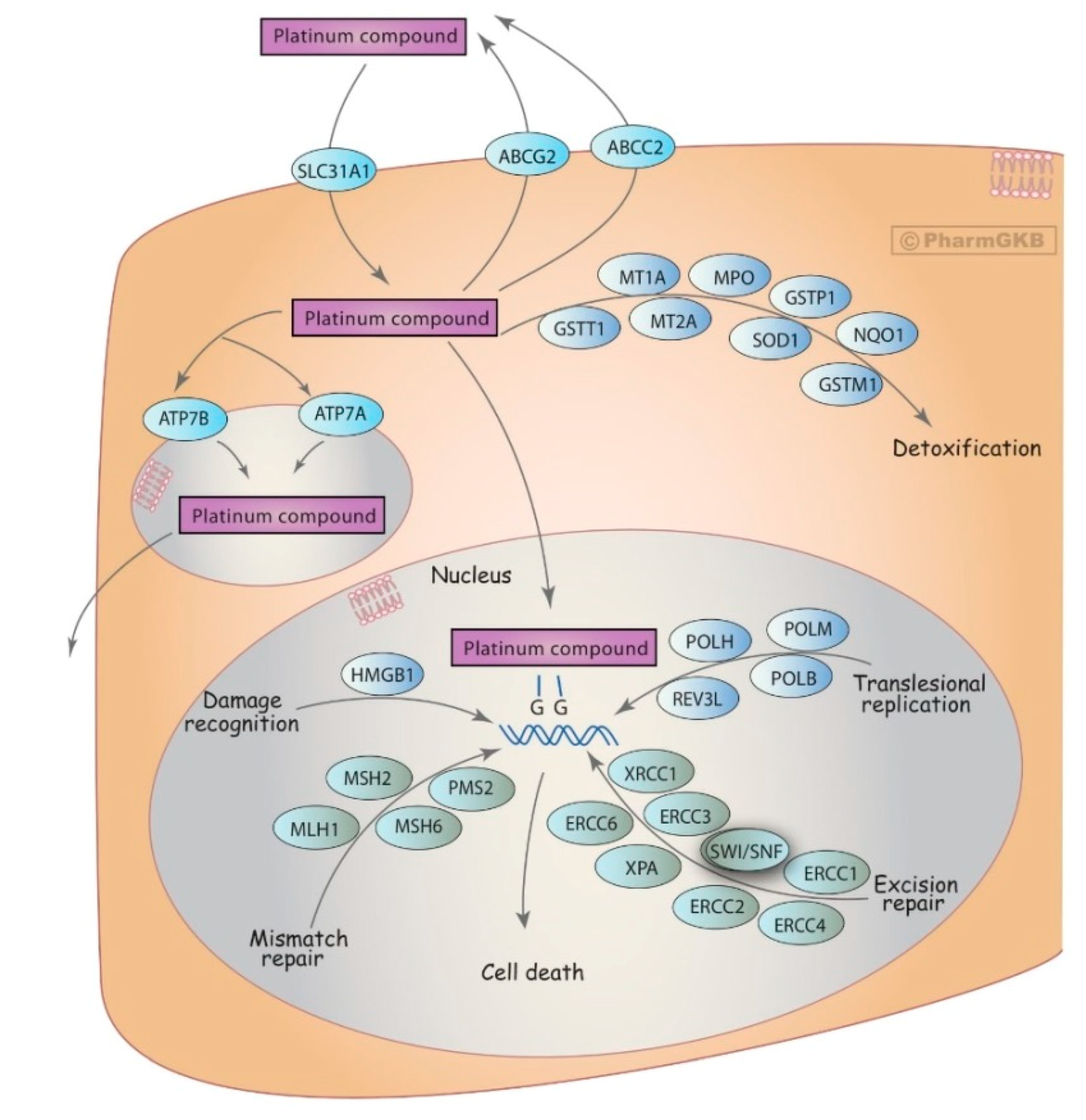
3.5. DNA Repair Genes
3.6. Glutathione S-Transferases (GSTs)
3.7. Adenosine-Triphosphate Binding Cassette (ABC) Transporters
3.8. Uridine Disphosphate Glucuronosyltransferases (UGTs)
3.9. Solute Carriers (SLCs)
3.10. Cytochrome p Gene Family (CYP)
3.11. Epidermal Growth Factor Receptor (EGFR)
3.12. Vascular Endothelial Growth Factor (VEGF)
3.13. Immunotherapy and Toxicity
4. Pharmacogenomic Approaches
4.1. Genome-Wide Association Studies (GWAS)
4.2. Next-Generation Sequencing (NGS)
5. Functional Assays
6. Cost-Effectiveness Analysis
7. Pharmacogenomic Testing Guidelines
8. Limitations in Pharmacogenomic Studies
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douillard, J.Y.; Cunningham, D.; Roth, A.D.; Navarro, M.; James, R.D.; Karasek, P.; Jandik, P.; Iveson, T.; Carmichael, J.; Alakl, M.; et al. Irinotecan combined with fluorouracil compared with fluorouracil alone. as first-line treatment for metastatic colorectal cancer: A multicentre randomised trial. Lancet 2000, 355, 1041–1047. [Google Scholar] [CrossRef]
- de Gramont, A.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Boni, C.; Cortes-Funes, H.; Cervantes, A.; Freyer, G.; et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Nordlinger, B.; Arnold, D. The ESMO Guidelines Working Group Metastatic colorectal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, iii1–iii9. [Google Scholar] [CrossRef]
- Iacovelli, R.; Pietrantonio, F.; Palazzo, A.; Maggi, C.; Ricchini, F.; De Braud, F.; Di Bartolomeo, M. Incidence and relative risk of grade 3 and 4 diarrhoea in patients treated with capecitabine or 5-fluorouracil: A meta-analysis of published trials. Br. J. Clin. Pharmacol. 2014, 78, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.H.; Litière, S.; De Vries, E.; Ford, R.; Gwyther, S.; Mandrekar, S.; Shankar, L.; Bogaerts, J.; Chen, A.; Dancey, J.; et al. RECIST 1.1-Update and clarification: From the RECIST committee. Eur. J. Cancer 2016, 62, 132–137. [Google Scholar] [CrossRef] [Green Version]
- Latremouille-Viau, D.; Chang, J.; Guerin, A.; Shi, S.; Wang, E.; Yu, J.; Ngai, C. The economic burden of common adverse events associated with metastatic colorectal cancer treatment in the United States. J. Med. Econ. 2016, 20, 54–62. [Google Scholar] [CrossRef]
- Peters, E.J.; Motsinger-Reif, A.; Havener, T.M.; Everitt, L.; Hardison, N.E.; Watson, V.G.; Wagner, M.; Richards, K.L.; Province, M.A.; McLeod, H.L. Pharmacogenomic characterization of US FDA-approved cytotoxic drugs. Pharmacogenomics 2011, 12, 1407–1415. [Google Scholar] [CrossRef] [Green Version]
- Watters, J.W.; Kraja, A.; Meucci, M.A.; Province, M.A.; McLeod, H.L. Genome-wide discovery of loci influencing chemotherapy cytotoxicity. Proc. Natl. Acad. Sci. USA 2004, 101, 11809–11814. [Google Scholar] [CrossRef] [Green Version]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Highlights of prescribing information. Capecitabine 2015. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/020896s037lbl.pdf (accessed on 13 November 2020).
- De Matti, E.; Roncato, R.; Dalle Fratte, C.; Ecca, F.; Toffoli, G.; Cecchin, E. The use of pharmacogenetics to increase the safety of colorectal cancer patients treatedwith fluoropyrimidines. Cancer Drug Resist. 2019, 2, 116–130. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsen, E.; Twelves, C.; Cassidy, J.; Allman, D.; Bajetta, E.; Boyer, M.; Bugat, R.; Findlay, M.; Frings, S.; Jahn, M.; et al. Oral capecitabine compared with intravenous fluorouracil plus leucovorin in patients with metastatic colorectal cancer: Results of a large phase III study. J. Clin. Oncol. 2001, 19, 4097–4106. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Highlights of prescribing information. Oxaliplatin 2002. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/021492s016lbl.pdf (accessed on 13 November 2020).
- Food and Drug Administration. Highlights of prescribing information. Irinotecan 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020571s048lbl.pdf (accessed on 13 November 2020).
- Food and Drug Administration. Highlights of prescribing information. Cetuximab 2009. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125084s273lbl.pdf (accessed on 13 November 2020).
- Food and Drug Administration. Highlights of prescribing information. Panitumumab 2015. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125147s200lbl.pdf (accessed on 13 November 2020).
- Food and Drug Administration. Highlights of prescribing information. Bevacizumab 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/125085s225lbl.pdf (accessed on 13 November 2020).
- Thorn, C.F.; Marsh, S.; Carrillo, M.W.; McLeod, H.L.; Klein, T.E.; Altman, R.B. Pharm GKB summary: Fluoropyrimidine pathways. Pharm. Genom. 2011, 21, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Diasio, R.B.; Harris, B.E. Clinical Pharmacokinetics; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 1989. [Google Scholar]
- Raymond, E.; Chaney, S.G.; Taamma, A.; Cvitkovic, E. Oxaliplatin: A review of preclinical and clinical studies. Ann. Oncol. 1998, 9, 1053–1071. [Google Scholar] [CrossRef]
- Marsh, S.; McLeod, H.; Dolan, E.; Shukla, S.J.; Rabik, C.A.; Gong, L.; Hernandez-Boussard, T.; Lou, X.J.; Klein, T.E.; Altman, R.B. Platinum pathway. Pharm. Genom. 2009, 19, 563–564. [Google Scholar] [CrossRef] [Green Version]
- Saltz, L.B.; Cox, J.V.; Blanke, C.; Rosen, L.S.; Fehrenbacher, L.; Moore, M.J.; Maroun, J.A.; Ackland, S.P.; Locker, P.K.; Pirotta, N.; et al. Irinotecan plus Fluorouracil and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2000, 343, 905–914. [Google Scholar] [CrossRef]
- Wall, M.E.; Wani, M.C. Discovery to Clinic. Ann. N. Y. Acad. Sci. 1996, 803, 1–12. [Google Scholar] [CrossRef]
- Rothenberg, M.L. Topoisomerase I inhibitors: Review and update. Ann. Oncol. 1997, 8, 837–855. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2016, 8, 3980–4000. [Google Scholar] [CrossRef] [Green Version]
- Toffoli, G.; Giodini, L.; Buonadonna, A.; Berretta, M.; De Paoli, A.; Scalone, S.; Miolo, G.; Mini, E.; Nobili, S.; Lonardi, S.; et al. Clinical validity of a DPYD-based pharmacogenetic test to predict severe toxicity to fluoropyrimidines. Int. J. Cancer 2015, 137, 2971–2980. [Google Scholar] [CrossRef] [PubMed]
- Offer, S.M.; Wegner, N.J.; Fossum, C.; Wang, K.; Diasio, R.B. Phenotypic profiling of DPYD variations relevant to 5-fluorouracil sensitivity using real-time cellular analysis and in vitro measurement of enzyme activity. Cancer Res. 2013, 73, 1958–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henricks, L.M.; Lunenburg†, C.A.; Meulendijks, D.; Gelderblom, H.; Cats, A.; Swen, J.J.; Schellens, J.H.; Guchelaar, H. TranslatingDPYDgenotype into DPD phenotype: Using theDPYDgene activity score. Pharmacogenomics 2015, 16, 1275–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offer, S.M.; Fossum, C.C.; Wegner, N.J.; Stuflesser, A.J.; Butterfield, G.L.; Diasio, R.B. Comparative Functional Analysis of DPYD Variants of Potential Clinical Relevance to Dihydropyrimidine Dehydrogenase Activity. Cancer Res. 2014, 74, 2545–2554. [Google Scholar] [CrossRef] [Green Version]
- Zaanan, A.; Dumont, L.M.; Loriot, M.A.; Taieb, J.; Narjoz, C. Clinical Pharmacology and Therapeutics; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Nie, Q.; Shrestha, S.; Tapper, E.E.; Trogstad-Isaacson, C.; Bouchonville, K.J.; Lee, A.M.; Wu, R.; Jerde, C.R.; Wang, Z.; Kubica, P.; et al. Quantitative Contribution of rs75017182 to Dihydropyrimidine Dehydrogenase mRNA Splicing and Enzyme Activity. Clin. Pharmacol. Ther. 2017, 102, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Meulendijks, D.; Henricks, L.M.; Sonke, G.S.; Deenen, M.J.; Froehlich, T.K.; Amstutz, U.; Largiadèr, C.R.; Jennings, B.A.; Marinaki, A.M.; Sanderson, J.D.; et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: A systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015, 16, 1639–1650. [Google Scholar] [CrossRef]
- Kouwaki, M.; Hamajima, N.; Sumi, S.; Nonaka, M.; Sasaki, M.; Dobashi, K.; Kidouchi, K.; Togari, H.; Wada, Y. Identification of novel mutations in the dihydropyrimidine dehydrogenase gene in a Japanese patient with 5-fluorouracil toxicity. Clin. Cancer Res. 1998, 4, 2999–3004. [Google Scholar]
- Falvella, F.S.; Cheli, S.; Martinetti, A.; Mazzali, C.; Iacovelli, R.; Maggi, C.; Gariboldi, M.; Pierotti, M.A.; Di Bartolomeo, M.; Sottotetti, E.; et al. DPD and UGT1A1 deficiency in colorectal cancer patients receiving triplet chemotherapy with fluoropyrimidines, oxaliplatin and irinotecan. Br. J. Clin. Pharmacol. 2015, 80, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Madi, A.; Fisher, D.; Maughan, T.S.; Colley, J.P.; Meade, A.M.; Maynard, J.; Humphreys, V.; Wasan, H.; A Adams, R.; Idziaszczyk, S.; et al. Pharmacogenetic analyses of 2183 patients with advanced colorectal cancer; potential role for common dihydropyrimidine dehydrogenase variants in toxicity to chemotherapy. Eur. J. Cancer 2018, 102, 31–39. [Google Scholar] [CrossRef]
- Vreken, P.; Van Kuilenburg, A.B.P.; Meinsma, R.; Van Gennip, A.H. Identification of novel point mutations in the dihydropyrimidine dehydrogenase gene. J. Inherit. Metab. Dis. 1997, 20, 335–338. [Google Scholar] [CrossRef]
- O’Donnell, P.H.; Trubetskoy, V.; Nurhussein-Patterson, A.; Hall, J.P.; Nath, A.; Huo, D.; Fleming, G.F.; Ingle, J.N.; Abramson, V.G.; Morrow, P.K.; et al. Clinical evaluation of germline polymorphisms associated with capecitabine toxicity in breast cancer: TBCRC-015. Breast Cancer Res. Treat. 2020, 181, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Ruzzo, A.; Graziano, F.; Galli, F.; Galli, F.; Rulli, E.; Lonardi, S.; Ronzoni, M.; Massidda, B.; Zagonel, V.; Pella, N.; et al. Dihydropyrimidine dehydrogenase pharmacogenetics for predicting fluoropyrimidine-related toxicity in the randomised, phase III adjuvant TOSCA trial in high-risk colon cancer patients. Br. J. Cancer 2017, 117, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Rosmarin, D.; Palles, C.; Pagnamenta, A.; Kaur, K.; Pita, G.; Martin, M.; Domingo, E.; Jones, A.; Howarth, K.; Freeman-Mills, L.; et al. A candidate gene study of capecitabine-related toxicity in colorectal cancer identifies new toxicity variants at DPYD and a putative role for ENOSF1 rather than TYMS. Gut 2015, 64, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellicer, M.; García-González, X.; García, M.I.; Robles, L.; Grávalos, C.; García-Alfonso, P.; Pachón, V.; Longo, F.; Martínez, V.; Blanco, C.; et al. Identification of new SNPs associated with severe toxicity to capecitabine. Pharmacol. Res. 2017, 120, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Meulendijks, D.; Jacobs, B.A.; Aliev, A.; Pluim, D.; Van Werkhoven, E.; Deenen, M.J.; Beijnen, J.H.; Cats, A.; Schellens, J.H. Increased risk of severe fluoropyrimidine-associated toxicity in patients carrying a G to C substitution in the first 28-bp tandem repeat of the thymidylate synthase 2R allele. Int. J. Cancer 2015, 138, 245–253. [Google Scholar] [CrossRef]
- Kristensen, M.H.; Pedersen, P.L.; Melsen, G.V.; Ellehauge, J.; Mejer, J. Variants in the dihydropyrimidine dehydrogenase, methylenetetrahydrofolate reductase and thymidylate synthase genes predict early toxicity of 5-fluorouracil in colorectal cancer patients. J. Int. Med. Res. 2010, 38, 870–883. [Google Scholar] [CrossRef] [Green Version]
- Rosmarin, D.; Palles, C.; Church, D.; Domingo, E.; Jones, A.; Johnstone, E.; Wang, H.; Love, S.; Julier, P.; Scudder, C.; et al. Genetic markers of toxicity from capecitabine and other fluorouracil-based regimens: Investigation in the QUASAR2 study, systematic review, and meta-analysis. J. Clin. Oncol. 2014, 32, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Tsunoda, A.; Nakao, K.; Watanabe, M.; Matsui, N.; Ooyama, A.; Kusano, M. Associations of various gene polymorphisms with toxicity in colorectal cancer patients receiving oral uracil and tegafur plus leucovorin: A prospective study. Ann. Oncol. 2011, 22, 355–361. [Google Scholar] [CrossRef]
- Loganayagam, A.; Arenas Hernandez, M.; Corrigan, A.; Fairbanks, L.; Lewis, C.M.; Harper, P.; Maisey, N.; Ross, P.; Sanderson, J.D.; Marinaki, A.M. Pharmacogenetic variants in the DPYD, TYMS, CDA and MTHFR genes are clinically significant predictors of fluoropyrimidine toxicity. Br. J. Cancer 2013, 108, 2505–2515. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Chang, H.J.; Han, S.W.; Oh, D.Y.; Im, S.A.; Bang, Y.J.; Kim, S.Y.; Lee, K.W.; Kim, J.H.; Hong, Y.S.; et al. Pharmacogenetic analysis of adjuvant FOLFOX for Korean patients with colon cancer. Cancer Chemother. Pharm. 2013, 71, 843–851. [Google Scholar] [CrossRef]
- Jennings, B.A.; Loke, Y.K.; Skinner, J.; Keane, M.; Chu, G.S.; Turner, R.; Epurescu, D.; Barrett, A.; Willis, G. Evaluating Predictive Pharmacogenetic Signatures of Adverse Events in Colorectal Cancer Patients Treated with Fluoropyrimidines. PLoS ONE 2013, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meulendijks, D.; Henricks, L.M.; Amstutz, U.; Froehlich, T.K.; Largiadèr, C.R.; Beijnen, J.H.; de Boer, A.; Deenen, M.J.; Cats, A.; Schellens, J.H.M. Rs895819 in MIR27A improves the predictive value of DPYD variants to identify patients at risk of severe fluoropyrimidine-associated toxicity. Int. J. Cancer 2016, 138, 2752–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magdy, T.; Arlanov, R.; Winter, S.; Lang, T.; Klein, K.; Toyoda, Y.; Ishikawa, T.; Schwab, M.; Zanger, U.M. ABCC11/MRP8 polymorphisms affect 5-fluorouracil-induced severe toxicity and hepatic expression. Pharmacogenomics 2013, 14, 1433–1448. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Haba, E.; García, M.I.; Cortejoso, L.; López-Lillo, C.; Barrueco, N.; García-Alfonso, P.; Alvarez, S.; Jiménez, J.L.; Martín, M.L.; Muñóz-Fernández, M.A.; et al. ABCB1 gene polymorphisms are associated with adverse reactions in fluoropyrimidine-treated colorectal cancer patients. Pharmacogenomics 2010, 11, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- García-González, X.; Cortejoso, L.; García, M.I.; García-Alfonso, P.; Robles, L.; Grávalos, C.; González-Haba, E.; Marta, P.; Sanjurjo, M.; López-Fernández, L.A. Variants in CDA and ABCB1 are predictors of capecitabinerelated adverse reactions in colorectal cancer. Oncotarget 2015, 6, 6422–6430. [Google Scholar] [CrossRef] [Green Version]
- Ribelles, N.; Lopez-Siles, J.; Sanchez, A.; Gonzalez, E.; Sanchez, M.; Carabantes, F.; Sanchez-Rovira, P.; Marquez, A.; Duenas, R.; Sevilla, I.; et al. A Carboxylesterase 2 Gene Polymorphism as Predictor of Capecitabine on Response and Time to Progression. Curr. Drug Metab. 2008, 9, 336–343. [Google Scholar] [CrossRef]
- Hamzic, S.; Kummer, D.; Milesi, S.; Mueller, D.; Joerger, M.; Aebi, S.; Amstutz, U.; Largiadèr, C.R. Novel Genetic Variants in Carboxylesterase 1 Predict Severe Early-Onset Capecitabine-Related Toxicity. Clin. Pharm. Ther. 2017, 102, 796–804. [Google Scholar] [CrossRef]
- Wheeler, H.E.; González-Neira, A.; Pita, G.; De La Torre-Montero, J.C.; Alonso, R.; Lopez-Fernandez, L.A.; Alba, E.; Martín, M.; Dolan, M.E. Identification of genetic variants associated with capecitabine-induced hand-foot syndrome through integration of patient and cell line genomic analyses. Pharm. Genom. 2014, 24, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Rozadilla, C.; Cazier, J.B.; Moreno, V.; Crous-Bou, M.; Guinó, E.; Durán, G.; Lamas, M.J.; López, R.; Candamio, S.; Gallardo, E.; et al. Pharmacogenomics in colorectal cancer: A genome-wide association study to predict toxicity after 5-fluorouracil or FOLFOX administration. Pharm. J. 2013, 13, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Cecchin, E.; D’Andrea, M.; Lonardi, S.; Zanusso, C.; Pella, N.; Errante, D.; De Mattia, E.; Polesel, J.; Innocenti, F.; Toffoli, G. A prospective validation pharmacogenomic study in the adjuvant setting of colorectal cancer patients treated with the 5-fluorouracil/leucovorin/oxaliplatin (FOLFOX4) regimen. Pharm. J. 2012, 13, 403–409. [Google Scholar] [CrossRef]
- Custodio, A.; Moreno-Rubio, J.; Aparicio, J.; Gallego-Plazas, J.; Yaya, R.; Maurel, J.; Higuera, O.; Burgos, E.; Ramos, D.; Calatrava, A.; et al. Pharmacogenetic predictors of severe peripheral neuropathy in colon cancer patients treated with oxaliplatin-based adjuvant chemotherapy: A GEMCAD group study. Ann. Oncol. 2014, 25, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Stoehlmacher, J.; Park, D.J.; Zhang, W.; Yang, D.; Groshen, S.; Zahedy, S.; Lenz, H.J. A multivariate analysis of genomic polymorphisms: Prediction of clinical outcome to 5-FU/oxaliplatin combination chemotherapy in refractory colorectal cancer. Br. J. Cancer 2004, 91, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Ruzzo, A.; Graziano, F.; Galli, F.; Giacomini, E.; Floriani, I.; Galli, F.; Rulli, E.; Lonardi, S.; Ronzoni, M.; Massidda, B.; et al. Genetic markers for toxicity of adjuvant oxaliplatin and fluoropyrimidines in the phase III TOSCA trial in high-risk colon cancer patients. Sci. Rep. 2014, 4, 1–7. [Google Scholar] [CrossRef]
- Boige, V.; Mendiboure, J.; Pignon, J.P.; Loriot, M.A.; Castaing, M.; Barrois, M.; Malka, D.; Trégouët, D.A.; Bouché, O.; Le Corre, D.; et al. Pharmacogenetic assessment of toxicity and outcome in patients with metastatic colorectal cancer treated with LV5FU2, FOLFOX, and FOLFIRI: FFCD 2000-05. J. Clin. Oncol. 2010, 28, 2556–2564. [Google Scholar] [CrossRef] [PubMed]
- Salimzadeh, H.; Lindskog, E.B.; Gustavsson, B.; Wettergren, Y.; Ljungman, D. Association of DNA repair gene variants with colorectal cancer: Risk, toxicity, and survival. BMC Cancer 2020, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Saka, H.; Ando, M.; Sawa, T.; Muro, K.; Ueoka, H.; Yokoyama, A.; Saitoh, S.; Shimokata, K.; Hasegawa, Y. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: A pharmacogenetic analysis. Cancer Res. 2000, 60, 6921–6926. [Google Scholar] [PubMed]
- Onoue, M.; Terada, T.; Kobayashi, M.; Katsura, T.; Matsumoto, S.; Yanagihara, K.; Nishimura, T.; Kanai, M.; Teramukai, S.; Shimizu, A.; et al. UGT1A1*6 polymorphism is most predictive of severe neutropenia induced by irinotecan in Japanese cancer patients. Int. J. Clin. Oncol. 2009, 14, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Laverdiere, I.; Tourancheau, A.; Jonker, D.; Couture, F.; Cecchin, E.; Villeneuve, L.; Harvey, M.; Court, M.H.; Innocenti, F.; et al. A novel UGT1 marker associated with better tolerance against irinotecan-induced severe neutropenia in metastatic colorectal cancer patients. Pharm. J. 2015, 15, 513–520. [Google Scholar] [CrossRef]
- Innocenti, F.; Undevia, S.D.; Iyer, L.; Chen, P.X.; Das, S.; Kocherginsky, M.; Karrison, T.; Janisch, L.; Ramírez, J.; Rudin, C.M.; et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J. Clin. Oncol. 2004, 22, 1382–1388. [Google Scholar] [CrossRef]
- Côté, J.F.; Kirzin, S.; Kramar, A.; Mosnier, J.F.; Diebold, M.D.; Soubeyran, I.; Thirouard, A.S.; Selves, J.; Laurent-Puig, P.; Ychou, M. UGT1A1 polymorphism can predict hematologic toxicity in patients treated with irinotecan. Clin. Cancer Res. 2007, 13, 3269–3275. [Google Scholar] [CrossRef] [Green Version]
- Lévesque, É.; Bélanger, A.S.; Harvey, M.; Couture, F.; Jonker, D.; Innocenti, F.; Cecchin, E.; Toffoli, G.; Guillemette, C. Refining the UGT1A haplotype associated with irinotecan-induced hematological toxicity in metastatic colorectal cancer patients treated with 5-fluorouracil/irinotecan-based regimens. J. Pharm. Exp. Ther. 2013, 345, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Han, J.-Y.; Lim, H.-S.; Park, Y.H.; Lee, S.Y.; Lee, J.S. Integrated pharmacogenetic prediction of irinotecan pharmacokinetics and toxicity in patients with advanced non-small cell lung cancer. Lung Cancer 2009, 63, 115–120. [Google Scholar] [CrossRef]
- Chen, S.; Villeneuve, L.; Jonker, D.; Couture, F.; Laverdière, I.; Cecchin, E.; Innocenti, F.; Toffoli, G.; Lévesque, E.; Guillemette, C. ABCC5 and ABCG1 polymorphisms predict irinotecan-induced severe toxicity in metastatic colorectal cancer patients. Pharm. Genom. 2015, 25, 573–583. [Google Scholar] [CrossRef]
- Di Martino, M.T.; Arbitrio, M.; Leone, E.; Guzzi, P.H.; Rotundo, M.S.; Ciliberto, D.; Tomaino, V.; Fabiani, F.; Talarico, D.; Sperlongano, P.; et al. Single nucleotide polymorphisms of ABCC5 and ABCG1 transporter genes correlate to irinotecan-associated gastrointestinal toxicity in colorectal cancer patients: A DMET microarray profiling study. Cancer Biol. Ther. 2011, 12, 780–787. [Google Scholar] [CrossRef]
- Li, M.; Seiser, E.L.; Baldwin, R.M.; Ramirez, J.; Ratain, M.J.; Innocenti, F.; Kroetz, D.L. ABC transporter polymorphisms are associated with irinotecan pharmacokinetics and neutropenia. Pharm. J. 2018, 18, 35–42. [Google Scholar] [CrossRef]
- De Mattia, E.; Toffoli, G.; Polesel, J.; D’Andrea, M.; Corona, G.; Zagonel, V.; Buonadonna, A.; Dreussi, E.; Cecchin, E. Pharmacogenetics of ABC and SLC transporters in metastatic colorectal cancer patients receiving first-line FOLFIRI treatment. Pharm. Genom. 2013, 23, 549–557. [Google Scholar] [CrossRef]
- Hoskins, J.M.; Marcuello, E.; Altes, A.; Marsh, S.; Maxwell, T.; Van Booven, D.J.; Paré, L.; Culverhouse, R.; McLeod, H.L.; Baiget, M. Irinotecan Pharmacogenetics: Influence of Pharmacodynamic Genes. Clin. Cancer Res. 2008, 14, 1788–1796. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yu, Q.; Fu, S.; Xu, M.; Zhang, T.; Xie, C.; Feng, J.; Chen, J.; Zang, A.; Cai, Y.; et al. A novel genetic score model of UGT1A1 and TGFB pathway as predictor of severe irinotecan-related diarrhea in metastatic colorectal cancer patients. J. Cancer Res. Clin. Oncol. 2016, 142, 1621–1628. [Google Scholar] [CrossRef]
- Takahashi, H.; Sai, K.; Saito, Y.; Kaniwa, N.; Matsumura, Y.; Hamaguchi, T.; Shimada, Y.; Ohtsu, A.; Yoshino, T.; Doi, T.; et al. Application of a Combination of a Knowledge-Based Algorithm and 2-Stage Screening to Hypothesis-Free Genomic Data on Irinotecan-Treated Patients for Identification of a Candidate Single Nucleotide Polymorphism Related to an Adverse Effect. PLoS ONE 2014, 9, e105160. [Google Scholar] [CrossRef]
- Won, H.H.; Lee, J.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Kim, J.W.; Lee, S.Y.; Park, S.H. Polymorphic markers associated with severe oxaliplatin-induced, chronic peripheral neuropathy in colon cancer patients. Cancer 2012, 118, 2828–2836. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Grimaldi, M.C.; Bennouna, J.; Formento, J.L.; Douillard, J.Y.; Francoual, M.; Hennebelle, I.; Chatelut, E.; Francois, E.; Faroux, R.; El Hannani, C.; et al. Multifactorial pharmacogenetic analysis in colorectal cancer patients receiving 5-fluorouracil-based therapy together with cetuximab-irinotecan. Br. J. Clin. Pharm. 2012, 73, 776–785. [Google Scholar] [CrossRef] [Green Version]
- Parmar, S.; Schumann, C.; Rüdiger, S.; Boeck, S.; Heinemann, V.; Kächele, V.; Seeringer, A.; Paul, T.; Seufferlein, T.; Stingl, J.C. Pharmacogenetic predictors for EGFR-inhibitor-associated skin toxicity. Pharm. J. 2013, 13, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Dahan, L.; Norguet, E.; Etienne-Grimaldi, M.-C.; Formento, J.-L.; Gasmi, M.; Nanni, I.; Gaudart, J.; Garcia, S.; Ouafik, L.; Seitz, J.-F.; et al. Pharmacogenetic profiling and cetuximab outcome in patients with advanced colorectal cancer. BMC Cancer 2011, 11, 496. [Google Scholar] [CrossRef] [Green Version]
- Baas, J.; Krens, L.; Bohringer, S.; Mol, L.; Punt, C.; Guchelaar, H.; Gelderblom, H. Genome wide association study to identify predictors for severe skin toxicity in colorectal cancer patients treated with cetuximab. PLoS ONE 2018, 13, e0208080. [Google Scholar] [CrossRef]
- Sibertin-Blanc, C.; Mancini, J.; Fabre, A.; Lagarde, A.; Del Grande, J.; Levy, N.; Seitz, J.F.; Olschwang, S.; Dahan, L. Vascular Endothelial Growth Factor A c.*237C>T polymorphism is associated with bevacizumab efficacy and related hypertension in metastatic colorectal cancer. Dig. Liver Dis. 2015, 47, 331–337. [Google Scholar] [CrossRef]
- Schneider, B.P.; Wang, M.; Radovich, M.; Sledge, G.W.; Badve, S.; Thor, A.; Flockhart, D.A.; Hancock, B.; Davidson, N.; Gralow, J.; et al. Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. J. Clin. Oncol. 2008, 26, 4672–4678. [Google Scholar] [CrossRef]
- Morita, S.; Uehara, K.; Nakayama, G.; Shibata, T.; Oguri, T.; Inada-Inoue, M.; Shimokata, T.; Sugishita, M.; Mitsuma, A.; Ando, Y. Association between bevacizumab-related hypertension and vascular endothelial growth factor (VEGF) gene polymorphisms in Japanese patients with metastatic colorectal cancer. Cancer Chemother. Pharm. 2013, 71, 405–411. [Google Scholar] [CrossRef]
- Ma, T.; Zhu, Z.G.; Ji, Y.B.; Zhang, Y.; Yu, Y.Y.; Liu, B.Y.; Yin, H.R.; Lin, Y.Z. Correlation of thymidylate synthase, thymidine phosphorylase and dihydropyrimidine dehydrogenase with sensitivity of gastrointestinal cancer cells to 5-fluorouracil and 5-fluoro-2′-deoxyuridine. World J. Gastroenterol. 2004, 10, 172–176. [Google Scholar] [CrossRef]
- Lecomte, T.; Ferraz, J.M.; Zinzindohoué, F.; Loriot, M.A.; Tregouet, D.A.; Landi, B.; Berger, A.; Cugnenc, P.H.; Jian, R.; Beaune, P.; et al. Thymidylate synthase gene polymorphism predicts toxicity in colorectal cancer patients receiving 5-fluorouracil-based chemotherapy. Clin. Cancer Res. 2004, 10, 5880–5888. [Google Scholar] [CrossRef] [Green Version]
- Popat, S.; Matakidou, A.; Houlston, R.S. Thymidylate-synthase expression and prognosis in colorectal cancer: A systematic review and meta-analysis. J. Clin. Oncol. 2004, 22, 529–536. [Google Scholar] [CrossRef]
- Danenberg, P.V.; Leichman, L.; Lenz, H.J.; Leichman, C.G.; Danenberg, K.D. Thymidylate Synthase Gene and Protein Expression Correlate and Are Associated with Response to 5-Fluorouracil in Human Colorectal and Gastric Tumors. Cancer Res. 1995, 55, 1407–1412. [Google Scholar]
- De Mattia, E.; Toffoli, G. C677T and A1298C MTHFR polymorphisms, a challenge for antifolate and fluoropyrimidine-based therapy personalisation. Eur. J. Cancer 2009, 45, 1333–1351. [Google Scholar] [CrossRef] [PubMed]
- Chua, W.; Goldstein, D.; Lee, C.K.; Dhillon, H.; Michael, M.; Mitchell, P.; Clarke, S.J.; Iacopetta, B. Molecular markers of response and toxicity to FOLFOX chemotherapy in metastatic colorectal cancer. Br. J. Cancer 2009, 101, 998–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvador-Martín, S.; García-González, X.; García, M.I.; Blanco, C.; García-Alfonso, P.; Robles, L.; Grávalos, C.; Pachón, V.; Longo, F.; Martínez, V.; et al. Clinical utility of ABCB1 genotyping for preventing toxicity in treatment with irinotecan. Pharm. Res. 2018, 136, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Glimelius, B.; Garmo, H.; Berglund, Å.; Fredriksson, L.A.; Berglund, M.; Kohnke, H.; Byström, P.; Sørbye, H.; Wadelius, M. Prediction of irinotecan and 5-fluorouracil toxicity and response in patients with advanced colorectal cancer. Pharm. J. 2011, 11, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M.; Martinez, N.; Ramos, M.; Calvo, L.; Lluch, A.; Zamora, P.; Munoz, M.; Carrasco, E.; Caballero, R.; Garcia-Saenz, J.A.; et al. Standard Versus Continuous Administration of Capecitabine in Metastatic Breast Cancer (GEICAM/2009-05): A Randomized, Noninferiority Phase II Trial With a Pharmacogenetic Analysis. Oncologist 2015, 20, 111–112. [Google Scholar] [CrossRef]
- Rhodes, K.E.; Zhang, W.; Yang, D.; Press, O.A.; Gordon, M.; Vallböhmer, D.; Schultheis, A.M.; Lurje, G.; Ladner, R.D.; Fazzone, W.; et al. ABCB1, SLCO1B1 and UGT1A1 gene polymorphisms are associated with toxicity in metastatic colorectal cancer patients treated with first-line irinotecan. Drug Metab. Lett. 2007, 1, 23–30. [Google Scholar] [CrossRef]
- Mathijssen, R.H.J.; Marsh, S.; Karlsson, M.O.; Xie, R.; Baker, S.D.; Verweij, J.; Sparreboom, A.; McLeod, H.L. Irinotecan pathway genotype analysis to predict pharmacokinetics. Clin. Cancer Res. 2003, 9, 3246–3253. [Google Scholar]
- Teft, W.A.; Welch, S.; Lenehan, J.; Parfitt, J.; Choi, Y.H.; Winquist, E.; Kim, R.B. OATP1B1 and tumour OATP1B3 modulate exposure, toxicity, and survival after irinotecan-based chemotherapy. Br. J. Cancer 2015, 112, 857–865. [Google Scholar] [CrossRef]
- Cecchin, E.; Corona, G.; Masier, S.; Biason, P.; Cattarossi, G.; Frustaci, S.; Buonadonna, A.; Colussi, A.; Toffoli, G. Carboxylesterase isoform 2 mRNA expression in peripheral blood mononuclear cells is a predictive marker of the irinotecan to SN38 activation step in colorectal cancer patients. Clin. Cancer Res. 2005, 11, 6901–6907. [Google Scholar] [CrossRef] [Green Version]
- Cortejoso, L.; López-Fernández, L.A. Pharmacogenetic markers of toxicity for chemotherapy in colorectal cancer patients. Pharmacogenomics 2012, 13, 1173–1191. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.-Y.; Liu, X.-Y.; Wu, Q.; Song, X.; Chen, X.-F.; Liu, Y.-Q.; Pei, D.; Shen, L.-Z.; Shu, Y.-Q. The ERCC1 C118T Polymorphism Predicts Clinical Outcomes of Colorectal Cancer Patients Receiving Oxaliplatin-Based Chemotherapy: A Meta-analysis Based on 22 Studies. Asian Pac. J. Cancer Prev. 2014, 15, 8383–8390. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Grimaldi, M.C.; Milano, G.; Maindrault-Gœbel, F.; Chibaudel, B.; Formento, J.L.; Francoual, M.; Lledo, G.; André, T.; Mabro, M.; Mineur, L.; et al. Methylenetetrahydrofolate reductase (MTHFR) gene polymorphisms and FOLFOX response in colorectal cancer patients. Br. J. Clin. Pharm. 2010, 69, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLeod, H.L.; Sargent, D.J.; Marsh, S.; Green, E.M.; King, C.R.; Fuchs, C.S.; Ramanathan, R.K.; Williamson, S.K.; Findlay, B.P.; Thibodeau, S.N.; et al. Pharmacogenetic predictors of adverse events and response to chemotherapy in metastatic colorectal cancer: Results from North American Gastrointestinal Intergroup Trial N9741. J. Clin. Oncol. 2010, 28, 3227–3233. [Google Scholar] [CrossRef]
- Gamelin, L.; Capitain, O.; Morel, A.; Dumont, A.; Traore, S.; Anne, L.B.; Gilles, S.; Boisdron-Celle, M.; Gamelin, E. Predictive factors of oxaliplatin neurotoxicity: The involvement of the oxalate outcome pathway. Clin. Cancer Res. 2007, 13, 6359–6368. [Google Scholar] [CrossRef] [Green Version]
- Massacesi, C.; Terrazzino, S.; Marcucci, F.; Rocchi, M.B.; Lippe, P.; Bisonni, R.; Lombardo, M.; Pilone, A.; Mattioli, R.; Leon, A. Uridine diphosphate glucuronosyl transferase 1A1 promoter polymorphism predicts the risk of gastrointestinal toxicity and fatigue induced by irinotecan-based chemotherapy. Cancer 2006, 106, 1007–1016. [Google Scholar] [CrossRef]
- Iyer, L.; Das, S.; Janisch, L.; Wen, M.; Ramírez, J.; Karrison, T.; Fleming, G.F.; Vokes, E.E.; Schilsky, R.L.; Ratain, M.J. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharm. J. 2002, 2, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.Y.; Chen, P.M.; Chiou, T.J.; Liu, J.H.; Lin, J.K.; Lin, T.C.; Chen, W.S.; Jiang, J.K.; Wang, H.S.; Wang, W.S. UGT1A1*28 polymorphism predicts irinotecan-induced severe toxicities without affecting treatment outcome and survival in patients with metastatic colorectal carcinoma. Cancer 2008, 112, 1932–1940. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, D.; Kuang, Q.; Liu, G.; Xu, W. Association of UGT1A1*28 polymorphisms with irinotecan-induced toxicities in colorectal cancer: A meta-analysis in Caucasians. Pharm. J. 2014, 14, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Toffoli, G.; Cecchin, E.; Gasparini, G.; D’Andrea, M.; Azzarello, G.; Basso, U.; Mini, E.; Pessa, S.; De Mattia, E.; Re, G.L.; et al. Genotype-driven phase I study of irinotecan administered in combination with fluorouracil/leucovorin in patients with metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 866–871. [Google Scholar] [CrossRef] [Green Version]
- Marcuello, E.; Páez, D.; Paré, L.; Salazar, J.; Sebio, A.; Del Rio, E.; Baiget, M. A genotype-directed phase I-IV dose-finding study of irinotecan in combination with fluorouracil/leucovorin as first-line treatment in advanced colorectal cancer. Br. J. Cancer 2011, 105, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Treenert, A.; Areepium, N.; Tanasanvimon, S. Effects of ABCC2 and SLCO1B1 Polymorphisms on treatment responses in Thai metastatic colorectal cancer patients treated with Irinotecan-based chemotherapy. Asian Pac. J. Cancer Prev. 2018, 19, 2757–2764. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, F.; Kroetz, D.L.; Schuetz, E.; Dolan, M.E.; Ramírez, J.; Relling, M.; Chen, P.; Das, S.; Rosner, G.L.; Ratain, M.J. Comprehensive pharmacogenetic analysis of irinotecan neutropenia and pharmacokinetics. J. Clin. Oncol. 2009, 27, 2604–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riera, P.; Salazar, J.; Virgili, A.C.; Tobeña, M.; Sebio, A.; Gallano, P.; Barnadas, A.; Páez, D. Relevance of CYP3A4*20, UGT1A1*37 and UGT1A1*28 variants in irinotecan-induced severe toxicity. Br. J. Clin. Pharm. 2018, 84, 1389–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujikura, K.; Ingelman-Sundberg, M.; Lauschke, V.M. Genetic variation in the human cytochrome P450 supergene family. Pharm. Genom. 2015, 25, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Paulík, A.; Grim, J.; Filip, S. Predictors of irinotecan toxicity and efficacy in treatment of metastatic colorectal cancer. Acta Medica 2012, 55, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Sunakawa, Y.; Yang, D.; Moran, M.; Astrow, S.H.; Tsuji, A.; Stephens, C.; Zhang, W.; Cao, S.; Takahashi, T.; Denda, T.; et al. Combined assessment of EGFR-related molecules to predict outcome of 1st-line cetuximab-containing chemotherapy for metastatic colorectal cancer. Cancer Biol. Ther. 2016, 17, 751–759. [Google Scholar] [CrossRef] [Green Version]
- Pander, J.; Gelderblom, H.; Antonini, N.F.; Tol, J.; van Krieken, J.H.J.M.; van der Straaten, T.; Punt, C.J.A.; Guchelaar, H.J. Correlation of FCGR3A and EGFR germline polymorphisms with the efficacy of cetuximab in KRAS wild-type metastatic colorectal cancer. Eur. J. Cancer 2010, 46, 1829–1834. [Google Scholar] [CrossRef]
- Lambrechts, D.; Moisse, M.; Delmar, P.; Miles, D.W.; Leighl, N.; Escudier, B.; Van Cutsem, E.; Bansal, A.T.; Carmeliet, P.; Scherer, S.J.; et al. Genetic markers of bevacizumab-induced hypertension. Angiogenesis 2014, 17, 685–694. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Grimaldi, M.C.; Formento, P.; Degeorges, A.; Pierga, J.Y.; Delva, R.; Pivot, X.; Dalenc, F.; Espié, M.; Veyret, C.; Formento, J.L.; et al. Prospective analysis of the impact of VEGF-A gene polymorphisms on the pharmacodynamics of bevacizumab-based therapy in metastatic breast cancer patients. Br. J. Clin. Pharm. 2011, 71, 921–928. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Food and Drug Administration. FDA Grants Accelerated Approval to Ipilimumab for MSI-H or dMMR Metastatic Colorectal Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-ipilimumab-msi-h-or-dmmr-metastatic-colorectal-cancer (accessed on 30 October 2020).
- Food and Drug Administration. FDA Approves Pembrolizumab for First-Line Treatment of MSI-H/dMMR Colorectal Cancer. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-first-line-treatment-msi-hdmmr-colorectal-cancer (accessed on 30 October 2020).
- Bins, S.; Basak, E.A.; El Bouazzaoui, S.; Koolen, S.L.W.; De Hoop, E.O.; Van Der Leest, C.H.; Van Der Veldt, A.A.M.; Sleijfer, S.; Debets, R.; Van Schaik, R.H.N.; et al. Association between single-nucleotide polymorphisms and adverse events in nivolumab-treated non-small cell lung cancer patients. Br. J. Cancer 2018, 118, 1296–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Samkari, H.; Snyder, G.D.; Nikiforow, S.; Tolaney, S.M.; A Freedman, R.; Losman, J.-A. Haemophagocytic lymphohistiocytosis complicating pembrolizumab treatment for metastatic breast cancer in a patient with the PRF1A91V gene polymorphism. J. Med. Genet. 2018, 56, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Queirolo, P.; Dozin, B.; Morabito, A.; Banelli, B.; Carosio, R.; Fontana, V.; Ferrucci, P.F.; Martinoli, C.; Cocorocchio, E.; Ascierto, P.A.; et al. CTLA-4 gene variant-1661A>G may predict the onset of endocrine adverse events in metastatic melanoma patients treated with ipilimumab. Eur. J. Cancer 2018, 97, 59–61. [Google Scholar] [CrossRef]
- Oguri, T.; Mitsuma, A.; Inada-Inoue, M.; Morita, S.; Shibata, T.; Shimokata, T.; Sugishita, M.; Nakayama, G.; Uehara, K.; Hasegawa, Y.; et al. Genetic polymorphisms associated with oxaliplatin-induced peripheral neurotoxicity in Japanese patients with colorectal cancer. Int. J. Clin. Pharm. Ther. 2013, 51, 475–481. [Google Scholar] [CrossRef]
- Terrazzino, S.; Argyriou, A.A.; Cargnin, S.; Antonacopoulou, A.G.; Briani, C.; Bruna, J.; Velasco, R.; Alberti, P.; Campagnolo, M.; Lonardi, S.; et al. Genetic determinants of chronic oxaliplatin-induced peripheral neurotoxicity: A genome-wide study replication and meta-analysis. J. Peripher. Nerv. Syst. 2015, 20, 15–23. [Google Scholar] [CrossRef]
- Motsinger-Reif, A.A.; Jorgenson, E.; Relling, M.V.; Kroetz, D.L.; Weinshilboum, R.; Cox, N.J.; Roden, D.M. Genome-wide association studies in pharmacogenomics:successes and lessons. Pharm. Genom. 2013, 23, 1744–6872. [Google Scholar] [CrossRef] [Green Version]
- Singleton, A.B.; Hardy, J.; Traynor, B.J.; Houlden, H. Towards a complete resolution of the genetic architecture of disease. Trends Genet. 2010, 26, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Scientific, T.F. Axiom Spain Biobank Array. Available online: http://www.usc.es/cegen/wp-content/uploads/2019/08/COL32017-1217-Axiom-Spain-EN_FLR_FINAL.pdf. (accessed on 7 October 2020).
- Mizzi, C.; Peters, B.; Mitropoulou, C.; Mitropoulos, K.; Katsila, T.; Agarwal, M.R.; Van Schaik, R.H.N.; Drmanac, R.; Borg, J.; Patrinos, G.P. Personalized pharmacogenomics profiling using whole-genome sequencing. Pharmacogenomics 2014, 15, 1223–1234. [Google Scholar] [CrossRef]
- Apellániz-Ruiz, M.; Lee, M.Y.; Sánchez-Barroso, L.; Gutiérrez-Gutiérrez, G.; Calvo, I.; García-Estévez, L.; Sereno, M.; García-Donás, J.; Castelo, B.; Guerra, E.; et al. Whole-exome sequencing reveals defective CYP3A4 variants predictive of paclitaxel dose-limiting neuropathy. Proc. Clin. Cancer Res. 2015, 21, 322–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozyra, M.; Ingelman-Sundberg, M.; Lauschke, V.M. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet. Med. 2017, 19, 20–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, M.; Niemi, M.; Hiratsuka, M.; Kumondai, M.; Ingelman-Sundberg, M.; Lauschke, V.M.; Rodríguez-Antona, C. Novel copy-number variations in pharmacogenes contribute to interindividual differences in drug pharmacokinetics. Genet. Med. 2018, 20, 622–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingelman-Sundberg, M.; Mkrtchian, S.; Zhou, Y.; Lauschke, V.M. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum. Genom. 2018, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schaller, L.; Lauschke, V.M. The genetic landscape of the human solute carrier (SLC) transporter superfamily. Hum. Genet. 2019, 138, 1359–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Staveren, M.C.; Jan Guchelaar, H.; Van Kuilenburg, A.B.P.; Gelderblom, H.; Maring, J.G. Evaluation of predictive tests for screening for dihydropyrimidine dehydrogenase deficiency. Pharm. J. 2013, 13, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Van Kuilenburg, A.B.P.; Häusler, P.; Schalhorn, A.; Tanck, M.W.T.; Proost, J.H.; Terborg, C.; Behnke, D.; Schwabe, W.; Jabschinsky, K.; Maring, J.G. Evaluation of 5-fluorouracil pharmacokinetics in cancer patients with a c.19051G>A Mutation in DPYD by means of a bayesian limited sampling strategy. Clin. Pharm. 2012, 51, 163–174. [Google Scholar] [CrossRef]
- Maring, J.G.; Van Kuilenburg, A.B.P.; Haasjes, J.; Piersma, H.; Groen, H.J.M.; Uges, D.R.A.; Van Gennip, A.H.; De Vries, E.G.E. Reduced 5-FU clearance in a patient with low DPD activity due to heterozygosity for a mutant allele of the DPYD gene. Br. J. Cancer 2002, 86, 1028–1033. [Google Scholar] [CrossRef]
- Offer, S.M.; Lee, A.M.; Mattison, L.K.; Fossum, C.; Wegner, N.J.; Diasio, R.B. A DPYD Variant (Y186C) in Individuals of African Ancestry Is Associated With Reduced DPD Enzyme Activity. Clin. Pharm. Ther. 2013, 94, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Morel, A.; Boisdron-Celle, M.; Fey, L.; Soulie, P.; Craipeau, M.C.; Traore, S.; Gamelin, E. Clinical relevance of different dihydropyrimidine dehydrogenase gene single nucleotide polymorphisms on 5-fluorouracil tolerance. Mol. Cancer Ther. 2006, 5, 2895–2904. [Google Scholar] [CrossRef] [Green Version]
- Van Kuilenburg, A.; Hausler, P.; Schalhorn, A.; Tanck, M.; Proost, J.H.; Terborg, C.; Behnke, D.; Schwabe, W.; Jabschinsky, K.; Maring, J.G. Evaluation of 5-FU pharmacokinetics in cancer patients with DPD deficiency using a Bayesian limited sampling strategy. Ther. Drug Monit. 2011, 33, 478. [Google Scholar]
- Zhang, H.; Li, Y.-M.; Zhang, H.; Jin, X. DPYD*5 gene mutation contributes to the reduced DPYD enzyme activity and chemotherapeutic toxicity of 5-FU. Med. Oncol. 2007, 24, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Belló, M.; Mangas-Sanjuán, V.; Martínez-Gómez, M.A.; López-Montenegro Soria, M.Á.; Climente-Martí, M.; Merino-Sanjuán, M. Evaluation of ABC gene polymorphisms on the pharmacokinetics and pharmacodynamics of capecitabine in colorectal patients: Implications for dosing recommendations. Br. J. Clin. Pharm. 2020, online. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, G.; Cecchin, E.; Corona, G.; Russo, A.; Buonadonna, A.; D’Andrea, M.; Pasetto, L.M.; Pessa, S.; Errante, D.; De Pangher, V.; et al. The role of UGT1A1*28 polymorphism in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J. Clin. Oncol. 2006, 24, 3061–3068. [Google Scholar] [CrossRef] [PubMed]
- Cecchin, E.; Innocenti, F.; D’Andrea, M.; Corona, G.; De Mattia, E.; Biason, P.; Buonadonna, A.; Toffoli, G. Predictive role of the UGT1A1, UGT1A7, and UGT1A9 genetic variants and their haplotypes on the outcome of metastatic colorectal cancer patients treated with fluorouracil, leucovorin, and irinotecan. J. Clin. Oncol. 2009, 27, 2457–2465. [Google Scholar] [CrossRef]
- Labriet, A.; De Mattia, E.; Cecchin, E.; Lévesque, É.; Jonker, D.; Couture, F.; Buonadonna, A.; D’Andrea, M.; Villeneuve, L.; Toffoli, G.; et al. Improved progression-free survival in irinotecan-treated metastatic colorectal cancer patients carrying the HNF1A coding variant p.I27L. Front. Pharm. 2017, 8, 712. [Google Scholar] [CrossRef]
- Fujita, K.I.; Nagashima, F.; Yamamoto, W.; Endo, H.; Sunakawa, Y.; Yamashita, K.; Ishida, H.; Mizuno, K.; Matsunaga, M.; Araki, K.; et al. Association of ATP-binding cassette, sub-family C, number 2 (ABCC2) genotype with pharmacokinetics of irinotecan in Japanese patients with metastatic colorectal cancer treated with irinotecan plus infusional 5-fluorouracil/leucovorin (FOLFIRI). Biol. Pharm. Bull. 2008, 31, 2137–2142. [Google Scholar] [CrossRef] [Green Version]
- Paoluzzi, L.; Singh, A.S.; Price, D.K.; Danesi, R.; Mathijssen, R.H.J.; Verweij, J.; Figg, W.D.; Sparreboom, A. Influence of genetic variants in UGT1A1 and UGT1A9 on the in vivo glucuronidation of SN-38. J. Clin. Pharm. 2004, 44, 854–860. [Google Scholar] [CrossRef]
- Sai, K.; Saeki, M.; Saito, Y.; Ozawa, S.; Katori, N.; Jinno, H.; Hasegawa, R.; Kaniwa, N.; Sawada, J.; Komamura, K.; et al. UGT1A1 haplotypes associated with reduced glucuronidation and increased serum bilirubin in irinotecan-administered Japanese patients with cancer*1. Clin. Pharm. Ther. 2004, 75, 501–515. [Google Scholar] [CrossRef]
- De Jong, F.A.; Scott-Horton, T.J.; Kroetz, D.L.; McLeod, H.L.; Friberg, L.E.; Mathijssen, R.H.; Verweij, J.; Marsh, S.; Sparreboom, A. Irinotecan-induced diarrhea: Functional significance of the polymorphic ABCC2 transporter protein. Clin. Pharm. Ther. 2007, 81, 42–49. [Google Scholar] [CrossRef]
- de Jong, F.A.; Kehrer, D.F.S.; Mathijssen, R.H.J.; Creemers, G.; de Bruijn, P.; van Schaik, R.H.N.; Planting, A.S.T.; van der Gaast, A.; Eskens, F.A.L.M.; Janssen, J.T.P.; et al. Prophylaxis of Irinotecan-Induced Diarrhea with Neomycin and Potential Role for UGT1A1*28 Genotype Screening: A Double-Blind, Randomized, Placebo-Controlled Study. Oncologist 2006, 11, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Minami, H.; Sai, K.; Saeki, M.; Saito, Y.; Ozawa, S.; Suzuki, K.; Kaniwa, N.; Sawada, J.I.; Hamaguchi, T.; Yamamoto, N.; et al. Irinotecan pharmacokinetics/pharmacodynamics and UGT1A genetic polymorphisms in Japanese: Roles of UGT1A1*6 and *28. Pharm. Genom. 2007, 17, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Jada, S.R.; Lim, R.; Wong, C.I.; Shu, X.; Lee, S.C.; Zhou, Q.; Goh, B.C.; Chowbay, B. Role of UGT1A1*6, UGT1A1*28 and ABCG2 c.421C>A polymorphisms in irinotecan-induced neutropenia in Asian cancer patients. Cancer Sci. 2007, 98, 1461–1467. [Google Scholar] [CrossRef] [PubMed]
- Sai, K.; Kaniwa, N.; Itoda, M.; Saito, Y.; Hasegawa, R.; Komamura, K.; Ueno, K.; Kamakura, S.; Kitakaze, M.; Shirao, K.; et al. Haplotype analysis of ABCB1/MDR1 blocks in a Japanese population reveals genotype-dependent renal clearance of irinotecan. Pharmacogenetics 2003, 13, 741–757. [Google Scholar] [CrossRef]
- Henricks, L.M.; Lunenburg, C.A.T.C.; de Man, F.M.; Meulendijks, D.; Frederix, G.W.J.; Kienhuis, E.; Creemers, G.-J.; Baars, A.; Dezentjé, V.O.; Imholz, A.L.T.; et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: A prospective safety analysis. Lancet Oncol. 2018, 19, 1459–1467. [Google Scholar] [CrossRef]
- Kleibl, Z.; Fidlerova, J.; Kleiblova, P.; Kormunda, S.; Bilek, M.; Bouskova, K.; Sevcik, J.; Novotny, J. Influence of dihydropyrimidine dehydrogenase gene (DPYD) coding sequence variants on the development of fluoropyrimidine-related toxicity in patients with high-grade toxicity and patients with excellent tolerance of fluoropyrimidine-based chemotherapy. Neoplasma 2009, 56, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Deenen, M.J.; Meulendijks, D.; Cats, A.; Sechterberger, M.K.; Severens, J.L.; Boot, H.; Smits, P.H.; Rosing, H.; Mandigers, C.M.; Soesan, M.; et al. Upfront Genotyping of DPYD*2A to Individualize Fluoropyrimidine Therapy: A Safety and Cost Analysis. J. Clin. Oncol. 2016, 34, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Cortejoso, L.; García-González, X.; García, M.I.; García-Alfonso, P.; Sanjurjo, M.; López-Fernández, L.A. Cost-effectiveness of screening for DPYD polymorphisms to prevent neutropenia in cancer patients treated with fluoropyrimidines. Pharmacogenomics 2016, 17, 979–984. [Google Scholar] [CrossRef]
- Murphy, C.; Byrne, S.; Ahmed, G.; Kenny, A.; Gallagher, J.; Harvey, H.; O’Farrell, E.; Bird, B. Cost Implications of Reactive Versus Prospective Testing for Dihydropyrimidine Dehydrogenase Deficiency in Patients with Colorectal Cancer: A Single-Institution Experience. Dose-Response 2018, 16, 16. [Google Scholar] [CrossRef] [Green Version]
- Henricks, L.M.; Lunenburg, C.A.; De Man, F.M.; Meulendijks, D.; Frederix, G.W.; Kienhuis, E.; Creemers, G.-J.; Baars, A.; Dezentjé, V.O.; Imholz, A.L.; et al. A cost analysis of upfront DPYD genotype–guided dose individualisation in fluoropyrimidine-based anticancer therapy. Eur. J. Cancer 2019, 107, 60–67. [Google Scholar] [CrossRef]
- Gold, H.T.; Hall, M.J.; Blinder, V.; Schackman, B.R. Cost effectiveness of pharmacogenetic testing for uridine diphosphate glucuronosyltransferase 1A1 before irinotecan administration for metastatic colorectal cancer. Cancer 2009, 115, 3858–3867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obradovic, M.; Mrhar, A.; Kos, M. Cost-effectiveness of UGT1A1 genotyping in second-line, high-dose, once every 3 weeks irinotecan monotherapy treatment of colorectal cancer. Pharmacogenomics 2008, 9, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Butzke, B.; Oduncu, F.S.; Severin, F.; Pfeufer, A.; Heinemann, V.; Giesen-Jung, C.; Stollenwerk, B.; Rogowski, W.H. The cost-effectiveness of UGT1A1 genotyping before colorectal cancer treatment with irinotecan from the perspective of the German statutory health insurance. Acta Oncol. 2016, 55, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Roncato, R.; Cecchin, E.; Montico, M.; De Mattia, E.; Giodini, L.; Buonadonna, A.; Solfrini, V.; Innocenti, F.; Toffoli, G. Cost Evaluation of Irinotecan-Related Toxicities Associated With the UGT1A1*28 Patient Genotype. Clin. Pharm. Ther. 2017, 102, 123–130. [Google Scholar] [CrossRef]
- Agencia Española de Medicamentosy Produtos Sanitarios Fluorouracilo, Capecitabina, Tegafury Flucitosina en Pacientes con Déficit de Dihidropirimidina Deshidrogenasa. Available online: https://www.aemps.gob.es/informa/notasinformativas/medicamentosusohumano-3/seguridad-1/2020-seguridad-1/fluorouracilo-capecitabina-tegafur-y-flucitosina-en-pacientes-con-deficit-de-dihidropirimidina-deshidrogenasa/ (accessed on 24 September 2020).
- Amstutz, U.; Henricks, L.M.; Offer, S.M.; Barbarino, J.; Schellens, J.H.; Swen, J.J.; Klein, T.E.; McLeod, H.L.; Caudle, K.E.; Diasio, R.B.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin. Pharm. Ther. 2018, 103, 210–216. [Google Scholar] [CrossRef]
- The Pharmacogenetics Working Group. UGT1A1: Irinotecan. Available online: https://www.g-standaard.nl/risicoanalyse/B0001694.PDF (accessed on 24 September 2020).
- European Medicines Agency. Recommendations on DPD Testing Prior to Treatment with Fluorouracil, Capecitabine, Tegafur and Flucytosine. Available online: https://www.ema.europa.eu/en/news/ema-recommendations-dpd-testing-prior-treatment-fluorouracil-capecitabine-tegafur-flucytosine (accessed on 24 September 2020).
- Food and Drug Administration. Table of Pharmacogenomic Biomarkers in Drug Labeling. Available online: https://www.fda.gov/media/124784/download (accessed on 24 September 2020).
- European Medicines Agency. Erbitux (cetuximab). Am. J. Neuroradiol. 2010, 31, 626–627. [Google Scholar]
- Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events (CTCAE).v.5.0. Available online: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcae_v5_quick_reference_5x7.pdf (accessed on 23 July 2020).
- Kerns, S.L.; Fachal, L.; Dorling, L.; Barnett, G.C.; Baran, A.; Peterson, D.R.; Hollenberg, M.; Hao, K.; Narzo, A.D.; Ahsen, M.E.; et al. Radiogenomics Consortium Genome-Wide Association Study Meta-Analysis of Late Toxicity After Prostate Cancer Radiotherapy. J. Natl. Cancer Inst. 2020, 112, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Barnett, G.C.; Thompson, D.; Fachal, L.; Kerns, S.; Talbot, C.; Elliott, R.M.; Dorling, L.; Coles, C.E.; Dearnaley, D.P.; Rosenstein, B.S.; et al. A genome wide association study (GWAS) providing evidence of an association between common genetic variants and late radiotherapy toxicity. Radiother. Oncol. 2014, 111, 178–185. [Google Scholar] [CrossRef]
- Fachal, L.; Gómez-Caamaño, A.; Barnett, G.C.; Peleteiro, P.; Carballo, A.M.; Calvo-Crespo, P.; Kerns, S.L.; Sánchez-García, M.; Lobato-Busto, R.; Dorling, L.; et al. A three-stage genome-wide association study identifies a susceptibility locus for late radiotherapy toxicity at 2q24.1. Nat. Genet. 2014, 46, 891–894. [Google Scholar] [CrossRef]
- Kerns, S.L.; Stock, R.G.; Stone, N.N.; Blacksburg, S.R.; Rath, L.; Vega, A.; Fachal, L.; Gómez-Caamaño, A.; De Ruysscher, D.; Lammering, G.; et al. Genome-wide association study identifies a region on chromosome 11q14.3 associated with late rectal bleeding following radiation therapy for prostate cancer. Radiother. Oncol. 2013, 107, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Kerns, S.L.; Dorling, L.; Fachal, L.; Bentzen, S.; Pharoah, P.D.; Barnes, D.R.; Gómez-Caamaño, A.; Carballo, A.M.; Dearnaley, D.P.; Peleteiro, P.; et al. Meta-analysis of Genome Wide Association Studies Identifies Genetic Markers of Late Toxicity Following Radiotherapy for Prostate Cancer. EBioMedicine 2016, 10, 150–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, M.; Kirkham, J.; Dwan, K.; Sloan, D.J.; Davies, G.R.; Jorgensen, A.L. STrengthening the Reporting Of Pharmacogenetic Studies: Development of the STROPS guideline. PLoS Med. 2020, 17, e1003344. [Google Scholar] [CrossRef]
- Hegde, M.; Santani, A.; Mao, R.; Ferreira-Gonzalez, A.; Weck, K.E.; Voelkerding, K.V. Development and validation of clinical whole-exome and whole-genome sequencing for detection of germline variants in inherited disease. Arch. Pathol. Lab. Med. 2017, 141, 798–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Fujikura, K.; Mkrtchian, S.; Lauschke, V.M. Computational methods for the pharmacogenetic interpretation of next generation sequencing data. Front. Pharm. 2018, 9, 1–17. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| CRC Stage | Treatment | |
|---|---|---|
| Surgery | Pharmacological Treatment | |
| I | Wide surgical resection and anastomosis | No adjuvant chemotherapy recommended |
| II | Wide surgical resection and anastomosis | Adjuvant chemotherapy for high-risk could be considered |
| III | Wide surgical resection and anastomosis | Adjuvant administration of oxaliplatin plus 5-FU or capecitabine |
| IV | The majority of patients have metastases that initially are not suitable for potentially curative resection. Revaluate after chemotherapy | Cytotoxic agents: 1st line: 5-FU or capecitabine alone or in combination either with oxaliplatin or irinotecan 2nd line: if refractory to irinotecan-based treatment, FOLFOX is recommended; and if refractory to oxaliplatin-based treatment, FOLFIRI is recommended |
| Biological targeted agents: 1st line: monoclonal antibodies against VEGF (bevacizumab, aflibercept) and/or EGFR (cetuximab, panitumumab), if RAS mutation excluded Multi-kinase inhibitor: regorafenib | ||
| Treatment | Significant ADRs (According to FDA Labels) * | ADR Incidence (% Patients) | Ref. |
|---|---|---|---|
| 5-Fluororacil | Diarrhoea, neutropenia, mucositis, nausea/vomiting, stomatitis, asthenia, leukopenia, anaemia. | 94% | [12] |
| Capecitabine | Hand-and-foot syndrome, diarrhoea, nausea/vomiting, abdominal pain, fatigue, hyperbilirubinemia. | 96% | [12] |
| Oxaliplatin | Peripheral sensory neuropathy, neutropenia, thrombocytopenia, anaemia, nausea/vomiting, increase in transaminases and alkaline phosphatase, diarrhoea, fatigue, stomatitis. | >92% | [15] |
| Irinotecan | Nausea/vomiting, diarrhoea, neutropenia, alopecia, abdominal pain, constipation, anorexia, leukopenia, anaemia, asthenia, fever, body weight decreasing. | 100% | [16] |
| Cetuximab | Cutaneous adverse reactions, headache, diarrhoea, infection. | >87% | [17] |
| Panitumumab | Skin rash, paronychia, fatigue, nausea, diarrhoea. | >90% | [18] |
| Bevacizumab | Haemorrhage, hypertension, headache, rhinitis, proteinuria, taste alteration, dry skin, lacrimation disorder, back pain, exfoliative dermatitis. | >60% | [19] |
| Drug | Gene | SNP (rsID) | Change | Alternative Nomenclature | Frequency of Risk Allele a | Associated ADR | OR (95% CI) | Evidence Level b | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Fluoropyrimidines | DPYD | rs55886062 | NM_000110.3:c.1679T>G; NP_000101.2:p.Ile560Ser | DPYD*13 | 3 × 10−4 (C) | Global toxicity | 6.0 (0.6–61) | 1A | [28] |
| rs3918290 | NM_000110.4:c.1905+1G>A (Splice donor) | DPYD*2A | 0.007 (T) | Global toxicity | 8.5 (1.8–40.9) | 1A | [29] | ||
| rs67376798 | NM_000110.3:c.2846A>T; NP_000101.2:p.Asp949Val | 0.003 (A) | Global toxicity | 7.8 (1.6–39.2) | 1A | [31] | |||
| rs115232898 | NM_000110.3:c.557A>G; NP_000101.2:p.Tyr186Cys | 0.002 (Afr: 0.023) (C) | Neutropenia, mucositis, alopecia | - | 1A | [32] | |||
| rs75017182 | NM_000110.4:c.1129-5923C>G (Intronic) | 0.013 (C) | Global toxicity | 6.8 (2.0–23) | 1A | [33] | |||
| rs56038477 | NM_000110.3:c.1236G>A; NP_000101.2:p.Glu412= | 0.014 (T) | Gastrointestinal; haematological | 2.0 (1.5–2.8) 2.8 (1.2–3.7) | 3 | [34] | |||
| rs72549303 c | NM_000110.4:c.1898del; NP_000101.2:p.Pro633fs | DPYD*3 | NA | NA | NA | 1A | [31] | ||
| rs72549309 c | NM_000110.4:c.295_298TCAT [1]; NP_000101.2:p.Phe100fs | DPYD*7 | 6 × 10−5 (delATGA) | NA | NA | 1A | [31] | ||
| rs1801266 c | NM_000110.4:c.703C>T; NP_000101.2:p.Arg235Trp | DPYD*8 | 3 × 10−5 (A) | NA | NA | 1A | [31] | ||
| rs1801268 c | NM_000110.4:c.2983G>T; NP_000101.2:p.Val995Phe | DPYD*10 | NA | NA | NA | 1A | [31] | ||
| rs78060119 | NM_000110.3:c.1156G>T; NP_000101.2:p.Glu386Ter | DPYD*12 | 8 × 10−6 (A) | Leucopenia, thrombocytopenia, mucositis | NA | 1A | [35] | ||
| rs2297595 | NM_000110.3:c.496A>G; NP_000101.2:p.Met166Val | 0.085(C) | Global toxicity | 5.9 (1.3–27.2) | 3 | [36] | |||
| rs1801265 | NM_000110.3:c.85T>C; NP_000101.2:p.Cys29Arg | DPYD*9A | 0.228 (G) | Diarrhoea | 0.8 (0.7–1) | 3 | [37] | ||
| rs1801267 c | NM_000110.4:c.2657G>A; NP_000101.2:p.Arg886His | DPYD*9B | 1 × 10−4 (T) | NA | NA | NA | [38] | ||
| rs1801159 | NM_000110.3:c.1627A>G; NP_000101.2:p.Ile543Val | DPYD*5 | 0.198 (C) | Diarrhoea | 4.9 (-) | 3 | [39] | ||
| rs1801158 | NM_000110.3:c.1601G>A; NP_000101.2:p.Ser534Asn | DPYD*4 | 0.015 (T) | Global toxicity | 1.7 (1.1–2.6) | 3 | [37] | ||
| rs17376848 | NM_000110.3:c.1896T>C; NP_000101.2:p.Phe632= | 0.051 (G) | Global toxicity | 14.5 (1.4–155.2) | 3 | [36] | |||
| rs1801160 | NM_000110.3:c.2194G>A; NP_000101.2:p.Val732Ile | DPYD*6 | 0.048 (T) | Global toxicity | 2.1 (1.5–3.0) | 3 | [40] | ||
| rs12022243 | NM_000110.4:c.1906-14763G>A (Intronic) | 0.181 (T) | Global toxicity | 1.7 (1.5- 1.9) | 3 | [41] | |||
| rs12119882 | NM_000110.4:c.680+2545T>C (Intronic) | 0.075 (G) | Hyperbilirubinemia | 4.9 (1.2–20.8) | 3 | [42] | |||
| rs76387818 | Intergenic | 0.019 (A) | Global toxicity | 4.1 (3.5–4.6) | 3 | [41] | |||
| rs12132152 | Intergenic | 0.020 (A) | HFS;global toxicity | 6.1 (5.5–6.8); 1.6 (1.4–1.8) | 3 | [41] | |||
| TYMS | rs183205964 | NM_001071.4:c.-86= (5′ UTR) | 3 × 10−5 (C) | Global toxicity | 3.0 (1.1- 8.4) | 3 | [43] | ||
| rs2853741 | NM_001071.4:c. (Upstream transcript) | 0.322 (T) | Diarrhoea | 0.3 (0.1–0.7) | 3 | [42] | |||
| rs699517 | NM_017512.7:c.*1289= (3′ UTR) | 0.379 (T) | Nausea/vomiting;asthenia | 7.9 (1.5–41.6); 0.3 (0.1–0.8) | 3 | [42] | |||
| rs45445694 | NM_001071.4:c. (5′ UTR) | 0.007 (2R2R) | Global toxicity | 1.7 (-) | 3 | [44] | |||
| rs2853542 | NM_001071.4:c.-58= (5′ UTR) | Global toxicity; HFS | 1.5 (1.2–1.8); 1.4 (1.2–1.8) | NA | [45] | ||||
| rs11280056 | NM_017512.7:c.*853_*861= (3′ UTR) | Global toxicity | 1.7 (1.2–2.2) | NA | [45] | ||||
| ENOSF1 | rs2612091 | NM_017512.7:c.742-227G>C (Intronic) | 0.373 (C) | Global toxicity | 1.6 (1.4–1.8) | 3 | [41] | ||
| UMPS | rs2279199 | NM_000373.4:c. (Genic upstream transcript) | 0.556 (T) | Nausea/vomiting | 0.2 (0.1–1.0) | 3 | [42] | ||
| rs4678145 | NM_000373.4:c.156+607G>C (Intronic) | 0.096 (C) | Asthenia | 4.5 (1.6–13.2) | 3 | [42] | |||
| rs1801019 d | NM_000373.4:c.638G>C; NP_000364.1:p.Gly213Ala | 0.169 (C) | Global toxicity | 17.6 (1.6–195.9) | 3 | [46] | |||
| MTHFR | rs1801131 | NM_001330358.1:c.1409A>C; NP_001317287.1:p.Glu470Ala | 0.289 (G) | HFS | 10.0 (3.8–27.8) | 3 | [47] | ||
| rs1801133 | NM_001330358.1:c.788C>T; NP_001317287.1:p.Ala263Val | 0.315 (A) | Neutropenia | 2.3 (1.2–4.6) | 3 | [48] | |||
| TYMP | rs11479 | NM_001113755.3:c.1412C>T; NP_001244917.1:p.Ser471Leu | 0.094 (A) | Global toxicity | 2.7 (1.2–5.9) | 3 | [49] | ||
| MIR27A | rs895819 | NR_029501.1:n.40A>G (Non-coding transcript) | 0.335 (C) | Global toxicity | 1.6 (1.1–2.2) | 3 | [50] | ||
| ABCC1 | rs7194667 | NM_032583.4:c.1609-491A>C (Intronic) | 0.063 (G) | Leucopenia | 3.31 (1.3–8.7) | 3 | [51] | ||
| ABCB1 | rs1045642 | NM_001348945.1:c.3645T>C; NP_001335874.1:p.Ile1215= | ABCB1*6 | 0.504 (G) | HFS | NA | 3 | [52] | |
| rs2032582 | NM_001348945.1:c.2887T>G; NP_001335874.1:p.Ser963Ala | ABCB1*7 | 0.637 (C) | HFS | NA | 3 | [52] | ||
| rs1128503 | NM_001348945.1:c.1446T>C; NP_001335874.1:p.Gly482= | ABCB1*8 | 0.614 (G) | Neutropenia | NA | 3 | [52] | ||
| SLC22A7 | rs2270860 | NM_006672.3:c.1269C>T; NP_006663.2:p.Ser423= | 0.368 (T) | Global toxicity | 17.1 (1.7–170.3) | 3 | [42] | ||
| rs4149178 | NM_006672.3:c.1586+206A>G (Intronic) | 0.795 (A) | Diarrhoea | 0.3 (0.1–0.9) | 3 | [42] | |||
| CDA | rs2072671 | NM_001785.3:c.79A>C; NP_001776.1:p.Lys27Gln | 0.279 (C) | Global toxicity | 1.8 (1.1–3.0) | 3 | [53] | ||
| rs1048977 | NM_001785.3: c.435C>T; NP_001776.1:p.Thr145= | 0.307 (T) | Hyperbilirubinemia | 8.6 (1.1–70.3) | 3 | [42] | |||
| rs602950 | NM_001785.3:c. (Upstream transcript) | 0.224 (G) | Diarrhoea | 2.3 (1.3–4.2) | 3 | [47] | |||
| rs3215400 | NM_001785.3:c.-33_-31= (5′ UTR) | 0.555 (delC) | HFS | 0.5 (0.3–1.0) | 3 | [54] | |||
| rs532545 | NM_001785.3:c. (Upstream transcript) | 0.220 (T) | Diarrhoea | 2.3 (1.3–4.2) | NA | [47] | |||
| CES1 | rs3217164 | NM_001025195.2:c.693+129del (Intronic) | 0.607 (G) | Global toxicity | 4.1 (1.8–9.0) | 3 | [55] | ||
| rs2244614 | NM_001025195.2:c.1171-41C>T (Intronic) | 0.482 (G) | Global toxicity | 4.7 (1.9–12.0) | 3 | [55] | |||
| rs2244613 | NM_001025195.2:c.1171-33C>T (Intronic) | 0.232 (G) | Global toxicity | 6.4 (1.5–27.7) | 3 | [55] | |||
| CES1P1 | rs7187684 | NR_003276.2:n. (Intronic) | 0.278 (T) | Global toxicity | 6.5 (1.5–28.0) | 3 | [55] | ||
| rs11861118 | NR_003276.2:n. (Upstream transcript) | 0.161 (G) | Global toxicity | 6.5 (1.5–28.0) | 3 | [55] | |||
| Intergenic | rs9936750 | Intergenic | 0.161 C | Global toxicity | 4.6 (1.5–13.9) | 3 | [56] | ||
| Intergenic | rs10876844 | Intergenic | 0.439 (A) | Diarrhoea | 6.5 (1.6–27.2) | NA | [57] | ||
| Oxaliplatin | ABCC2 | rs717620 | NM_000392.5:c.-24= (5‘ UTR) | 0.171 (T) | Neuropathy | 14.4 (1.6–127.0) | 3 | [58] | |
| rs3740066 | NM_000392.5:c.3972C>TNP_000383.2:p.Ile1324= | Neuropathy | 3.0 (1.2–7.7) | NA | [58] | ||||
| rs1885301 | NM_000392.5:c. (Upstream Transcript) | 0.413 (A) | Neuropathy | 3.1 (1.4–6.9) | NA | [58] | |||
| rs4148396 | NM_000392.5:c.3258+56T>C (Intronic) | 0.347 (T) | Neuropathy | 4.7 (1.6–13.7) | NA | [58] | |||
| ABCG2 | rs3114018 | NM_004827.3:c.-19-3415T>G (Intronic) | 0.516 (A) | Neuropathy | 2.7 (1.0–4.4) | NA | [59] | ||
| GSTP1 | rs1695 | NM_000852.3:c.313A>G; NP_000843.1:p.Ile105Val | GSTP1*B | 0.339 (G) | Dying | 3.0 (1.2–7.6) | 3 | [60] | |
| GSTM1 | Null genotype | - | GSTM1*0 | Neutropenia | 2.0 (1.1–3.7) | NA | [61] | ||
| GSTT1 | Null genotype | - | Neutropenia | 2.0 (1.1–3.7) | NA | [61] | |||
| ERCC1 | rs11615 | NM_202001.3:c.354T>C; NP_001356337.1:p.Asn118= | 0.498 (A) | Neutropenia | 4.6 (1.2–17.4) | 3 | [48] | ||
| ERCC2 | rs13181 | NM_000400.3:c.2251A>C; NP_000391.1:p.Lys751Gln | 0.323 (G) | Haematological | 2.2 (1.3–3.8) | 3 | [62] | ||
| rs238406 | NM_000400.4:c.468A>C NP_000391.1:p.Arg156= | 0.645 (C) | Thrombocytopenia | NA | NA | [63] | |||
| PARD3B | rs17626122 | NM_001302769.2:c.3261-6168T>C (Intronic) | 0.550 (T) | Global toxicity | 3.4 (1.9–6.8) | 3 | [57] | ||
| Intergenic | rs7325568 | Intergenic | 0.409 (T) | Haematological | 1.8 (1.3–2.4) | 3 | [57] | ||
| Irinotecan | UGT1A1 | rs3064744 | NM_000463.3:c.(Upstream transcript) | UGT1A1*28 | 0.347 (dupTA) (EAS:0.122) | Global toxicity | 7.2 (2.5–22.3) | 2A | [64] |
| rs4148323 c | NM_000463.2:c.211G>A; NP_000454.1:p.Gly71Arg | UGT1A1*6 | 0.014 (EAS: 0.144) (A) | NA | NA | 2A | [65] | ||
| rs11563250 | NM_001367507.1:c. (Genic upstream transcript) | 0.893 (A) | Neutropenia | 0.3 (0.2–0.6) | 3 | [66] | |||
| rs4124874 | NM_001072.3:c.862-10021T>G (Intronic) | UGT1A1*60 | 0.452 (T) | Neutropenia | NA | 3 | [67] | ||
| rs10929302 | NM_019075.2:c.856-9898G>A (Intronic) | UGT1A1*93 | 0.299 (A) | Global toxicity | 8.4 (1.9–37.2) | 3 | [68] | ||
| UGT1A9 | rs11692021 | NM_021027.3:c.855+9770T>C (Intronic) | 0.349 (C) | Global toxicity | 2.0 (1.1–3.6) | 3 | [69] | ||
| rs3832043 e | NM_021027.3:c. (Upstream Transcript) | 0.609 (delT) | Diarrhoea | 6.3 (1.3–31.7) | 3 | [70] | |||
| UGT1A6 | rs2070959 | NM_001072.4:c.541A>G (Intronic) | 0.689 (A) | Global toxicity | 2.1 (1.1–3.9) | 3 | [69] | ||
| ABCG1 | rs225440 | NM_016818.3:c.286+7029C>T (Intronic) | 0.428 (T) | Neutropenia | 3.1 (1.1–8.6) | 3 | [71] | ||
| rs425215 | NM_016818.3:c.974-898C>G (Intronic) | 0.623 (G) | Gastrointestinal | 11.4 (1.7–78.4) | NA | [72] | |||
| ABCB1 | rs12720066 | NM_001348945.1:c.2529+971T>G (Intronic) | 0.035 (C) | Neutropenia | NA | 3 | [73] | ||
| ABCC1 | rs17501331 | NM_004996.4:c.49-12232A>G (Intronic) | 0.928 (A) | Neutropenia | NA | 3 | [73] | ||
| rs3743527 | NM_004996.4:c.*543= (3′ UTR) | 0.774 (C) | Neutropenia | NA | 3 | [73] | |||
| ABCC5 | rs2292997 | NM_005688.4:c.129+7980C>T (Intronic) | 0.126 (A) | Neutropenia | 3.2 (1.3–7.9) | 3 | [71] | ||
| rs10937158 | NM_005688.4:c.130-1268A>T (Intronic) | 0.612 (C) | Diarrhoea | 0.4 (0.2–0.8) | 3 | [71] | |||
| rs3749438 | NM_005688.4:c.591+374C>T (Intronic) | 0.324 (A) | Diarrhoea | 5.9 (1.3–26.3) | 3 | [71] | |||
| rs562 | NM_005688.4:c.*1243= (3′ UTR) | 0.515 (C) | Gastrointestinal | 32.0 (2.8–370.8 | NA | [72] | |||
| ABCG2 | rs7699188 | NM_004827.3:c. (Genic upstream transcript) | 0.227 (A) | Global toxicity; non-haematological | 7.3 (1.5–34.5); 15.2 (2.5–78.2) | 3 | [74] | ||
| SLCO1B1 | rs2306283 | NM_006446.5:c.388A>G NP_006437.3:p.Asn130Asp | SLCO1B1*1b | 0.529 (G) | Gastrointestinal | 2.3 (0.4–15.1) | NA | [72] | |
| TOP1 | rs6072262 | NM_003286.4:c.279+61G>A (Intronic) | 0.144 (A) | Neutropenia | NA | 3 | [75] | ||
| TGFBR2 | rs3087465 | NM_001024847.2:c. (2KB upstream) | 0.659 (G) | Diarrhoea | 3.7 (1.0–13.3) | 3 | [76] | ||
| TGFB1 | rs1800469 | NM_000660.7:c. (Upstream transcript) | 0.701 (G) | Diarrhoea | 4.4 (1.0–18.9) | 3 | [76] | ||
| KCNQ5 | rs9351963 | NM_019842.4:c.490-1798A>C (Intronic) | 0.178 (C) | Diarrhoea | 3.3 (1.8–5.6) | 3 | [77] | ||
| Intergenic | rs10486003 | Intergenic | 0.913 (C) | Neuropathy | 0.3 (0.2–0.5) | NA | [78] | ||
| Intergenic | rs2338 | Intergenic | 0.275 (A) | Neuropathy | 2.3 (1.6–3.3) | NA | [78] | ||
| Intergenic | rs830884 | Intergenic | 0.92 (T) | Neuropathy | 0.3 (0.2–0.5) | NA | [78] | ||
| ACYP2 | rs843748 | NM_001320586.2:c.405-28913G>A (Intronic) | 0.379 (A) | Neuropathy | 2.4 (1.6–3.7) | NA | [78] | ||
| DLEU7 | rs797519 | NC_000013.11:g.50656996G>C (Intronic) | 0.548 (G) | Neuropathy | 0.5 (045–0.7) | NA | [78] | ||
| FARS2 | rs17140129 | NM_001318872.2:c.-22+36771A>G (Intronic) | 0.158 (G) | Neuropathy | 3.3 (1.8–6.4) | NA | [78] | ||
| Cetuximab | EGFR | rs712830 | NM_005228.5:c.-191= (5′ UTR) | 0.894 (C) | Global toxicity | 6.1 (1.6–23.8) | 3 | [79] | |
| rs2227983 | NM_005228.5:c.1562G>A NP_005219.2:p.Arg521Lys | 0.768 (G) | Skin toxicity | 3.2 (1.3–8.3) | 3 | [80] | |||
| rs11568315 | NM_005228.5:c.88+1195AC [10] (Intronic) | 3.9 × 10−4 (CA > 35) | Skin toxicity | 2.9 (1.0–8.9) | NA | [81] | |||
| RPS7 | rs10203413 | NC_000002.12:g.3581588G>A (Regulatory region) | 0.776 (G) | Skin toxicity | 0.1 (0.1–0.4) | NA | [82] | ||
| ZNF827 | rs12646351 | NC_000002.12:g.3581588G>A (Intronic) | 0.815 (G) | Skin toxicity | 0.04 (0.01–0.3) | NA | [82] | ||
| rs17806780 | NM_001306215.2:c.2383+11920A>T (Intronic) | 0.818 (T) | Skin toxicity | 0.04 (0.01–0.4) | NA | [82] | |||
| EPHA5 | rs7692430 | NM_004439.8:c.2237-1876A>G (Intronic) | 0.156 (G) | Skin toxicity | 4.6 (2.5–8.5) | NA | [82] | ||
| Bevacizumab | VEGF | rs3025039 | NM_001171623.1:c.*237= (3′ UTR) | 0.134 (T) | Hypertension | 0.2 (0.03–0.8) | NA | [83] | |
| rs2010963 | NM_001171623.1:c.-634= (5′ UTR) | 0.698 (G) | Hypertension | NA | NA | [84] | |||
| rs833061 | NM_001025366.3:c. (Upstream transcript) | 0.452 (C) | Hypertension | 0.2 (0.03–0.8) | NA | [85] | |||
| rs699947 | NM_001025366.3:c. (Upstream transcript) | 0.414 (A) | Hypertension | 0.1 (0.01–0.6) | NA | [85] |
| Approach | Advantages | Disadvantages |
|---|---|---|
| Candidate genes |
|
|
| SNP arrays (GWAS) |
|
|
| SNP arrays (targeted fine-mapping approaches) |
|
|
| NGS (targeted panels, WES, WGS) |
|
|
| Functional assays |
|
|
| N | Cohort | Method | Genes | Results | Ref. |
|---|---|---|---|---|---|
| 482 | Genomes Data, Wellderly Study | WGS or SNP array genotyping | 231 pharmacogenes | ≈17,733 (WGS) vs. 249.5 (SNP array) UGT1A1 (WGS): 254 of 332 variants were novel, 31 functional and 26 with frequency < 1%. | [131] |
| >6500 | 1KG phase 3; ESP | WES and WGS | 146 pharmacogenes | 19,328 SNV, 62.9% exonic 6225 and 6258 variants in ABC transporter (22 genes) and SLC genes (49), respectively, 253 variants in UGTs (16) and GTSs (14) 92.9% rare, 82.7% very rare 56.2% missenses ≈30–40% of the functional variability in pharmacogenes | [133] |
| 141,456 | gnomAD v2.1 a | WES and WGS | SLC genes | 204,287 SNVs and indels, 56.9% missenses, 2.5% frameshifts, 1.7% stop-gains and 1.5% variations in canonical splice sites Each individual had ≈29.7 putatively functional SLC variants, 18% of functional variability due to rare variants | [136] |
| 100 | QUASAR | Amplicon sequencing | DPYD and TYMS coding regions | Novel rare independent DPYD variant (c.1651G>A; p.Ala551Thr)—classified as strongly damaging | [41] |
| 62,402 | 1 KG phase 3; ExAC b | WES and WGS | 208 pharmacogenes | 201 (97%) genes had 5589 novel CNVs, 47% deletions and 54% duplications Novel deletions responsible for >5% of loss-of-function alleles in 87, 25, 49, 48, 59 and 51 genes in non-Finnish Europeans, Finnish, East Asians, South Asians, Africans and admixed Americans, respectively | [134] |
| Genes | Significant Variants | N | Pharmacokinetic Results | Ref. |
|---|---|---|---|---|
| Fluoropyrimidines | ||||
| DPYD | rs3918290 | 1 case (heterozygous for IVS14+1G>A) vs. 6 controls (CRC) | inactivation of one DPYD allele: strong ↓CL5-FU: severe toxicity | [139] |
| DPYD | rs1801265 rs115232898 rs55886062 | 175 CRC patients | rs55886062: lowest activity (p = 0.014) rs115232898: 46% ↓activity (p = 0.026) rs1801265: 27% ↑activity (p = 0.013) | [140] |
| DPYD | rs3918290 rs67376798 rs55886062 | 487 advanced carcinoma patients | rs3918290, rs67376798, or rs55886062: ↓CL5-FU (p < 0.001) | [141] |
| DPYD | rs3918290 | 30 patients (heterozygous for IVS14+1G>A) and 18 controls | rs3918290: 40% ↓Vmax (p < 0.001) | [142] |
| DPYD | rs1801159 | 112 gastric or colon cancer patients | rs1801159: ↓k (p = 0.022) and nausea/vomiting (p = 0.005) | [143] |
| DPYD | rs55886062 rs1801265 rs1801158 | Expression vector | rs1801158: 36% ↑activity (p = 3.4 × 10−7) rs1801265: 13% ↑activity (p = 0.0013) rs55886062: 75% ↓activity (p = 5.2 × 10−9) | [29] |
| DPYD | rs141044036 rs72549308 rs1801268 rs145773863 rs55674432 rs72547601 rs137999090 rs59086055 rs1801266 rs111858276 rs183385770 rs72549307 rs138616379 rs72549304 rs112766203 rs183105782 rs143986398 rs115232898 rs2297595 | Expression vector | rs141044036, rs72549308, rs1801268, rs145773863, rs55674432, rs137999090, rs72547601, rs59086055: <12.5% activity (p < 3.5 × 10−4) rs1801266, rs72549307, rs111858276, rs138616379, rs183385770, rs72549304: 12.5–25% activity (p < 0.0021) rs112766203, rs143986398, rs183105782, rs115232898: >25% ↓activity (p < 0.05) rs2297595: 120% ↑activity (p = 0.025) | [31] |
| ABC | rs2271862 | 48 CRC patients | ABCA2 rs2271862: ↑CL5-FU | [144] |
| ABCB1 ABCC1 ABCG2 UGT1A1 | rs12720066 rs6498588 rs10929302 | 85 advanced cancer patients | ABCB1 rs12720066 (p = 6.24 × 10−4) and rs6498588 (p = 9.50 × 10−4), and UGT1A1 rs10929302 (p = 9.00 × 10−5): ↑AUCSN-38 ↑AUCSN-38: G ≥ 3 neutropenia (p = 0.0001) | [73] |
| ABCG2 SLCO1B1 ABCB1 ABCC1 ABCC2 UGT1A1 UGT1A9 UGT1A7 CES CYP3A4 CYP3A5 HNF1A | rs717620 rs1169288 rs4149056 rs35605 rs1092302 rs3740066 | 85 advanced cancer patients | ABCC2 rs717620 (p = 0.002), HNF1A rs1169288 (p = 0.007), SLCO1B1 rs4149056 (p = 0.015): ↑AUCCPT-11 ABCC1 rs35605 (p = 0.031), UGT1A1 rs1092302 (p = 0.007): ↑AUCSN-38 ABCC2 rs3740066: ↑AUCSN-38G and ↑AUCAPC (p = 0.012) ABCC1 rs35605 (p = 0.023), rs3064744 (p < 0.0001): ↓GR | [111] |
| UGT1A1 | rs3064744 | 250 mCRC patients | ↓GR (p = 0.01) and ↑BI (p = 0.003): G ≥ 3 toxicity rs3064744: ↓GR (p = 0.01) and ↑BI (p = 0.007) | [145] |
| UGT1A1 UGT1A7 UGT1A9 | rs4124874 rs10929302 UGT1A7*3 | Subset of 71 patients | UGT1A1 rs4124874 and rs10929302: ↑BI (p = 0.03 and p = 0.04, respectively) UGT1A7*3: ↓GR (p = 0.02) and ↑BI (p = 0.007) | [146] |
| HNF1A | rs2244608 | Subset of 49 patients | rs2244608: ↑AUCSN-38 (p = 0.032), ↑BI (p = 0.021) and ↓GR (p = 0.035) | [147] |
| ABCC2 | rs2273697 rs17216114 rs1885301 rs2804402 rs717620 rs3740066 | 31 mCRC patients | rs2273697: ↓AUCCPT-11 (p = 0.011) rs17216114: ↓AUCSN-38 rs1885301, rs2804402, rs717620 and rs3740066: ↑AUNSN-38 (p < 0.03) | [148] |
| ABCB1 ABCC1 ABCC2 ABCG2 CES1 CES2 CYP3A4 CYP3A5 UGT1A XRCC1 | rs1128503 | 65 solid tumour patients | ABCB1 rs1128503: ↑AUCCPT-11 (p = 0.038), AUCSN-38 (p = 0.031) and ↓CLSN-38 (p = 0.015) | [96] |
| UGT1A1 | rs3064744 | 20 solid tumour patients | rs3064744 ↓GR (p = 0.001) and ↑BI (p = 0.001) AUCSN-38: neutropenia (p < 0.0001) | [105] |
| UGT1A1 UGT1A9 | rs3064744 | 94 solid tumour patients | rs3064744: ↓GR (p = 0.022) | [149] |
| UGT1A1 | rs4148323 rs4124874 rs3064744 | 85 solid tumour patients | rs4148323: ↓GR (p = 0.0372) rs4124874: ↑BI (p = 0.0048) rs3064744: ↑BI (p = 0.0007) | [150] |
| ABCC2 UGT1A1 | ABCC2*2 rs306474 | 167 solid tumour patients | ABCC2*2: ↓CLCPT-11 (p = 0.020) rs3064744: ↓CLSN-38 (p < 0.001), GR and BI (p = 0.014) | [151] |
| UGT1A1 | rs3064744 | 62 solid tumour patients | rs3064744: ↓CLSN-38 (p < 0.01) ↑SN-38 exposure: G2–3 diarrhoea (p = 0.03) | [152] |
| UGT1A1 | rs3064744 | 65 solid tumour or lymphoma patients | rs3064744: ↑BI (p = 0.0003) and ↓GR (p = 0.03) ↑BI: G4 neutropenia (p = 0.001) | [67] |
| UGT1A1 UGT1A7 UGT1A9 UGT1A10 | rs3064744 rs4148323 | 176 cancer patients | rs3064744 or rs4148323: ↓GR (p < 0.0001) | [153] |
| UGT1A1 ABCG2 | rs4148323 | 45 cancer patients | rs4148323: ↑AUCSN-38 (p = 0.018), ↓GR (p = 0.006) and 61% ↑BI (p = 0.003) | [154] |
| ABCB1 | ABCB1*2 | 49 cancer patients | ABCB1*2: ↓CLCPT-11, SN-38, APC (p = 0.0154, 0.0043, 0.0169, respectively) | [155] |
| Drug | Gene | Annotation by Drug Regulatory Agencies and Guidelines Recommendations | ||||
|---|---|---|---|---|---|---|
| FDA | CPIC | AEMPS | EMA | DPWG | ||
| Fluoropyrimidines | DPYD | Actionable PGx a Withhold or permanently discontinue treatment in patients with evidence of acute early-onset or severe toxicity, which may indicate near complete or absence of DPD activity. No dose has been proven safe for patients with no DPD activity. There is insufficient data to recommend a specific dose in patients with partial DPD activity [170] | Actionable PGx a Intermediate metaboliser (individual with one normal function allele plus one no function allele or one decreased function allele, or with two decreased function alleles)—decreased DPD activity and increased risk for severe/fatal ADR. Reduce starting dose based on AS followed by titration of dose based on toxicity or therapeutic drug monitoring (if available). AS 1: Reduce dose by 50% AS 1.5: Reduce dose by 25–50% Poor metaboliser (individual with two no function alleles, or with one no function plus one decreased function allele)—complete DPD deficiency and increased risk for severe/fatal ADR. AS 0.5: Avoid treatment, and in case alternative agents are not suitable, strongly reduce starting dose with early therapeutic drug monitoring. AS 0: Avoid treatment [167] | Testing required b Test for DPYD genotype (c.1905+1G>A, c.1679T>G, c.2846A>T and c.1236G>A/HapB3) and/or DPD deficiency (measure blood uracil level) before treatment. Treatment is contraindicated in patients with complete DPD deficiency. In case of partial DPD deficiency with no suitable alternative agents, reduce initial dose and monitor levels. No concrete reduction has been established [166] | Testing required b Test for the lack of DPD activity before treatment (measure blood uracil level, or check for DPYD variants—c.1905+1G>A, c.1679T>G, c.2846A>T y c.1236G>A/HapB3). Treatment is contraindicated in patients with complete DPD deficiency. A reduced starting dose should be considered in patients with partial DPD deficiency [169] | - |
| Irinotecan | UGT1A1 | Actionable PGx a Consider reduction in starting dose for patients homozygous for the UGT1A1*28 allele. The precise dose reduction is not known and subsequent dose modifications should be considered based on individual patient tolerance to treatment [170] | - | - | - | Actionable PGx a Start with 70% of standard dose. If the patient tolerates it, the dose can be increased, guided by the neutrophil count [168] |
| Cetuximab/panitumumab | EGFR KRAS NRAS | Testing required b Determine EGFR expression status and confirm the absence of an RAS mutation before treatment [170] | - | - | Testing required b Test RAS status (KRAS and NRAS exons 2, 3 and 4) before treatment [171] | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simões, A.R.; Fernández-Rozadilla, C.; Maroñas, O.; Carracedo, Á. The Road so Far in Colorectal Cancer Pharmacogenomics: Are We Closer to Individualised Treatment? J. Pers. Med. 2020, 10, 237. https://doi.org/10.3390/jpm10040237
Simões AR, Fernández-Rozadilla C, Maroñas O, Carracedo Á. The Road so Far in Colorectal Cancer Pharmacogenomics: Are We Closer to Individualised Treatment? Journal of Personalized Medicine. 2020; 10(4):237. https://doi.org/10.3390/jpm10040237
Chicago/Turabian StyleSimões, Ana Rita, Ceres Fernández-Rozadilla, Olalla Maroñas, and Ángel Carracedo. 2020. "The Road so Far in Colorectal Cancer Pharmacogenomics: Are We Closer to Individualised Treatment?" Journal of Personalized Medicine 10, no. 4: 237. https://doi.org/10.3390/jpm10040237