A Genotyping/Phenotyping Approach with Careful Clinical Monitoring to Manage the Fluoropyrimidines-Based Therapy: Clinical Cases and Systematic Review of the Literature

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Case Series
2.2. Systematic Review
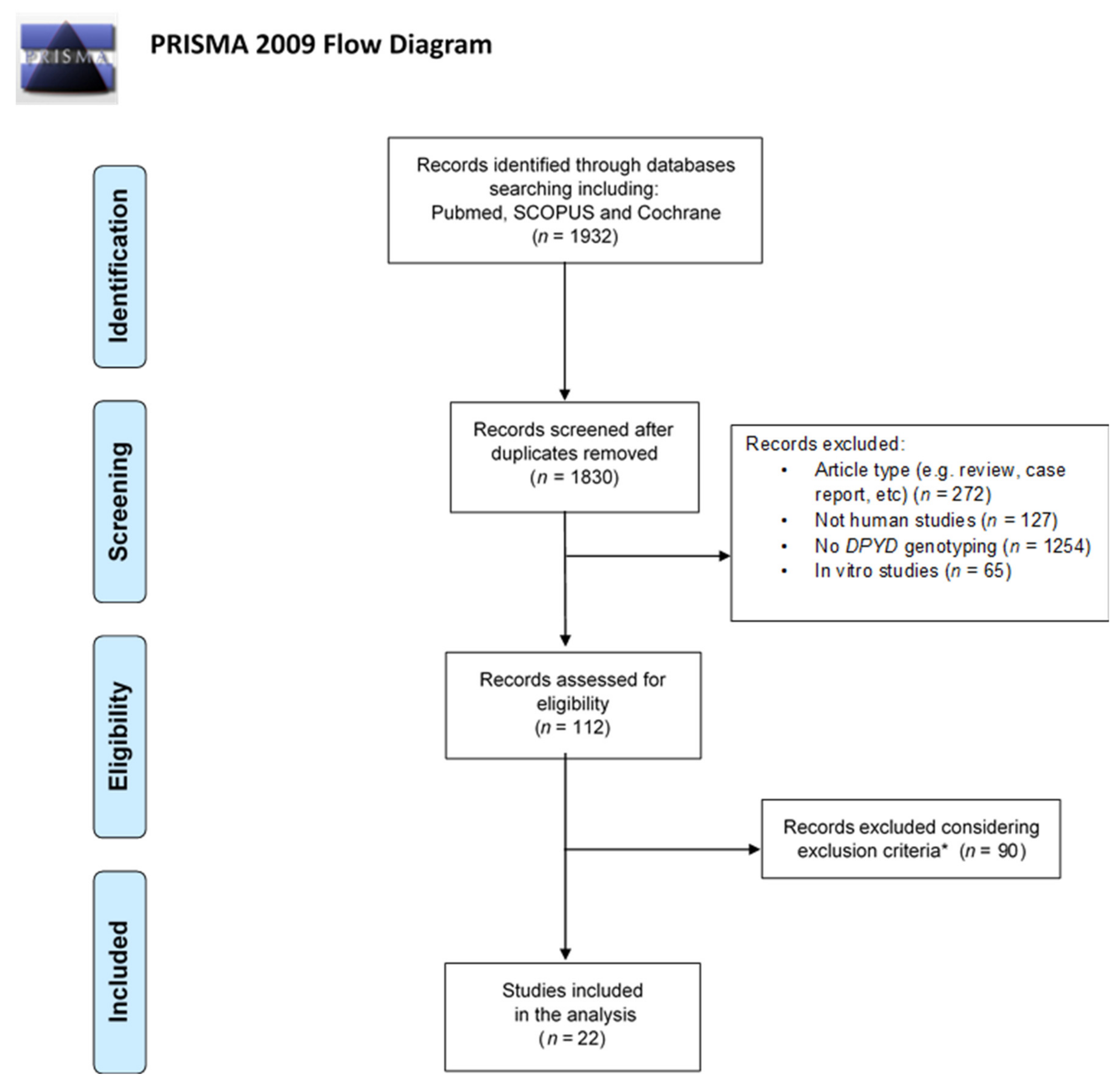
2.2.1. Search Strategy
2.2.2. Eligibility Criteria
2.2.3. Article Selection
2.2.4. Data Extraction
3. Results
3.1. Cases Presentation
3.2. Systematic Review
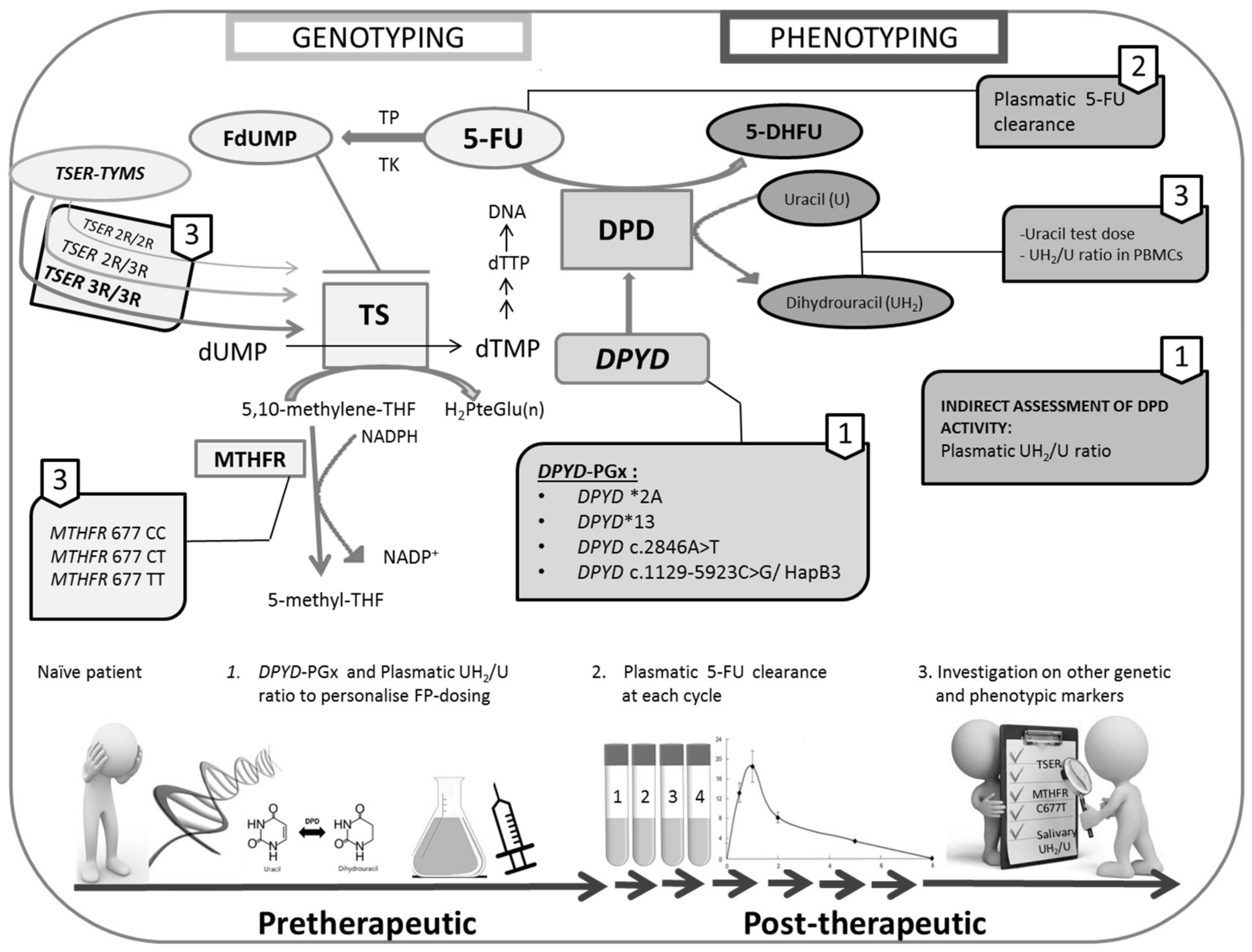
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FP | Fluoropyrimidines |
| DPD | Dihydropyrimidine Dehydrogenase |
| DPYD-PGx | DPYD-Pharmacogenomics |
| PCR | Polymerase Chain Reaction |
| 5-FU | 5-Fluorouracil |
| ADR | Adverse Drug Reaction |
| HFS | Hand-Foot Syndrome |
| SNPs | Single-Nucleotide Polymorphisms |
| CPIC | The Clinical Pharmacogenetics Implementation Consortium |
| DPWG | Dutch Pharmacogenetics Working Group |
| DPYD-AS | DPYD-Activity Score |
| TDM | Therapeutic Drug Monitoring |
| MTHFR | Methylene-Tetrahydrofolate Reductase |
| TYMS-TSER | Thymidylate Synthase-Thymidylate Synthase Enhancer Region |
| GSTP1 | Glutathione S-Trasferase-p1 |
| UH2/U ratio | Dihydrouracil/Uracil Ratio |
| CTC-AE | Common Terminology Criteria for Adverse Events |
| RECIST | Response Evaluation Criteria in Solid Tumours |
| HPLC | High-Performance Liquid Chromatography |
| UHPLC-MS/MS | Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry |
| MeSH | Medical Subject Heading |
| PRISMA | Preferred Reporting Items for Systematic reviews and Meta-Analyses |
| FOLFOX | 5-FU, Leucovorin and Oxaliplatin |
| CT scan | Computed Tomography Scan |
| FOLFIRI | 5-FU, Leucovorin and Irinotecan |
| PRAC | Pharmacovigilance Risk Assessment Committee |
| EMA | European Medicines Agency |
| CHMP | Committee for Medicinal Products for Human Use |
| AUC | Area Under the Curve |
| LC-MS/MS | Liquid Chromatography-Tandem Mass Spectrometry |
| PBMCs | Peripheral Blood Mononuclear Cells |
References
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Venook, A.P.; Niedzwiecki, D.; Lenz, H.-J.; Innocenti, F.; Fruth, B.; Meyerhardt, J.A.; Schrag, D.; Greene, C.; O’Neil, B.H.; Atkins, J.N.; et al. Effect of First-Line Chemotherapy Combined with Cetuximab or Bevacizumab on Overall Survival in Patients With KRAS Wild-Type Advanced or Metastatic Colorectal Cancer: A Randomized Clinical Trial. JAMA 2017, 317, 2392–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoff, P.M.; Ansari, R.; Batist, G.; Cox, J.; Kocha, W.; Kuperminc, M.; Maroun, J.; Walde, D.; Weaver, C.; Harrison, E.; et al. Comparison of Oral Capecitabine versus Intravenous Fluorouracil plus Leucovorin as First-Line Treatment in 605 Patients with Metastatic Colorectal Cancer: Results of a Randomized Phase III Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2001, 19, 2282–2292. [Google Scholar] [CrossRef] [PubMed]
- Van Kuilenburg, A.B.P.; Dobritzsch, D.; Meijer, J.; Meinsma, R.; Benoist, J.-F.; Assmann, B.; Schubert, S.; Hoffmann, G.F.; Duran, M.; de Vries, M.C.; et al. Dihydropyrimidinase Deficiency: Phenotype, Genotype and Structural Consequences in 17 Patients. Biochim. Biophys. Acta 2010, 1802, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Amstutz, U.; Henricks, L.M.; Offer, S.M.; Barbarino, J.; Schellens, J.H.M.; Swen, J.J.; Klein, T.E.; McLeod, H.L.; Caudle, K.E.; Diasio, R.B.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin. Pharmacol. Ther. 2018, 103, 210–216. [Google Scholar] [CrossRef]
- Lunenburg, C.A.T.C.; van der Wouden, C.H.; Nijenhuis, M.; Crommentuijn-van Rhenen, M.H.; de Boer-Veger, N.J.; Buunk, A.M.; Houwink, E.J.F.; Mulder, H.; Rongen, G.A.; van Schaik, R.H.N.; et al. Dutch Pharmacogenetics Working Group (DPWG) Guideline for the Gene–Drug Interaction of DPYD and Fluoropyrimidines. Eur. J. Hum. Genet. 2020, 28, 508–517. [Google Scholar] [CrossRef]
- Del Re, M.; Cinieri, S.; Michelucci, A.; Salvadori, S.; Loupakis, F.; Schirripa, M.; Cremolini, C.; Crucitta, S.; Barbara, C.; Di Leo, A.; et al. DPYD*2A and c.2846A>T: A Comprehensive Analysis in 1254 Patients. Pharm. J. 2019, 19, 556–563. [Google Scholar] [CrossRef]
- Van Kuilenburg, A.B.P.; Meijer, J.; Mauer, D.; Dobritzsch, D.; Meinsma, R.; Los, M.; Knegt, L.C.; Zoetekouw, L.; Jansen, R.L.H.; Dezentjé, V.; et al. Severe Fluoropyrimidine Toxicity Due to Novel and Rare DPYD Missense Mutations, Deletion and Genomic Amplification Affecting DPD Activity and MRNA Splicing. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 721–730. [Google Scholar] [CrossRef]
- Coenen, M.J.H.; Paulussen, A.D.C.; Breuer, M.; Lindhout, M.; Tserpelis, D.C.J.; Steyls, A.; Bierau, J.; van den Bosch, B.J.C. Evolution of Dihydropyrimidine Dehydrogenase Diagnostic Testing in a Single Center during an 8-Year Period of Time. Curr. Ther. Res. Clin. Exp. 2019, 90, 1–7. [Google Scholar] [CrossRef]
- Ulvik, A.; Ueland, P.M.; Fredriksen, A.; Meyer, K.; Vollset, S.E.; Hoff, G.; Schneede, J. Functional Inference of the Methylenetetrahydrofolate Reductase 677C > T and 1298A > C Polymorphisms from a Large-Scale Epidemiological Study. Hum. Genet. 2007, 121, 57–64. [Google Scholar] [CrossRef]
- Lecomte, T.; Ferraz, J.-M.; Zinzindohoué, F.; Loriot, M.-A.; Tregouet, D.-A.; Landi, B.; Berger, A.; Cugnenc, P.-H.; Jian, R.; Beaune, P.; et al. Thymidylate Synthase Gene Polymorphism Predicts Toxicity in Colorectal Cancer Patients Receiving 5-Fluorouracil-Based Chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 5880–5888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLeod, H.L.; Sargent, D.J.; Marsh, S.; Green, E.M.; King, C.R.; Fuchs, C.S.; Ramanathan, R.K.; Williamson, S.K.; Findlay, B.P.; Thibodeau, S.N.; et al. Pharmacogenetic Predictors of Adverse Events and Response to Chemotherapy in Metastatic Colorectal Cancer: Results from North American Gastrointestinal Intergroup Trial N9741. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3227–3233. [Google Scholar] [CrossRef] [PubMed]
- Boisdron-Celle, M.; Remaud, G.; Traore, S.; Poirier, A.L.; Gamelin, L.; Morel, A.; Gamelin, E. 5-Fluorouracil-Related Severe Toxicity: A Comparison of Different Methods for the Pretherapeutic Detection of Dihydropyrimidine Dehydrogenase Deficiency. Cancer Lett. 2007, 249, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Van Staveren, M.C.; Jan Guchelaar, H.; Van Kuilenburg, A.B.P.; Gelderblom, H.; Maring, J.G. Evaluation of Predictive Tests for Screening for Dihydropyrimidine Dehydrogenase Deficiency. Pharm. J. 2013, 13, 389–395. [Google Scholar] [CrossRef]
- National Cancer Institute Home Page. Available online: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_5x7.pdf (accessed on 11 July 2020).
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New Response Evaluation Criteria in Solid Tumours: Revised RECIST Guideline (Version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Svobaite, R.; Solassol, I.; Pinguet, F.; Ivanauskas, L.; Brès, J.; Bressolle, F.M.M. HPLC with UV or Mass Spectrometric Detection for Quantifying Endogenous Uracil and Dihydrouracil in Human Plasma. Clin. Chem. 2008, 54, 1463–1472. [Google Scholar] [CrossRef]
- Büchel, B.; Rhyn, P.; Schürch, S.; Bühr, C.; Amstutz, U.; Largiadèr, C.R. LC-MS/MS Method for Simultaneous Analysis of Uracil, 5,6-Dihydrouracil, 5-Fluorouracil and 5-Fluoro-5,6-Dihydrouracil in Human Plasma for Therapeutic Drug Monitoring and Toxicity Prediction in Cancer Patients. Biomed. Chromatogr. 2013, 27, 7–16. [Google Scholar] [CrossRef]
- Shamseer, L.; Moher, D.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A. Preferred Reporting Items for Systematic Review and Meta-Analysis Protocols (PRISMA-P) 2015: Elaboration and Explanation. BMJ 2015, 350, g7647. [Google Scholar] [CrossRef] [Green Version]
- Van Kuilenburg, A.B.; Haasjes, J.; Richel, D.J.; Zoetekouw, L.; Van Lenthe, H.; De Abreu, R.A.; Maring, J.G.; Vreken, P.; van Gennip, A.H. Clinical Implications of Dihydropyrimidine Dehydrogenase (DPD) Deficiency in Patients with Severe 5-Fluorouracil-Associated Toxicity: Identification of New Mutations in the DPD Gene. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 4705–4712. [Google Scholar]
- Schwab, M.; Zanger, U.M.; Marx, C.; Schaeffeler, E.; Klein, K.; Dippon, J.; Kerb, R.; Blievernicht, J.; Fischer, J.; Hofmann, U.; et al. Role of Genetic and Nongenetic Factors for Fluorouracil Treatment-Related Severe Toxicity: A Prospective Clinical Trial by the German 5-FU Toxicity Study Group. J. Clin. Oncol. 2008, 26, 2131–2138. [Google Scholar] [CrossRef]
- Kristensen, M.H.; Pedersen, P.; Mejer, J. The Value of Dihydrouracil/Uracil Plasma Ratios in Predicting 5-Fluorouracilrelated Toxicity in Colorectal Cancer Patients. J. Int. Med. Res. 2010, 38, 1313–1323. [Google Scholar] [CrossRef]
- Deenen, M.J.; Tol, J.; Burylo, A.M.; Doodeman, V.D.; De Boer, A.; Vincent, A.; Guchelaar, H.-J.; Smits, P.H.M.; Beijnen, J.H.; Punt, C.J.A.; et al. Relationship between Single Nucleotide Polymorphisms and Haplotypes in DPYD and Toxicity and Efficacy of Capecitabine in Advanced Colorectal Cancer. Clin. Cancer Res. 2011, 17, 3455–3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deenen, M.J.; Meulendijks, D.; Cats, A.; Sechterberger, M.K.; Severens, J.L.; Boot, H.; Smits, P.H.; Rosing, H.; Mandigers, C.M.P.W.; Soesan, M.; et al. Upfront Genotyping of DPYD*2A to Individualize Fluoropyrimidine Therapy: A Safety and Cost Analysis. J. Clin. Oncol. 2016, 34, 227–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sistonen, J.; Büchel, B.; Froehlich, T.K.; Kummer, D.; Fontana, S.; Joerger, M.; Van Kuilenburg, A.B.P.; Largiadèr, C.R. Predicting 5-Fluorouracil Toxicity: DPD Genotype and 5,6-Dihydrouracil:Uracil Ratio. Pharmacogenomics 2014, 15, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.M.; Shi, Q.; Pavey, E.; Alberts, S.R.; Sargent, D.J.; Sinicrope, F.A.; Berenberg, J.L.; Goldberg, R.M.; Diasio, R.B. DPYD variants as predictors of 5-fluorouracil toxicity in adjuvant colon cancer treatment (NCCTG N0147). J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Gentile, G.; Botticelli, A.; Lionetto, L.; Mazzuca, F.; Simmaco, M.; Marchetti, P.; Borro, M. Genotype-Phenotype Correlations in 5-Fluorouracil Metabolism: A Candidate DPYD Haplotype to Improve Toxicity Prediction. Pharm. J. 2016, 16, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Joerger, M.; Huitema, A.D.R.; Boot, H.; Cats, A.; Doodeman, V.D.; Smits, P.H.M.; Vainchtein, L.; Rosing, H.; Meijerman, I.; Zueger, M.; et al. Germline TYMS Genotype Is Highly Predictive in Patients with Metastatic Gastrointestinal Malignancies Receiving Capecitabine-Based Chemotherapy. Cancer Chemother. Pharmacol. 2015, 75, 763–772. [Google Scholar] [CrossRef]
- Lunenburg, C.A.T.C.; Van Staveren, M.C.; Gelderblom, H.; Guchelaar, H.-J.; Swen, J.J. Evaluation of Clinical Implementation of Prospective DPYD Genotyping in 5-Fluorouracil- or Capecitabine-Treated Patients. Pharmacogenomics 2016, 17, 721–729. [Google Scholar] [CrossRef] [Green Version]
- Galarza, A.F.A.; Linden, R.; Antunes, M.V.; Hahn, R.Z.; Raymundo, S.; da Silva, A.C.C.; Staggemeier, R.; Spilki, F.R.; Schwartsmann, G. Endogenous Plasma and Salivary Uracil to Dihydrouracil Ratios and DPYD Genotyping as Predictors of Severe Fluoropyrimidine Toxicity in Patients with Gastrointestinal Malignancies. Clin. Biochem. 2016, 49, 1221–1226. [Google Scholar] [CrossRef]
- Milano, G. Highlight on DPYD Gene Polymorphisms and Treatment by Capecitabine*. Scand. J. Clin. Lab. Invest. 2016, 76, S30–S33. [Google Scholar] [CrossRef]
- Boisdron-Celle, M.; Metges, J.P.; Capitain, O.; Adenis, A.; Raoul, J.L.; Lecomte, T.; Lam, Y.H.; Faroux, R.; Masliah, C.; Poirier, A.L.; et al. A Multicenter Phase II Study of Personalized FOLFIRI-Cetuximab for Safe Dose Intensification. Semin. Oncol. 2017, 44, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Grimaldi, M.-C.; Boyer, J.-C.; Beroud, C.; Mbatchi, L.; Van Kuilenburg, A.; Bobin-Dubigeon, C.; Thomas, F.; Chatelut, E.; Merlin, J.-L.; Pinguet, F.; et al. New Advances in DPYD Genotype and Risk of Severe Toxicity under Capecitabine. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Li, J.; Gao, J.; Li, Y.; Yang, R.; Shen, L. Examination of Multiple UGT1A and DPYD Polymorphisms Has Limited Ability to Predict the Toxicity and Efficacy of Metastatic Colorectal Cancer Treated with Irinotecan-Based Chemotherapy: A Retrospective Analysis. BMC Cancer 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Henricks, L.M.; Lunenburg, C.A.T.C.; de Man, F.M.; Meulendijks, D.; Frederix, G.W.J.; Kienhuis, E.; Creemers, G.-J.; Baars, A.; Dezentjé, V.O.; Imholz, A.L.T.; et al. DPYD Genotype-Guided Dose Individualisation of Fluoropyrimidine Therapy in Patients with Cancer: A Prospective Safety Analysis. Lancet Oncol. 2018, 19, 1459–1467. [Google Scholar] [CrossRef]
- Cremolini, C.; Del Re, M.; Antoniotti, C.; Lonardi, S.; Bergamo, F.; Loupakis, F.; Borelli, B.; Marmorino, F.; Citi, V.; Cortesi, E.; et al. DPYD and UGT1A1 Genotyping to Predict Adverse Events during First-Line FOLFIRI or FOLFOXIRI plus Bevacizumab in Metastatic Colorectal Cancer. Oncotarget 2018, 9, 7859–7866. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, B.A.W.; Deenen, M.J.; Joerger, M.; Rosing, H.; de Vries, N.; Meulendijks, D.; Cats, A.; Beijnen, J.H.; Schellens, J.H.M.; Huitema, A.D.R. Pharmacokinetics of Capecitabine and Four Metabolites in a Heterogeneous Population of Cancer Patients: A Comprehensive Analysis. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 940–950. [Google Scholar] [CrossRef] [Green Version]
- Iachetta, F.; Bonelli, C.; Romagnani, A.; Zamponi, R.; Tofani, L.; Farnetti, E.; Nicoli, D.; Damato, A.; Banzi, M.; Casali, B.; et al. The Clinical Relevance of Multiple DPYD Polymorphisms on Patients Candidate for Fluoropyrimidine Based-Chemotherapy. An Italian Case-Control Study. Br. J. Cancer 2019, 120, 834–839. [Google Scholar] [CrossRef] [Green Version]
- Kleinjan, J.P.; Brinkman, I.; Bakema, R.; Van Zanden, J.J.; Van Rooijen, J.M. Tolerance-Based Capecitabine Dose Escalation after DPYD Genotype-Guided Dosing in Heterozygote DPYD Variant Carriers: A Single-Center Observational Study. Anticancer Drugs 2019, 30, 410–415. [Google Scholar] [CrossRef]
- Negarandeh, R.; Salehifar, E.; Saghafi, F.; Jalali, H.; Janbabaei, G.; Abdhaghighi, M.J.; Nosrati, A. Evaluation of Adverse Effects of Chemotherapy Regimens of 5-Fluoropyrimidines Derivatives and Their Association with DPYD Polymorphisms in Colorectal Cancer Patients. BMC Cancer 2020, 20, 560. [Google Scholar] [CrossRef]
- Pallet, N.; Hamdane, S.; Garinet, S.; Blons, H.; Zaanan, A.; Paillaud, E.; Taieb, J.; Laprevote, O.; Loriot, M.-A.; Narjoz, C. A Comprehensive Population-Based Study Comparing the Phenotype and Genotype in a Pretherapeutic Screen of Dihydropyrimidine Dehydrogenase Deficiency. Br. J. Cancer 2020. [Google Scholar] [CrossRef]
- European Medicines Agency Home Page. Available online: https://www.ema.europa.eu/en/medicines/human/referrals/fluorouracil-fluorouracil-related-substances-capecitabine-tegafur-flucytosine-containing-medicinal (accessed on 30 April 2020).
- European Medicines Agency Home Page. Available online: https://www.ema.europa.eu/en/documents/referral/fluorouracil-fluorouracil-related-substances-article-31-referral-ema-recommendations-dpd-testing_en.pdf (accessed on 15 July 2020).
- Froehlich, T.K.; Amstutz, U.; Aebi, S.; Joerger, M.; Largiadèr, C.R. Clinical Importance of Risk Variants in the Dihydropyrimidine Dehydrogenase Gene for the Prediction of Early-Onset Fluoropyrimidine Toxicity. Int. J. Cancer 2015, 136, 730–739. [Google Scholar] [CrossRef]
- Toffoli, G.; Giodini, L.; Buonadonna, A.; Berretta, M.; De Paoli, A.; Scalone, S.; Miolo, G.; Mini, E.; Nobili, S.; Lonardi, S.; et al. Clinical Validity of a DPYD-Based Pharmacogenetic Test to Predict Severe Toxicity to Fluoropyrimidines. Int. J. Cancer 2015, 137, 2971–2980. [Google Scholar] [CrossRef]
- Meulendijks, D.; Jacobs, B.A.W.; Aliev, A.; Pluim, D.; van Werkhoven, E.; Deenen, M.J.; Beijnen, J.H.; Cats, A.; Schellens, J.H.M. Increased Risk of Severe Fluoropyrimidine-Associated Toxicity in Patients Carrying a G to C Substitution in the First 28-Bp Tandem Repeat of the Thymidylate Synthase 2R Allele. Int. J. Cancer 2016, 138, 245–253. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency Home Page. Available online: https://www.ema.europa.eu/en/documents/product-information/erbitux-epar-product-information_it.pdf (accessed on 1 August 2020).
- European Medicines Agency Home Page. Available online: https://www.ema.europa.eu/documents/overview/avastin-epar-summary-public_it.pdf (accessed on 1 August 2020).
- Agenzia Italiana del Farmaco Home Page. Available online: https://farmaci.agenziafarmaco.gov.it/aifa/servlet/PdfDownloadServlet%3FpdfFileName%3Dfooter_003142_039170_RCP.pdf%26retry%3D0%26sys%3Dm0b1l3&ved=2ahUKEwicnenw0MrrAhVI-aQKHc9iAT0QFjADegQIBBAB&usg=AOvVaw302NVA85pxPclL8QoyfW-R&cshid=1599056097201 (accessed on 1 August 2020).
- European Medicines Agency Home Page. Available online: https://www.ema.europa.eu/en/documents/product-information/onivyde-pegylated-liposomal-epar-product-information_it.pdf (accessed on 1 August 2020).
- Toffoli, G.; De Mattia, E. Pharmacogenetic Relevance of MTHFR Polymorphisms. Pharmacogenomics 2008, 9, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; He, X.; Zhang, Y.; Chuan, J.-L.; Chen, M.; Zhu, S.-M.; Peng, Q. Relevance of Methylenetetrahydrofolate Reductase Gene Variants C677T and A1298C with Response to Fluoropyrimidine-Based Chemotherapy in Colorectal Cancer: A Systematic Review and Meta-Analysis. Oncotarget 2018, 9, 31291–31301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecomte, T.; Landi, B.; Beaune, P.; Laurent-Puig, P.; Loriot, M.-A. Glutathione S-Transferase P1 Polymorphism (Ile105Val) Predicts Cumulative Neuropathy in Patients Receiving Oxaliplatin-Based Chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 3050–3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-P.; Ling, Y.; Qi, Q.-F.; Zhang, Y.-P.; Zhang, C.-S.; Zhu, C.-T.; Wang, M.-H.; Pan, Y.-D. Genetic Polymorphisms of ERCC1-118, XRCC1-399 and GSTP1-105 Are Associated with the Clinical Outcome of Gastric Cancer Patients Receiving Oxaliplatin-based Adjuvant Chemotherapy. Mol. Med. Rep. 2013, 7, 1904–1911. [Google Scholar] [CrossRef] [Green Version]
- Lv, F.; Ma, Y.; Zhang, Y.; Li, Z. Relationship between GSTP1 Rs1695 Gene Polymorphism and Myelosuppression Induced by Platinum-Based Drugs: A Meta-Analysis. Int. J. Biol. Markers 2018, 33, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Wang, Q.; Gao, J.; Ji, Z.; Yuan, J.; Tian, Y.; Shen, L. Association between GSTP1 Ile105Val Polymorphism and Oxaliplatin-Induced Neuropathy: A Systematic Review and Meta-Analysis. Cancer Chemother. Pharmacol. 2013, 72, 305–314. [Google Scholar] [CrossRef]
- Hamzic, S.; Kummer, D.; Froehlich, T.K.; Joerger, M.; Aebi, S.; Palles, C.; Thomlinson, I.; Meulendijks, D.; Schellens, J.H.M.; García-González, X.; et al. Evaluating the Role of ENOSF1 and TYMS Variants as Predictors in Fluoropyrimidine-Related Toxicities: An IPD Meta-Analysis. Pharmacol. Res. 2020, 152, 104594. [Google Scholar] [CrossRef]
- Gallegos-Arreola, M.P.; Zúñiga-González, G.M.; Sánchez-López, J.Y.; Cruz, A.Y.N.; Peralta-Leal, V.; Figuera, L.E.; Puebla-Pérez, A.M.; Ronquillo-Carreón, C.A.; Puebla-Mora, A.G. TYMS 2R3R Polymorphism and DPYD [IVS]14+1G > A Gene Mutation in Mexican Colorectal Cancer Patients. Acta Biochim. Pol. 2018, 65, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Gamelin, E.C.; Danquechin-Dorval, E.M.; Dumesnil, Y.F.; Maillart, P.J.; Goudier, M.J.; Burtin, P.C.; Delva, R.G.; Lortholary, A.H.; Gesta, P.H.; Larra, F.G. Relationship between 5-Fluorouracil (5-FU) Dose Intensity and Therapeutic Response in Patients with Advanced Colorectal Cancer Receiving Infusional Therapy Containing 5-FU. Cancer 1996, 77, 441–451. [Google Scholar] [CrossRef]
- Chazal, M.; Etienne, M.C.; Renée, N.; Bourgeon, A.; Richelme, H.; Milano, G. Link between Dihydropyrimidine Dehydrogenase Activity in Peripheral Blood Mononuclear Cells and Liver. Clin. Cancer Res. 1996, 2, 507–510. [Google Scholar] [PubMed]
- Gamelin, E.; Boisdron-Celle, M.; Guérin-Meyer, V.; Delva, R.; Lortholary, A.; Genevieve, F.; Larra, F.; Ifrah, N.; Robert, J. Correlation between Uracil and Dihydrouracil Plasma Ratio, Fluorouracil (5-FU) Pharmacokinetic Parameters, and Tolerance in Patients with Advanced Colorectal Cancer: A Potential Interest for Predicting 5-FU Toxicity and Determining Optimal 5-FU Dosage. J. Clin. Oncol. 1999, 17, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.; Hennebelle, I.; Delmas, C.; Lochon, I.; Dhelens, C.; Tixidre, C.G.; Bonadona, A.; Penel, N.; Goncalves, A.; Delord, J.P.; et al. Genotyping of a Family with a Novel Deleterious DPYD Mutation Supports the Pretherapeutic Screening of DPD Deficiency with Dihydrouracil/Uracil Ratio. Clin. Pharmacol. Ther. 2016, 99, 235–241. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Pt | Sex | Age (years) | Tumor Type and Stage | Chemotherapy Regimen | Pre-Therapeutic DPYD-PGx | Post-Therapeutic DPYD-PGx | DPYD Genotype | UH2/U Ratio | 5-FU Dosage | 5-FU Clearance | ADR ≥ 3 | Total Toxicity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 55 | CRC (metastatic) | Folfox plus cetuximab | / | Yes (C4) | Heterozygous for DPYD*2A | 4.52 | C1-C3: 100% C4-C6: 50% | 950 ng/mL (C4) 400 ng/mL (C6) | G3 mucositis (C3) G3 neutropenia (C3) (neuthrophils: 830.58 /mm3) | 7 |
| 2 | M | 48 | CRC (metastatic) | Folfox plus bevacizumab | Yes | / | Heterozygous for DPYD*2A | 3.22 | C1-C8: 50% | 474 ng/mL (C1) | / | 6 |
| 3 | M | 60 | Rectal cancer (metastatic) | Folfox plus cetuximab) | Yes | / | Heterozygous for c.2846A>T | 1.77 | C1-C3: 50% C4: 40% C5-C6: 40% (only bolus) | 811 ng/mL (C2) 1093 ng/mL (C3) 1048 ng/mL (C4) 934 ng/mL (C5) | G3 diarrhoea (C3) G4 diarrhoea (C4) G3 diarrhoea (C5) | 3 |
| Pt | Sex | Age (Years) | Tumor Type and Stage | Chemotherapy Regimen | Pre-Therapeutic DPYD-PGx | Post-Therapeutic DPYD-PGx | DPYD Genotype | UH2/U Ratio | 5-FU Dosage | ADR ≥3 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 63 | Stomach cancer (locally advanced) | Folfox | / | Yes (C2) | Heterozygous for DPYD*2A | 7.09 | C1-C2: 100% C3: 5-FU withdrawal | G3 vomit (C2) |
| 2 | M | 43 | CRC (metastatic) | Folfiri with bevacizumab | / | Yes (C8) | Heterozygous for c.2846A>T | 3.88 | C1-C8: 100% C9: 50% | G3 vomit (C8) |
| 3 | M | 63 | Kidney cancer (metastatic) | Xeloda | yes | / | Heterozygous for c.2846A>T | 6.57 | C1-C6: 50% | / |
| 4 | M | 68 | CRC (metastatic) | Xelox | yes | / | Heterozygous for c.2846A>T | 4.4 | C1-C7: 50% | / |
| 5 | M | 78 | CRC (local) | Xelox | yes | / | Heterozygous for c.2846A>T | 3.37 | C1-C2: 50% | / |
| 6 | M | 72 | CRC (locally advanced) | Xelox | yes | / | Heterozygous for DPYD*2A | 5.15 | C1-C8: 50% | / |
| 7 | F | 52 | Vulva carcinoma (local) | Xeloda with cisplatin | yes | / | Heterozygous for DPYD*2A | 7.38 | C1-C5: 50% | / |
| 8 | M | 76 | Rectosigmoid cancer (locally advanced) | Folfox | yes | / | Heterozygous for DPYD*2A | 2.44 | C1-C5: 50% | / |
| First Author‘s Name (Published Year) | Enrolled Patients (n) | Outcomes | DPYD-PGx/Clinical Monitoring | DPYD-PGx/Phenotyping | DPYD-PGx/Phenotyping/Clinical Monitoring |
|---|---|---|---|---|---|
| Kuilenburg et al. (2000) [20] | 37 | DPD activity and overall toxicity; DPYD genotyping in patients with reduced DPD activity. | DPYD*2A, c.2846A>T, DPYD*6, DPYD*9A, c.496A>G/ UH2/U ratio in PBMC/ADR until two treatment months. | ||
| Schwab et al. (2008) [21] | 683 | Overall toxicity; DPYD, TYMS, MTHFR genotyping; sequencing of DPYD exome; influence of sex and promoter methylation on DPD expression in human liver. | DPYD*2A, c.2846A>T, c.623G>T, DPYD*4, DPYD*6, and c.2858G>C/ ADR reported until the second cycle of treatment. | ||
| Kristensen et al.(2010) [22] | 68 | Relationship between UH2/U plasma ratio and 5-FU-related early toxicity; relationship between 5-FU concentration and toxicity; IVS14+1G>A mutation screening. | DPYD*2A/ UH2/U ratio in plasma 5-FU clearance by HPLC-UV/ ADR reported until the second cycle of treatment. | ||
| Deenen et al. (2011) [23] | 568 | Relationships between SNPs and toxicity, SNPs and dose modification of capecitabine, DPYD haplotypes and toxicity, DPYD SNPs and haplotypes and survival. | DPYD*2A, c.2846A>T and c.1236G>A [HapB3]/ ADR reported until the second cycle of treatment. | ||
| Deenen et al. (2016) [24] | 2038 | Feasibility, safety and cost of DPYD*2A genotype-guided dosing. | DPYD*2A/ADR reported until the sixth cycle of treatment. | ||
| Sistonen et al. (2014) [25] | 28 | Relationship between UH2/U plasma ratio and DPYD genetic variation; plasma concentration of 5-FU and corresponding AUC; toxicity. | c.234-123G>C, c.496A>G, c.775A>G, c.1129-5923C>G [Hap B3], DPYD*13, DPYD*2A and c.2846A>T/ UH2/U ratio in plasma- 5-FU clearance by LC-MS/MS/ ADR reported until the second cycle of treatment. | ||
| Lee et al.(2014) [26] | 2886 | Relationship between DPYD variants and toxicity. | DPYD*2A, DPYD*13, c.2846A>T/ ADR until the twelfth cycle of treatment. | ||
| Gentile et al. (2015) [27] | 156 | Correlation between degradation rate of 5-FU with detected SNPs. | DPYD*2A, DPYD*13, c.2846A>T/5-FUDR assay in PBMC by HPLC-MS/MS | ||
| Joerger et al. (2015) [28] | 140 | Quantitative effect of 44 gene polymorphism in 16 drug pathway associated genes on progression free survival (PFS), on chemotherapy toxicity, on objective response rate (ORR), on overall survival (OS). | DPYD*13, DPYD*2A, c.2846A>T, DPYD*9A, c.1896T>C/5-FU clearance by AAS and HPLC/ADR until disease progression. | ||
| Lunenburg et al. (2016) [29] | 275 | Evaluation of requests of prospective DPYD screening and results with a dose recommendation; estimation of the follow up of the dose recommendations. | DPYD*2A, DPYD*13, c.2846A>T, c.1236G>A [HapB3]/ ADR reported until the second cycle of treatment. | ||
| Galarza et al. (2016) [30] | 60 | Estimation of the use of plasma and saliva; Uracil to UH2 metabolic ratio and DPYD genotyping. | DPYD *2A, *13, c.557A>G, DPYD *7/ UH2/U ratio in plasma/ ADR reported until the third cycle of treatment. | ||
| Milano et al. (2016) [31] | 243 | Sequencing of DPYD exome and frequence of G3, G4 toxicity over cycle 1-2. | DPYD*2A, DPYD*13, c.2846A>T, c.1774C>T, c.1475C>T, D342G/ ADR reported until the second cycle of treatment. | ||
| Boisdron-Celle et al.(2017) [32] | 85 | UGT1A1 and DPYD genotyping; UH2/U ratio; follow up of efficacy and tolerance. | DPYD*2A, DPYD*13, c.2846A>T, DPYD*7/ UH2/U ratio in plasma/ADR every two weeks until three months. | ||
| Etienne-Grimaldi et al.(2017) [33] | 243 | DPYD sequencing; relationship between toxicity and DPYD variants; DPD phenotyping. | DPYD*2A, DPYD*13, c.2846A>T/ UH2/U ratio in plasma/ ADR reported until the second cycle of treatment. | ||
| Liu et al.(2017) [34] | 661 | Relationship between UGT1A1 and DPYD polymorphism and incidence of severe neutropenia and diarrhea; relationship between UGT1A1 and DPYD variants and objective response rate, disease control rate, overall and progression free survival. | DPYD*5, c.1896 T > C, and DPYD*2A/ ADR reported every two-three cycles or whenever patient’s condition changed. | ||
| Henricks et al.(2018) [35] | 1181 | Frequency of severe overall FP-related toxicity; pharmacokinetics of fluoropyrimidines in DPYD variant allele carriers; DPD enzyme activity; cost analysis on individualised dosing by upfront DPYD genotyping. | DPYD*2A, c.2846A>T, DPYD*13 and c.1236G>A [Hap B3]/ UH2/U ratio in PBMC/PK data by UHPLC-MS/MS/ADR until toxicity resolution. | ||
| Cremolini et al. (2018) [36] | 443 | Relationship between DPYD and UGT1A1 genotyping and toxicity. | DPYD*2A, *13, c.2846A>T/ADR reported until the fourth cycle of treatment. | ||
| Jacobs et al.(2019) [37] | 237 | Pharmacokinetics of capecitabine and 5-FU in DPYD variant allele carriers. | DPYD*2A, c.2846A>T, c.1236G>A [HapB3]/5-FU clearance by LC MS/MS. | ||
| Iachetta et al.(2019) [38] | 1827 | Relationship between DPYD and toxicity. | DPYD*13, DPYD*2A, c.2846A>T, DPYD*6 /ADR reported until the eleventh cycle of treatment. | ||
| Kleinjan et al. (2019) [39] | 185 | DPYD genotyping and toxicity. | DPYD*2A, c.2846A>T, DPYD*13 and c.1236G>A [HapB3] /ADR reported until the second cycle of treatment. | ||
| Negarandeh et al.(2020) [40] | 88 | Relationship between DPYD genotyping and toxicity. | DPYD*2A, c.2846A>T, DPYD*6/ADR reported following 227 cycles for 88 patients. | ||
| Nicolas Pallet et al.(2020) [41] | 5886 | Relationship between DPYD genotyping and [U] and UH2/U ratio in plasma. | DPYD*2A, DPYD*13, c.2846A>T, c.1236G>A [HapB3]/ [U] and UH2/U ratio in plasma. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conti, V.; De Bellis, E.; Manzo, V.; Sabbatino, F.; Iannello, F.; Dal Piaz, F.; Izzo, V.; Charlier, B.; Stefanelli, B.; Torsiello, M.; et al. A Genotyping/Phenotyping Approach with Careful Clinical Monitoring to Manage the Fluoropyrimidines-Based Therapy: Clinical Cases and Systematic Review of the Literature. J. Pers. Med. 2020, 10, 113. https://doi.org/10.3390/jpm10030113
Conti V, De Bellis E, Manzo V, Sabbatino F, Iannello F, Dal Piaz F, Izzo V, Charlier B, Stefanelli B, Torsiello M, et al. A Genotyping/Phenotyping Approach with Careful Clinical Monitoring to Manage the Fluoropyrimidines-Based Therapy: Clinical Cases and Systematic Review of the Literature. Journal of Personalized Medicine. 2020; 10(3):113. https://doi.org/10.3390/jpm10030113
Chicago/Turabian StyleConti, Valeria, Emanuela De Bellis, Valentina Manzo, Francesco Sabbatino, Francesco Iannello, Fabrizio Dal Piaz, Viviana Izzo, Bruno Charlier, Berenice Stefanelli, Martina Torsiello, and et al. 2020. "A Genotyping/Phenotyping Approach with Careful Clinical Monitoring to Manage the Fluoropyrimidines-Based Therapy: Clinical Cases and Systematic Review of the Literature" Journal of Personalized Medicine 10, no. 3: 113. https://doi.org/10.3390/jpm10030113