
Role of Hybrid Brain Imaging in Neuropsychiatric Disorders


{kind=link}
{kind=link}
{kind=link}
Abstract
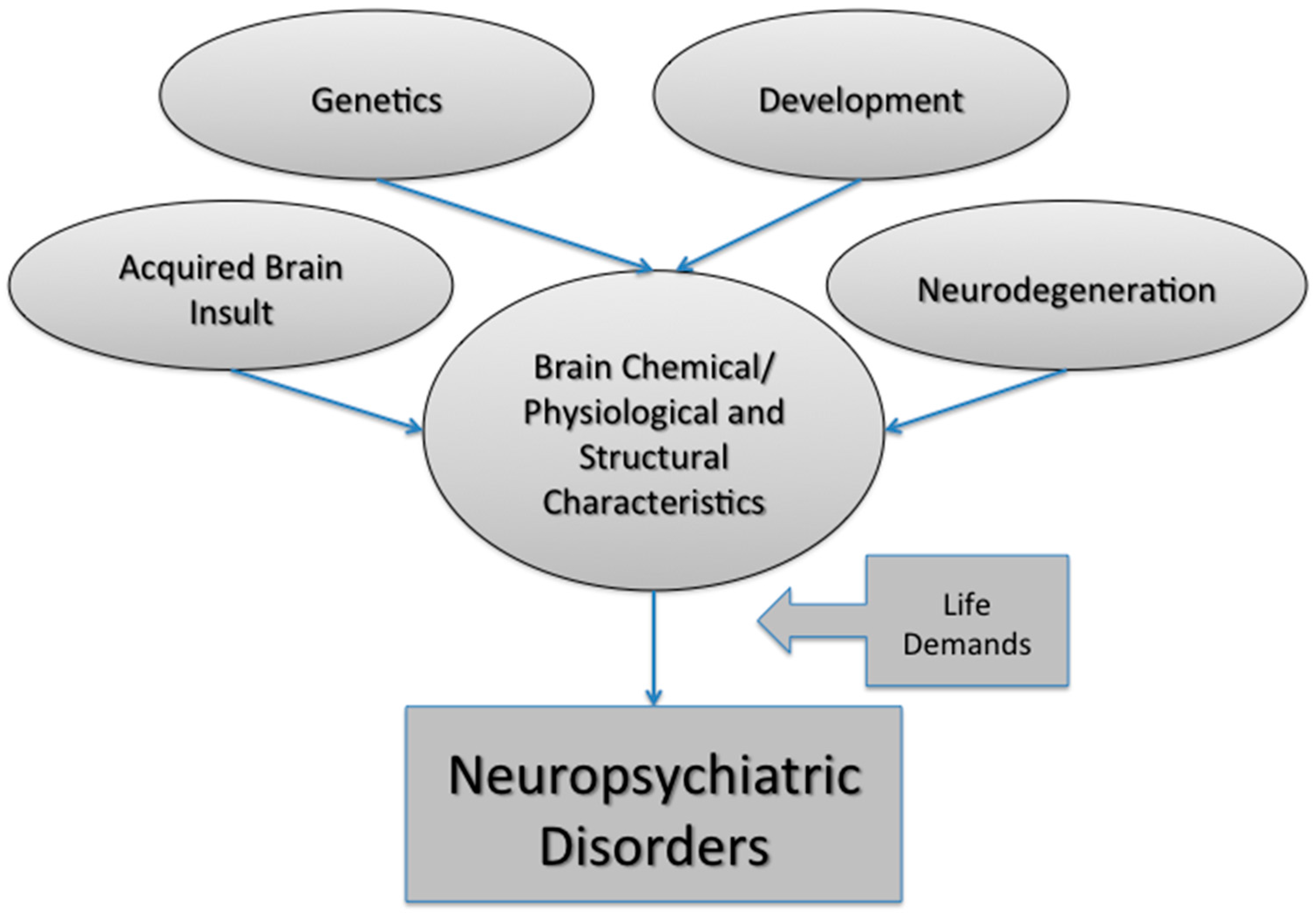
:1. Introduction
1.1. Issues with Clinical Classification
1.2. Brain Imaging as the Biomarker for Neuropsychiatric Disorders

1.3. Evolution of Network Models in Neuropsychiatric Disorders
2. Hybrid Imaging
2.1. Hybrid PET/CT: Development and Utility in Neuroimaging
2.2. Hybrid PET/MR: Development and Utility in Neuroimaging


2.3. Utility of Hybrid Imaging in Understanding Brain Connectivity in Neuropsychiatric Disorders
3. Alzheimer’s Disease as a Prototypical Neuropsychiatric Disorder
3.1. Large Investments Result in Tangible Progress for Neuropsychiatric Disorders
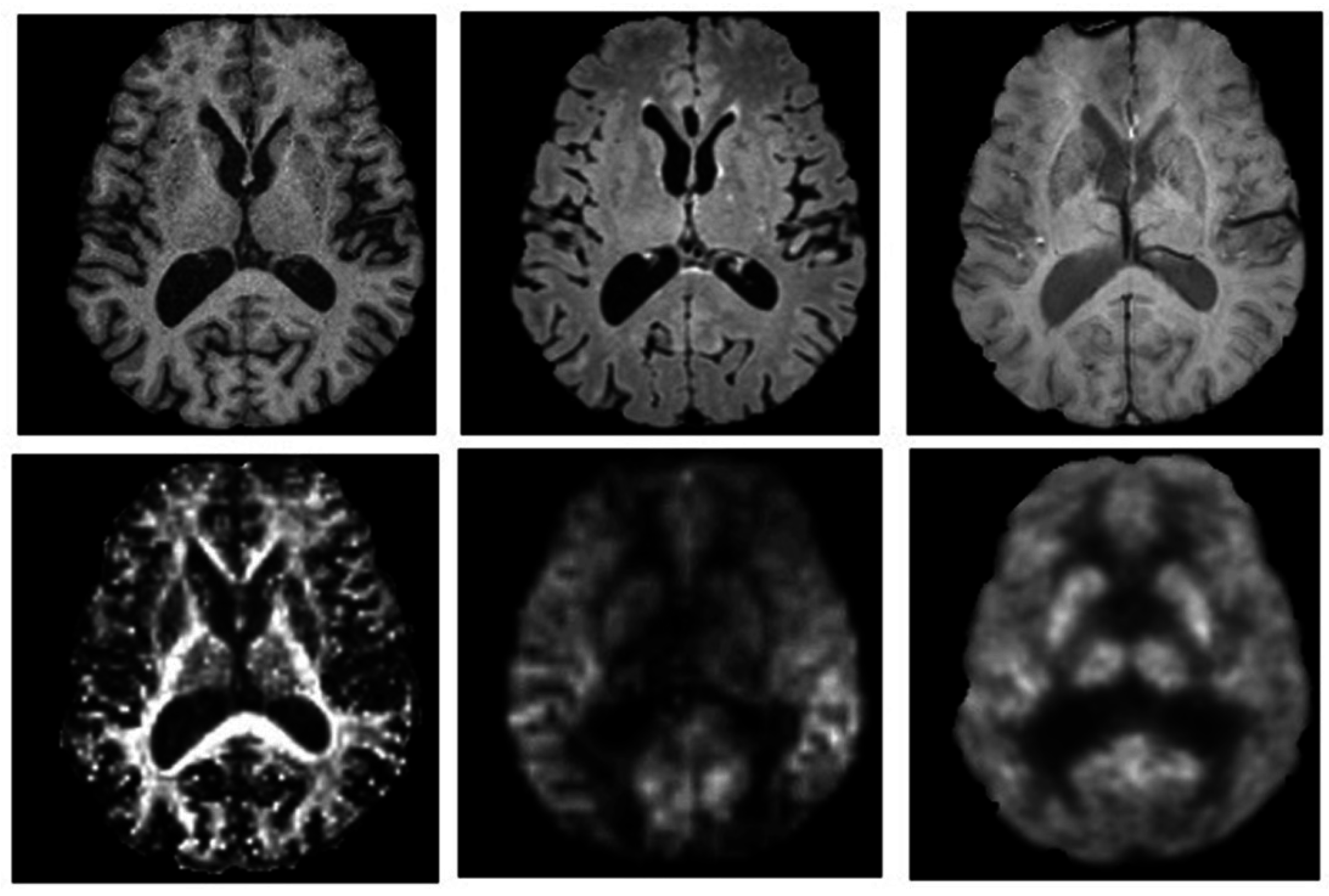
3.2. Neuroimaging the Neuropsychiatric Characteristics of Alzheimer’s Disease
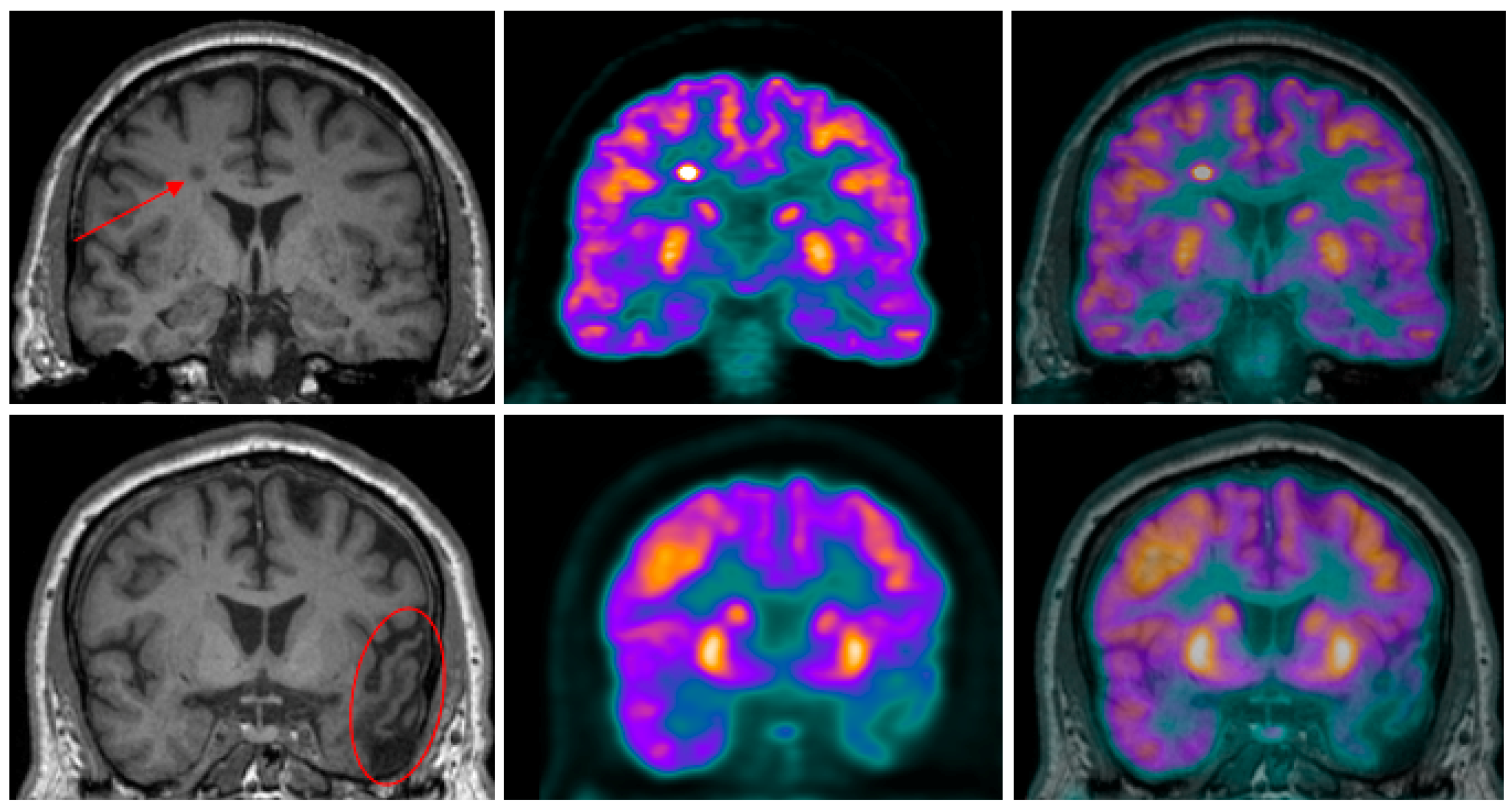
3.2.1. Psychosis and Alzheimer’s Disease
3.2.2. Agitation and Alzheimer’s Disease
3.2.3. Apathy and Alzheimer’s Disease
3.3. Network Neuroimaging Limitations in Alzheimer’s Disease
4. Psychoses
4.1. Network Abnormalities in Psychosis
4.2. Multimodal Imaging Opportunities in understanding Molecular and Functional Attributes of Psychosis
- (1)
- NMDA (N-Methyl-d-aspartate) encephalitis, an autoimmune inflammatory condition, mimics schizophrenia in many subjects and has been identified as an important, albeit small, contributor of first episode psychosis in clinical practice [115].
- (2)
- Several NSAID’s (nonsteroidal anti-inflammatory drugs) have shown to have a small but noticeable effect in reducing symptoms of schizophrenia [116].
5. Affective Disorders (Mood and Anxiety Disorders)
5.1. Anxiety Disorders
5.2. Anxiety Disorders and Network Abnormalities
5.3. Depression
5.4. Predictive Biomarkers for Depression
5.5. Network Abnormalities in Depression
5.6. Bipolar Disorder (BD)
5.7. Network Abnormalities in Bipolar Disorder
6. Discussion
6.1. Hybrid PET/MR Opportunities
6.2. Multifactorial Illnesses Require Multiple Modalities to Uncover Useful Biomarkers
6.3. Therapeutic Biomarker Discovery with Hybrid Imaging
6.4. Uncovering Molecular Mechanisms with Hybrid Imaging
6.5. Hybrid Imaging as a Potential Biomarker for Neuropsychiatric Disorders
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Global Burden of Disease. 2004. Available online: http://www.who.int/healthinfo/global_burden_disease/GBD_report_2004update_part3.pdf (accessed on 1 August 2015).
- World Health Organization. International Statistical Classification of Diseases and Related Health Problems; 10th revision for 2015; WHO: Geneva, Switzerland, 2015; Available online: http://apps.who.int/classifications/icd10/browse/2015/en (accessed 30 September 2015).
- American Psychiatric Association. DSM-5: Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Press: Arlington, VA, USA, 2013. [Google Scholar]
- Hyman, S.E. A glimmer of light for neuropsychiatric disorders. Nature 2008, 455, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Hirschfeld, R.M.; Lewis, L.; Vornik, L.A. Perceptions and impact of bipolar disorder: How far have we really come? Results of the national depressive and manic-depressive association 2000 survey of individuals with bipolar disorder. J. Clin. Psychiatry 2003, 64, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Kapur, S.; Phillips, A.G.; Insel, T.R. Why has it taken so long for biological psychiatry to develop clinical tests and what to do about it? Mol. Psychiatry 2012, 17, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Rohrer, J.D.; Guerreiro, R.; Vandrovcova, J.; Uphill, J.; Reiman, D.; Beck, J.; Isaacs, A.M.; Authier, A.; Ferrari, R.; Fox, N.C.; et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology 2009, 73, 1451–1456. [Google Scholar] [CrossRef] [PubMed]
- Serretti, A.; Olgiati, P.; de Ronchi, D. Genetics of Alzheimer’s disease. A rapidly evolving field. J. Alzheimer’s Dis. 2007, 12, 73–92. [Google Scholar]
- Brindle, N.; George-Hyslop, P.S. The genetics of Alzheimer’s disease. Methods Mol. Med. 2000, 32, 23–43. [Google Scholar] [PubMed]
- Sullivan, P.F.; Kendler, K.S.; Neale, M.C. Schizophrenia as a complex trait: Evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 2003, 60, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Bondy, B. Genetics in psychiatry: Are the promises met? World J. Biol. Psychiatry 2011, 12, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J.B.; Rauch, S.L.; Drevets, W.C. Clinical application of brain imaging for the diagnosis of mood disorders: The current state of play. Mol. Psychiatry 2013, 18, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr. Alzheimer disease: New concepts on its neurobiology and the clinical role imaging will play. Radiology 2012, 263, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Goodkind, M.; Eickhoff, S.B.; Oathes, D.J.; Jiang, Y.; Chang, A.; Jones-Hagata, L.B.; Ortega, B.N.; Zaiko, Y.V.; Roach, E.L.; Korgaonkar, M.S.; et al. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry 2015, 72, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Sporns, O. From simple graphs to the connectome: Networks in neuroimaging. NeuroImage 2012, 62, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Van Essen, D.C.; Ugurbil, K.; Auerbach, E.; Barch, D.; Behrens, T.E.; Bucholz, R.; Chang, A.; Chen, L.; Corbetta, M.; Curtiss, S.W.; et al. The human connectome project: A data acquisition perspective. NeuroImage 2012, 62, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- Menon, V. Large-scale brain networks and psychopathology: A unifying triple network model. Trends Cogn. Sci. 2011, 15, 483–506. [Google Scholar] [CrossRef] [PubMed]
- Williamson, P.C.; Allman, J.M. A framework for interpreting functional networks in schizophrenia. Front. Hum. Neurosci. 2012, 6, 184. [Google Scholar] [CrossRef] [PubMed]
- Raichle, M.E.; MacLeod, A.M.; Snyder, A.Z.; Powers, W.J.; Gusnard, D.A.; Shulman, G.L. A default mode of brain function. Proc. Natl. Acad. Sci. USA 2001, 98, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Seeley, W.W.; Menon, V.; Schatzberg, A.F.; Keller, J.; Glover, G.H.; Kenna, H.; Reiss, A.L.; Greicius, M.D. Dissociable intrinsic connectivity networks for salience processing and executive control. J. Neurosci. 2007, 27, 2349–2356. [Google Scholar] [CrossRef] [PubMed]
- Thomas Yeo, B.T.; Krienen, F.M.; Sepulcre, J.; Sabuncu, M.R.; Lashkari, D.; Hollinshead, M.; Roffman, J.L.; Smoller, J.W.; Zollei, L.; Polimeni, J.R.; et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J. Neurophysiol. 2011, 106, 1125–1165. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.; Uddin, L.Q. Saliency, switching, attention and control: A network model of insula function. Brain Struct. Funct. 2010, 15, 483–506. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.D.; Zhang, D.; Snyder, A.Z.; Raichle, M.E. The global signal and observed anticorrelated resting state brain networks. J. Neurophysiol. 2009, 101, 3270–3283. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.D.; Snyder, A.Z.; Vincent, J.L.; Corbetta, M.; van Essen, D.C.; Raichle, M.E. The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc. Natl. Acad. Sci. USA 2005, 102, 9673–9678. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, D.; Levitin, D.J.; Menon, V. A critical role for the right fronto-insular cortex in switching between central-executive and default-mode networks. Proc. Natl. Acad. Sci. USA 2008, 105, 12569–12574. [Google Scholar] [CrossRef] [PubMed]
- Hricak, H.; Choi, B.I.; Scott, A.M.; Sugimura, K.; Muellner, A.; von Schulthess, G.K.; Reiser, M.F.; Graham, M.M.; Dunnick, N.R.; Larson, S.M. Global trends in hybrid imaging. Radiology 2010, 257, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Hicks, R.; Lau, E.; Binns, D. Hybrid imaging is the future of molecular imaging. Biomed. Imaging Interv. J. 2007, 3, e49. [Google Scholar] [CrossRef] [PubMed]
- Anazodo, U.C.; Shoemaker, J.K.; Suskin, N.; Ssali, T.; Wang, D.J.; St Lawrence, K.S. Impaired Cerebrovascular function in coronary artery disease patients and recovery following cardiac rehabilitation. Front. Aging Neurosci. 2015, 7, 224. [Google Scholar] [CrossRef]
- Townsend, D.W. Multimodality imaging of structure and function. Phys. Med. Biol. 2008, 53, R1–R39. [Google Scholar] [CrossRef] [PubMed]
- Slomka, P.J. Software approach to merging molecular with anatomic information. J. Nucl. Med. 2004, 45 (Suppl. 1), 36S–45S. [Google Scholar] [PubMed]
- Beyer, T.; Townsend, D.W.; Brun, T.; Kinahan, P.E.; Charron, M.; Roddy, R.; Jerin, J.; Young, J.; Byars, L.; Nutt, R. A combined PET/CT scanner for clinical oncology. J. Nucl. Med. 2000, 41, 1369–1379. [Google Scholar] [PubMed]
- Cook, R.A.; Carnes, G.; Lee, T.Y.; Wells, R.G. Respiration-averaged CT for attenuation correction in canine cardiac PET/CT. J. Nucl. Med. 2007, 48, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Cuocolo, A.; Breatnach, E. Multimodality imaging in Europe: A survey by the European association of nuclear medicine (EANM) and the European society of radiology (ESR). Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, L.; Luxen, A. PET radiotracers for molecular imaging in the brain: Past, present and future. NeuroImage 2012, 61, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Bourgeat, P.; Chetelat, G.; Villemagne, V.L.; Fripp, J.; Raniga, P.; Pike, K.; Acosta, O.; Szoeke, C.; Ourselin, S.; Ames, D.; et al. Beta-amyloid burden in the temporal neocortex is related to hippocampal atrophy in elderly subjects without dementia. Neurology 2010, 74, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Rowe, C.C.; Ackerman, U.; Browne, W.; Mulligan, R.; Pike, K.L.; O’Keefe, G.; Tochon-Danguy, H.; Chan, G.; Berlangieri, S.U.; Jones, G.; et al. Imaging of amyloid beta in Alzheimer’s disease with 18F-BAY94-9172, a novel PET tracer: Proof of mechanism. Lancet Neurol. 2008, 7, 129–135. [Google Scholar] [CrossRef]
- Brix, G.; Beyer, T. PET/CT: Dose-escalated image fusion? Nucl. Med. 2005, 44 (Suppl. 1), S51–S57. [Google Scholar]
- Huettel, S.A.; Song, A.W.; McCarthy, G. Functional Magnetic Resonance Imaging, 2nd ed.; Sinauer Associates, Inc.: Sunderland, MA, USA, 2009. [Google Scholar]
- Logothetis, N.K.; Pfeuffer, J. On the nature of the bold fmri contrast mechanism. Magn. Reson. Imaging 2004, 22, 1517–1531. [Google Scholar] [CrossRef] [PubMed]
- Schlemmer, H.P.; Pichler, B.J.; Schmand, M.; Burbar, Z.; Michel, C.; Ladebeck, R.; Jattke, K.; Townsend, D.; Nahmias, C.; Jacob, P.K.; et al. Simultaneous MR/PET imaging of the human brain: Feasibility study. Radiology 2008, 248, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Disselhorst, J.A.; Bezrukov, I.; Kolb, A.; Parl, C.; Pichler, B.J. Principles of PET/MR imaging. J. Nucl. Med. 2014, 55, 2S–10S. [Google Scholar] [CrossRef] [PubMed]
- Pichler, B.J.; Judenhofer, M.S.; Pfannenberg, C. Multimodal imaging approaches: PET/CT and PET/MRI. In Handbook of Experimental Pharmacology; Springer Berlin Heidelberg: Tübingen, Germany, 2008; pp. 109–132. [Google Scholar]
- Garibotto, V.; Heinzer, S.; Vulliemoz, S.; Guignard, R.; Wissmeyer, M.; Seeck, M.; Lovblad, K.O.; Zaidi, H.; Ratib, O.; Vargas, M.I. Clinical applications of hybrid PET/MRI in neuroimaging. Clin. Nucl. Med. 2013, 38, e13–e18. [Google Scholar] [CrossRef] [PubMed]
- Catana, C.; Drzezga, A.; Heiss, W.D.; Rosen, B.R. PET/MRI for neurologic applications. J. Nucl. Med. 2012, 53, 1916–1925. [Google Scholar] [CrossRef] [PubMed]
- Brookes, M.J.; Hall, E.L.; Robson, S.E.; Price, D.; Palaniyappan, L.; Liddle, E.B.; Liddle, P.F.; Robinson, S.E.; Morris, P.G. Complexity measures in magnetoencephalography: Measuring “disorder” in schizophrenia. PLoS ONE 2015, 10, e0120991. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, R.M.; Hashemi, N.; Gati, J.S.; Menon, R.S.; Everling, S. Electrophysiological signatures of spontaneous bold fluctuations in macaque prefrontal cortex. NeuroImage 2015, 113, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Menon, R.S.; Tank, D.W.; Kim, S.G.; Merkle, H.; Ellermann, J.M.; Ugurbil, K. Functional brain mapping by blood oxygenation level-dependent contrast magnetic resonance imaging. A comparison of signal characteristics with a biophysical model. Biophys. J. 1993, 64, 803–812. [Google Scholar] [CrossRef]
- Logothetis, N.K. What we can do and what we cannot do with fMRI. Nature 2008, 453, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.; Sydekum, E.; Krueppel, R.; Engelbrecht, C.J.; Schlegel, F.; Schröter, A.; Rudin, M.; Helmchen, F. Simultaneous bold fMRI and fiber-optic calcium recording in rat neocortex. Nat. Methods 2012, 9, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-G.; Ogawa, S. Biophysical and physiological origins of blood oxygenation level-dependent fmri signals. J. Cereb. Blood Flow Metab. 2012, 32, 1188–1206. [Google Scholar] [CrossRef] [PubMed]
- Van Berckel, B.N.; Bossong, M.G.; Boellaard, R.; Kloet, R.; Schuitemaker, A.; Caspers, E.; Luurtsema, G.; Windhorst, A.D.; Cahn, W.; Lammertsma, A.A.; et al. Microglia activation in recent-onset schizophrenia: A quantitative (R)-[11C]PK11195 positron emission tomography study. Biol. Psychiatry 2008, 64, 820–822. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2014 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2014, 10, 47–92. [Google Scholar]
- Hardy, J. Alzheimer’s disease: The amyloid cascade hypothesis: An update and reappraisal. J. Alzheimer’s Dis. 2006, 9, 151–153. [Google Scholar]
- Mueller, S.G.; Weiner, M.W.; Thal, L.J.; Petersen, R.C.; Jack, C.; Jagust, W.; Trojanowski, J.Q.; Toga, A.W.; Beckett, L. The Alzheimer’s disease neuroimaging initiative. Neuroimaging Clin. N. Am. 2005, 15, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Weiner, M.W.; Veitch, D.P.; Aisen, P.S.; Beckett, L.A.; Cairns, N.J.; Green, R.C.; Harvey, D.; Jack, C.R.; Jagust, W.; Liu, E.; et al. The Alzheimer’s disease neuroimaging initiative: A review of papers published since its inception. Alzheimer’s Dement. 2013, 9, e111–e194. [Google Scholar] [CrossRef] [PubMed]
- Soucy, J.P.; Bartha, R.; Bocti, C.; Borrie, M.; Burhan, A.M.; Laforce, R.; Rosa-Neto, P. Clinical applications of neuroimaging in patients with Alzheimer’s disease: A review from the fourth Canadian consensus conference on the diagnosis and treatment of dementia 2012. Alzheimer’s Res. Ther. 2013, 5, S3. [Google Scholar] [CrossRef] [PubMed]
- Burhan, A.M.; Bartha, R.; Bocti, C.; Borrie, M.; Laforce, R.; Rosa-Neto, P.; Soucy, J.P. Role of emerging neuroimaging modalities in patients with cognitive impairment: A review from the canadian consensus conference on the diagnosis and treatment of dementia 2012. Alzheimer’s Res. Ther. 2013, 5, S4. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Mintzer, J.; Weiner, M.F.; Cummings, J.L. Disease-modifying therapies in Alzheimer’s disease. Alzheimer’s Dement. 2008, 4, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.M.; Mallmann, J.; Santacruz, A.M.; Fuqua, A.; Carril, M.; Aisen, P.S.; Althage, M.C.; Belyew, S.; Benzinger, T.L.; Brooks, W.S.; et al. Preclinical trials in autosomal dominant AD: Implementation of the DIAN-TU trial. Rev. Neurol. 2013, 169, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Rentz, D.M.; Johnson, K.A.; Karlawish, J.; Donohue, M.; Salmon, D.P.; Aisen, P. The A4 study: Stopping AD before symptoms begin? Sci. Transl. Med. 2014, 6, 228fs213. [Google Scholar] [CrossRef] [PubMed]
- Price, J.C.; Klunk, W.E.; Lopresti, B.J.; Lu, X.; Hoge, J.A.; Ziolko, S.K.; Holt, D.P.; Meltzer, C.C.; DeKosky, S.T.; Mathis, C.A. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J. Cereb. Blood Flow Metab. 2005, 25, 1528–1547. [Google Scholar] [CrossRef] [PubMed]
- Ikonomovic, M.D.; Klunk, W.E.; Abrahamson, E.E.; Mathis, C.A.; Price, J.C.; Tsopelas, N.D.; Lopresti, B.J.; Ziolko, S.; Bi, W.; Paljug, W.R. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 2008, 131, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Lyketsos, C.G.; Lopez, O.; Jones, B.; Fitzpatrick, A.L.; Breitner, J.; DeKosky, S. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: Results from the cardiovascular health study. JAMA 2002, 288, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Seitz, D.; Purandare, N.; Conn, D. Prevalence of psychiatric disorders among older adults in long-term care homes: A systematic review. Int. Psychogeriatr. 2010, 22, 1025–1039. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Mega, M.; Gray, K.; Rosenberg-Thompson, S.; Carusi, D.A.; Gornbein, J. The neuropsychiatric inventory: Comprehensive assessment of psychopathology in dementia. Neurology 1994, 44, 2308–2314. [Google Scholar] [CrossRef] [PubMed]
- Geda, Y.E.; Schneider, L.S.; Gitlin, L.N.; Miller, D.S.; Smith, G.S.; Bell, J.; Evans, J.; Lee, M.; Porsteinsson, A.; Lanctot, K.L.; et al. Neuropsychiatric symptoms in Alzheimer’s disease: Past progress and anticipation of the future. Alzheimer’s Dement. 2013, 9, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Howard, R. Neuroleptic drugs in dementia: Benefits and harm. Nat. Rev. Neurosci. 2006, 7, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.E.; Ting, W.K.; Millikin, C.P.; Ismail, Z.; Schweizer, T.A.; Alzheimer’s Disease Neuroimaging Initiative. Gray matter atrophy in patients with mild cognitive impairment/Alzheimer’s disease over the course of developing delusions. Int. J. Geriatr. Psychiatry 2015. [Google Scholar] [CrossRef] [PubMed]
- Ting, W.K.; Fischer, C.E.; Millikin, C.P.; Ismail, Z.; Chow, T.W.; Schweizer, T.A. Grey matter atrophy in mild cognitive impairment/early Alzheimer’s disease associated with delusions: A voxel-based morphometry study. Curr. Alzheimer’s Res. 2015, 12, 165–172. [Google Scholar] [CrossRef]
- Rafii, M.S.; Taylor, C.S.; Kim, H.T.; Desikan, R.S.; Fleisher, A.S.; Katibian, D.; Brewer, J.B.; Dale, A.M.; Aisen, P.S. Neuropsychiatric symptoms and regional neocortical atrophy in mild cognitive impairment and Alzheimer’s disease. Am. J. Alzheimer’s Dis. Dement. 2014, 29, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Bruen, P.D.; McGeown, W.J.; Shanks, M.F.; Venneri, A. Neuroanatomical correlates of neuropsychiatric symptoms in Alzheimer’s disease. Brain 2008, 131, 2455–2463. [Google Scholar] [CrossRef] [PubMed]
- Ismail, Z.; Nguyen, M.Q.; Fischer, C.E.; Schweizer, T.A.; Mulsant, B.H. Neuroimaging of delusions in Alzheimer’s disease. Psychiatry Res. 2012, 202, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Sultzer, D.L.; Leskin, L.P.; Melrose, R.J.; Harwood, D.G.; Narvaez, T.A.; Ando, T.K.; Mandelkern, M.A. Neurobiology of delusions, memory, and insight in Alzheimer’s disease. Am. J. Geriatr. Psychiatry 2014, 22, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Blanc, F.; Noblet, V.; Philippi, N.; Cretin, B.; Foucher, J.; Armspach, J.P.; Rousseau, F.; Alzheimer’s Disease Neuroimaging Initiative. Right anterior insula: Core region of hallucinations in cognitive neurodegenerative diseases. PLoS ONE 2014, 9, e114774. [Google Scholar] [CrossRef] [PubMed]
- Donovan, N.J.; Wadsworth, L.P.; Lorius, N.; Locascio, J.J.; Rentz, D.M.; Johnson, K.A.; Sperling, R.A.; Marshall, G.A.; Alzheimer’s Disease Neuroimaging Initiative. Regional cortical thinning predicts worsening apathy and hallucinations across the Alzheimer’s disease spectrum. Am. J. Geriatr. Psychiatry 2014, 22, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Mintzer, J.; Brodaty, H.; Sano, M.; Banerjee, S.; Devanand, D.P.; Gauthier, S.; Howard, R.; Lanctot, K.; Lyketsos, C.G.; et al. Agitation in cognitive disorders: International psychogeriatric association provisional consensus clinical and research definition. Int. Psychogeriatr. 2015, 27, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Mansfield, J.; Billig, N. Agitated behaviors in the elderly. I. A conceptual review. J. Am. Geriatr. Soc. 1986, 34, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Trzepacz, P.T.; Yu, P.; Bhamidipati, P.K.; Willis, B.; Forrester, T.; Tabas, L.; Schwarz, A.J.; Saykin, A.J.; Alzheimer’s Disease Neuroimaging Initiative. Frontolimbic atrophy is associated with agitation and aggression in mild cognitive impairment and Alzheimer’s disease. Alzheimer’s Dement. 2013, 9, S95.e101–S104.e101. [Google Scholar] [CrossRef] [PubMed]
- Balthazar, M.L.; Pereira, F.R.; Lopes, T.M.; da Silva, E.L.; Coan, A.C.; Campos, B.M.; Duncan, N.W.; Stella, F.; Northoff, G.; Damasceno, B.P.; et al. Neuropsychiatric symptoms in Alzheimer’s disease are related to functional connectivity alterations in the salience network. Hum. Brain Mapp. 2014, 35, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.B.; Nowrangi, M.A.; Lyketsos, C.G. Neuropsychiatric symptoms in Alzheimer’s disease: What might be associated brain circuits? Mol. Asp. Med. 2015, 43–44, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Marin, R.S.; Wilkosz, P.A. Disorders of diminished motivation. J. Head Trauma Rehabil. 2005, 20, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Starkstein, S.E.; Mayberg, H.S.; Preziosi, T.J.; Andrezejewski, P.; Leiguarda, R.; Robinson, R.G. Reliability, validity, and clinical correlates of apathy in parkinson’s disease. J. Neuropsychiatry Clin. Neurosci. 1992, 4, 134–139. [Google Scholar] [PubMed]
- Delrieu, J.; Desmidt, T.; Camus, V.; Sourdet, S.; Boutoleau-Bretonniere, C.; Mullin, E.; Vellas, B.; Payoux, P.; Lebouvier, T.; Alzheimer’s Disease Neuroimaging Initiative. Apathy as a feature of prodromal Alzheimer’s disease: An FDG-PET ADNI study. Int. J. Geriatr. Psychiatry 2015, 30, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Lanctot, K.L.; Moosa, S.; Herrmann, N.; Leibovitch, F.S.; Rothenburg, L.; Cotter, A.; Black, S.E. A spect study of apathy in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2007, 24, 65–72. [Google Scholar] [PubMed]
- Murray, P.S.; Kirkwood, C.M.; Gray, M.C.; Fish, K.N.; Ikonomovic, M.D.; Hamilton, R.L.; Kofler, J.K.; Klunk, W.E.; Lopez, O.L.; Sweet, R.A. Hyperphosphorylated tau is elevated in Alzheimer’s disease with psychosis. J. Alzheimer’s Dis. 2014, 39, 759–773. [Google Scholar]
- Koppel, J.; Sunday, S.; Goldberg, T.E.; Davies, P.; Christen, E.; Greenwald, B.S.; Alzheimer’s Disease Neuroimaging Initiative. Psychosis in Alzheimer’s disease is associated with frontal metabolic impairment and accelerated decline in working memory: Findings from the Alzheimer’s disease neuroimaging initiative. Am. J. Geriatr. Psychiatry 2014, 22, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Neary, D.; Snowden, J.S.; Gustafson, L.; Passant, U.; Stuss, D.; Black, S.; Freedman, M.; Kertesz, A.; Robert, P.H.; Albert, M.; et al. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology 1998, 51, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Rascovsky, K.; Hodges, J.R.; Kipps, C.M.; Johnson, J.K.; Seeley, W.W.; Mendez, M.F.; Knopman, D.; Kertesz, A.; Mesulam, M.; Salmon, D.P.; et al. Diagnostic criteria for the behavioral variant of frontotemporal dementia (bvFTD): Current limitations and future directions. Alzheimer’s Dis. Assoc. Disord. 2007, 21, S14–S18. [Google Scholar] [CrossRef] [PubMed]
- Seeley, W.W. Selective functional, regional, and neuronal vulnerability in frontotemporal dementia. Curr. Opin. Neurol. 2008, 21, 701–707. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with lewy bodies: Third report of the dlb consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Leucht, S.; Tardy, M.; Komossa, K.; Heres, S.; Kissling, W.; Salanti, G.; Davis, J.M. Antipsychotic drugs versus placebo for relapse prevention in schizophrenia: A systematic review and meta-analysis. Lancet 2012, 379, 2063–2071. [Google Scholar] [CrossRef]
- Palaniyappan, L.; Liddle, P.F. Does the salience network play a cardinal role in psychosis? An emerging hypothesis of insular dysfunction. J. Psychiatry Neurosci. 2012, 37, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Manoliu, A.; Riedl, V.; Zherdin, A.; Mühlau, M.; Schwerthöffer, D.; Scherr, M.; Peters, H.; Zimmer, C.; Förstl, H.; Bäuml, J.; et al. Aberrant dependence of default mode/central executive network interactions on anterior insular salience network activity in schizophrenia. Schizophr. Bull. 2013. [Google Scholar] [CrossRef] [PubMed]
- Moran, L.V.; Tagamets, M.A.; Sampath, H.; O’Donnell, A.; Stein, E.A.; Kochunov, P.; Hong, L.E. Disruption of anterior insula modulation of large-scale brain networks in schizophrenia. Biol. Psychiatry 2013, 74, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Palaniyappan, L.; Simmonite, M.; White, T.P.; Liddle, E.B.; Liddle, P.F. Neural primacy of the salience processing system in schizophrenia. Neuron 2013, 79, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Wotruba, D.; Michels, L.; Buechler, R.; Metzler, S.; Theodoridou, A.; Gerstenberg, M.; Walitza, S.; Kollias, S.; Rössler, W.; Heekeren, K. Aberrant coupling within and across the default mode, task-positive, and salience network in subjects at risk for psychosis. Schizophr. Bull. 2014, 40, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Driesen, N.R.; McCarthy, G.; Bhagwagar, Z.; Bloch, M.; Calhoun, V.; D’Souza, D.C.; Gueorguieva, R.; He, G.; Ramachandran, R.; Suckow, R.F.; et al. Relationship of resting brain hyperconnectivity and schizophrenia-like symptoms produced by the NMDA receptor antagonist ketamine in humans. Mol. Psychiatry 2013, 18, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Fuchigami, T.; Nakayama, M.; Yoshida, S. Development of PET and SPECT probes for glutamate receptors. Sci. World J. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.H.; Liao, M.H.; Tseng, Y.C. Recent advances in imaging of dopaminergic neurons for evaluation of neuropsychiatric disorders. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Poels, E.M.; Kegeles, L.S.; Kantrowitz, J.T.; Slifstein, M.; Javitt, D.C.; Lieberman, J.A.; Abi-Dargham, A.; Girgis, R.R. Imaging glutamate in schizophrenia: Review of findings and implications for drug discovery. Mol. Psychiatry 2014, 19, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Agius, M.; Goh, C.; Ulhaq, S.; McGorry, P. The staging model in schizophrenia, and its clinical implications. Psychiatr. Danub. 2010, 22, 211–220. [Google Scholar] [PubMed]
- Marsman, A.; van den Heuvel, M.P.; Klomp, D.W.J.; Kahn, R.S.; Luijten, P.R.; Hulshoff Pol, H.E. Glutamate in schizophrenia: A focused review and meta-analysis of 1H-MRS studies. Schizophr. Bull. 2013, 39, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Fryer, S.L.; Woods, S.W.; Kiehl, K.A.; Calhoun, V.D.; Pearlson, G.D.; Roach, B.J.; Ford, J.M.; Srihari, V.H.; McGlashan, T.H.; Mathalon, D.H. Deficient suppression of default mode regions during working memory in individuals with early psychosis and at clinical high-risk for psychosis. Front. Psychiatry 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Qiu, L.; Yuan, L.; Ma, H.; Ye, R.; Yu, F.; Hu, P.; Dong, Y.; Wang, K. Evidence for progressive brain abnormalities in early schizophrenia: A cross-sectional structural and functional connectivity study. Schizophr. Res. 2014, 159, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M.; Berretta, S. Gabaergic interneurons: Implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 2001, 25, 1–27. [Google Scholar] [CrossRef]
- Lewis, D.A.; Curley, A.A.; Glausier, J.R.; Volk, D.W. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012, 35, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Goto, N.; Yoshimura, R.; Moriya, J.; Kakeda, S.; Ueda, N.; Ikenouchi-Sugita, A.; Umene-Nakano, W.; Hayashi, K.; Oonari, N.; Korogi, Y.; et al. Reduction of brain gamma-aminobutyric acid (GABA) concentrations in early-stage schizophrenia patients: 3T Proton MRS study. Schizophr. Res. 2009, 112, 192–193. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.F.; Tso, I.F. GABA abnormalities in schizophrenia: A methodological review of in vivo studies. Schizophr. Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Anticevic, A.; Cole, M.W.; Repovs, G.; Savic, A.; Driesen, N.R.; Yang, G.; Cho, Y.T.; Murray, J.D.; Glahn, D.C.; Wang, X.-J.; et al. Connectivity, pharmacology, and computation: Toward a mechanistic understanding of neural system dysfunction in schizophrenia. Front. Psychiatry 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Wiebking, C.; Duncan, N.W.; Qin, P.; Hayes, D.J.; Lyttelton, O.; Gravel, P.; Verhaeghe, J.; Kostikov, A.P.; Schirrmacher, R.; Reader, A.J.; et al. External awareness and GABA—A multimodal imaging study combining fMRI and [18F]flumazenil-PET. Hum. Brain Mapp. 2014, 35, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Monji, A.; Kato, T.A.; Mizoguchi, Y.; Horikawa, H.; Seki, Y.; Kasai, M.; Yamauchi, Y.; Yamada, S.; Kanba, S. Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 42, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Lennox, B.R.; Coles, A.J.; Vincent, A. Antibody-mediated encephalitis: A treatable cause of schizophrenia. Br. J. Psychiatry 2012, 200, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Sommer, I.E.; de Witte, L.; Begemann, M.; Kahn, R.S. Nonsteroidal anti-inflammatory drugs in schizophrenia: Ready for practice or a good start? A meta-analysis. J. Clin. Psychiatry 2012, 73, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Ory, D.; Celen, S.; Verbruggen, A.; Bormans, G. PET radioligands for in vivo visualization of neuroinflammation. Curr. Pharm. Des. 2014, 20, 5897–5913. [Google Scholar] [CrossRef] [PubMed]
- Hasler, G.; Drevets, W.C.; Manji, H.K.; Charney, D.S. Discovering endophenotypes for major depression. Neuropsychopharmacology 2004, 29, 1765–1781. [Google Scholar] [CrossRef] [PubMed]
- Ressler, K.J.; Mayberg, H.S. Targeting abnormal neural circuits in mood and anxiety disorders: From the laboratory to the clinic. Nat. Neurosci. 2007, 10, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, S.; Honig, A.; van Duinen, M.A.; Riedel, W.J. Serotonergic dysregulation in bipolar disorders: A literature review of serotonergic challenge studies. Bipolar Disord. 2002, 4, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Heiss, W.D.; Herholz, K. Brain receptor imaging. J. Nucl. Med. 2006, 47, 302–312. [Google Scholar] [PubMed]
- Ruhe, H.G.; Mason, N.S.; Schene, A.H. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: A meta-analysis of monoamine depletion studies. Mol. Psychiatry 2007, 12, 331–359. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.C.; Berglund, P.; Demler, O.; Jin, R.; Merikangas, K.R.; Walters, E.E. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the national comorbidity survey replication. Arch. Gen. Psychiatry 2005, 62, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Gershuny, B.S.; Baer, L.; Parker, H.; Gentes, E.L.; Infield, A.L.; Jenike, M.A. Trauma and posttraumatic stress disorder in treatment-resistant obsessive-compulsive disorder. Depression Anxiety 2008, 25, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M.J.; Resick, P.A.; Bryant, R.A.; Strain, J.; Horowitz, M.; Spiegel, D. Classification of trauma and stressor-related disorders in DSM-5. Depression Anxiety 2011, 28, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.; Thome, J.; Frewen, P.; Lanius, R.A. Resting-state neuroimaging studies: A new way of identifying differences and similarities among the anxiety disorders? Can. J. Psychiatry. 2014, 59, 294–300. [Google Scholar] [PubMed]
- Rabinak, C.A.; Angstadt, M.; Welsh, R.C.; Kenndy, A.E.; Lyubkin, M.; Martis, B.; Phan, K.L. Altered amygdala resting-state functional connectivity in post-traumatic stress disorder. Front. Psychiatry 2011, 2, 62. [Google Scholar] [CrossRef] [PubMed]
- Sripada, R.K.; King, A.P.; Welsh, R.C.; Garfinkel, S.N.; Wang, X.; Sripada, C.S.; Liberzon, I. Neural dysregulation in posttraumatic stress disorder: Evidence for disrupted equilibrium between salience and default mode brain networks. Psychosom. Med. 2012, 74, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Liao, W.; Ding, J.; Feng, Y.; Zhu, C.; Nie, X.; Zhang, W.; Chen, H.; Gong, Q. Regional homogeneity changes in social anxiety disorder: A resting-state fMRI study. Psychiatry Res. 2011, 194, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Stein, P.; Windischberger, C.; Weissenbacher, A.; Spindelegger, C.; Moser, E.; Kasper, S.; Lanzenberger, R. Reduced resting-state functional connectivity between amygdala and orbitofrontal cortex in social anxiety disorder. NeuroImage 2011, 56, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Qiu, C.; Gentili, C.; Walter, M.; Pan, Z.; Ding, J.; Zhang, W.; Gong, Q.; Chen, H. Altered effective connectivity network of the amygdala in social anxiety disorder: A resting-state fMRI study. PLoS ONE 2010, 5, e15238. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Shi, F.; Shi, C.; Yang, Q.; Chan, R.C.; Shen, D. Disrupted cortical network as a vulnerability marker for obsessive-compulsive disorder. Brain Struct. Funct. 2014, 219, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Stern, E.R.; Fitzgerald, K.D.; Welsh, R.C.; Abelson, J.L.; Taylor, S.F. Resting-state functional connectivity between fronto-parietal and default mode networks in obsessive-compulsive disorder. PLoS ONE 2012, 7, e36356. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.D.; Stern, E.R.; Angstadt, M.; Nicholson-Muth, K.C.; Maynor, M.R.; Welsh, R.C.; Hanna, G.L.; Taylor, S.F. Altered function and connectivity of the medial frontal cortex in pediatric obsessive-compulsive disorder. Biol. Psychiatry 2010, 68, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Gusnard, D.A.; Akbudak, E.; Shulman, G.L.; Raichle, M.E. Medial prefrontal cortex and self-referential mental activity: Relation to a default mode of brain function. Proc. Natl. Acad. Sci. USA 2001, 98, 4259–4264. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, S.; Dong, Z.; Luo, J.; Han, H.; Xiong, H.; Guo, Z.; Li, Z. Altered resting state functional connectivity patterns of the anterior prefrontal cortex in obsessive-compulsive disorder. NeuroReport 2012, 23, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Posner, J.; Hellerstein, D.J.; Gat, I.; Mechling, A.; Klahr, K.; Wang, Z.; McGrath, P.J.; Stewart, J.W.; Peterson, B.S. Antidepressants normalize the default mode network in patients with dysthymia. JAMA Psychiatry 2013, 70, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; Roberts, J.E. Memory and attention in obsessive-compulsive disorder: A review. J. Anxiety Disord. 2005, 19, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Hamner, M.B.; Robert, S.; Frueh, B.C. Treatment-resistant posttraumatic stress disorder: Strategies for intervention. CNS Spectr. 2004, 9, 740–752. [Google Scholar] [PubMed]
- Ammar, G.; Naja, W.J.; Pelissolo, A. Treatment-resistant anxiety disorders: A literature review of drug therapy strategies. L’Encephale 2015, 41, 260–265. (in French). [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, P.V.; Kapczinski, N.S.; Kapczinski, F. Correlates and impact of obsessive-compulsive comorbidity in bipolar disorder. Compr. Psychiatry 2010, 51, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Al-Asadi, A.M.; Klein, B.; Meyer, D. Comorbidity structure of psychological disorders in the online e-pass data as predictors of psychosocial adjustment measures: Psychological distress, adequate social support, self-confidence, quality of life, and suicidal ideation. J. Med. Internet Res. 2014, 16, e248. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Leon, S.; Janssens, A.C.; Gonzalez-Zuloeta Ladd, A.M.; Del-Favero, J.; Claes, S.J.; Oostra, B.A.; van Duijn, C.M. Meta-analyses of genetic studies on major depressive disorder. Mol. Psychiatry 2008, 13, 772–785. [Google Scholar] [CrossRef] [PubMed]
- Mill, J.; Petronis, A. Molecular studies of major depressive disorder: The epigenetic perspective. Mol. Psychiatry 2007, 12, 799–814. [Google Scholar] [CrossRef] [PubMed]
- Mulders, P.C.; van Eijndhoven, P.F.; Schene, A.H.; Beckmann, C.F.; Tendolkar, I. Resting-state functional connectivity in major depressive disorder: A review. Neurosci. Biobehav. Rev. 2015, 56, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Nestler, E.J. The molecular neurobiology of depression. Nature 2008, 455, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Park, Y.M.; Um, T.H.; Kim, S. Lower serum brain-derived neurotrophic factor levels are associated with failure to achieve remission in patients with major depression after escitalopram treatment. Neuropsychiatr. Dis. Treat. 2014, 10, 1393–1398. [Google Scholar] [PubMed]
- Miller, C.H.; Hamilton, J.P.; Sacchet, M.D.; Gotlib, I.H. Meta-analysis of functional neuroimaging of major depressive disorder in youth. JAMA Psychiatry 2015, 72, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Pizzagalli, D.A. Frontocingulate dysfunction in depression: Toward biomarkers of treatment response. Neuropsychopharmacology 2011, 36, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Drevets, W.C.; Price, J.L.; Furey, M.L. Brain structural and functional abnormalities in mood disorders: Implications for neurocircuitry models of depression. Brain Struct. Funct. 2008, 213, 93–118. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.H.; Andrews-Hanna, J.R.; Wager, T.D.; Pizzagalli, D.A. Large-scale network dysfunction in major depressive disorder: A meta-analysis of resting-state functional connectivity. JAMA Psychiatry 2015, 72, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Hasler, G.; Northoff, G. Discovering imaging endophenotypes for major depression. Mol. Psychiatry 2011, 16, 604–619. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, C.; Zhu, X.; Tan, Y.; Zhong, Y. Aberrant connectivity within the default mode network in first-episode, treatment-naive major depressive disorder. J. Affect. Disord. 2015, 183, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Liddle, E.B.; Iwabuchi, S.J.; Zhang, C.; Wu, Z.; Liu, J.; Jiang, K.; Xu, L.; Liddle, P.F.; Palaniyappan, L.; et al. Dissociated large-scale functional connectivity networks of the precuneus in medication-naive first-episode depression. Psychiatry Res. 2015, 232, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Yao, J.; Jiang, X.; Zhang, L.; Xu, L.; Feng, R.; Cai, L.; Liu, J.; Wang, J.; Chen, W. Sub-hubs of baseline functional brain networks are related to early improvement following two-week pharmacological therapy for major depressive disorder. Hum. Brain Mapp. 2015, 36, 2915–2927. [Google Scholar] [CrossRef] [PubMed]
- McGrath, C.L.; Kelley, M.E.; Holtzheimer, P.E.; Dunlop, B.W.; Craighead, W.E.; Franco, A.R.; Craddock, R.C.; Mayberg, H.S. Toward a neuroimaging treatment selection biomarker for major depressive disorder. JAMA Psychiatry 2013, 70, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Woods, S.W. The economic burden of bipolar disease. J. Clin. Psychiatry 2000, 61 (Suppl. 13), S38–S41. [Google Scholar]
- Oswald, P.; Souery, D.; Kasper, S.; Lecrubier, Y.; Montgomery, S.; Wyckaert, S.; Zohar, J.; Mendlewicz, J. Current issues in bipolar disorder: A critical review. Eur. Neuropsychopharmacol. 2007, 17, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Baldassano, C.F.; Ballas, C.A.; O’Reardon, J.P. Rethinking the treatment paradigm for bipolar depression: The importance of long-term management. CNS Spectr. 2004, 9, 11–18. [Google Scholar] [PubMed]
- Almeida, O.P.; Garrido, G.J.; Etherton-Beer, C.; Lautenschlager, N.T.; Arnolda, L.; Alfonso, H.; Flicker, L. Brain and mood changes over 2 years in healthy controls and adults with heart failure and ischaemic heart disease. Eur. J. Heart Fail. 2013, 15, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Belmaker, R.H. Bipolar disorder. N. Engl. J. Med. 2004, 351, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Lachaine, J.; Beauchemin, C.; Mathurin, K.; Gilbert, D.; Beillat, M. Cost-effectiveness of asenapine in the treatment of bipolar disorder in Canada. BMC Psychiatry 2014, 14, 16. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, J.R.; de Campos, B.K.; Galazzi, R.M.; Martinez, J.L.; Arruda, M.A. Bipolar disorder: Recent advances and future trends in bioanalytical developments for biomarker discovery. Anal. Bioanal. Chem. 2015, 407, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, V.D.; Maciejewski, P.K.; Pearlson, G.D.; Kiehl, K.A. Temporal lobe and “default” hemodynamic brain modes discriminate between schizophrenia and bipolar disorder. Hum. Brain Mapp. 2008, 29, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Magioncalda, P.; Martino, M.; Conio, B.; Escelsior, A.; Piaggio, N.; Presta, A.; Marozzi, V.; Rocchi, G.; Anastasio, L.; Vassallo, L.; et al. Functional connectivity and neuronal variability of resting state activity in bipolar disorder—Reduction and decoupling in anterior cortical midline structures. Hum. Brain Mapp. 2015, 36, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.M.; Sham, P.; van Os, J.; Zanelli, J.; Cannon, M.; McDonald, C. A developmental model for similarities and dissimilarities between schizophrenia and bipolar disorder. Schizophr. Res. 2004, 71, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Weiser, M.; Reichenberg, A.; Rabinowitz, J.; Kaplan, Z.; Mark, M.; Bodner, E.; Nahon, D.; Davidson, M. Association between nonpsychotic psychiatric diagnoses in adolescent males and subsequent onset of schizophrenia. Arch. Gen. Psychiatry 2001, 58, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.J.; Whitfield-Gabrieli, S.; Shinn, A.K.; Gabrieli, J.D.; Nieto Castanon, A.; McCarthy, J.M.; Cohen, B.M.; Ongur, D. Abnormal medial prefrontal cortex resting-state connectivity in bipolar disorder and schizophrenia. Neuropsychopharmacology 2011, 36, 2009–2017. [Google Scholar] [CrossRef] [PubMed]
- Ongur, D.; Lundy, M.; Greenhouse, I.; Shinn, A.K.; Menon, V.; Cohen, B.M.; Renshaw, P.F. Default mode network abnormalities in bipolar disorder and schizophrenia. Psychiatry Res. 2010, 183, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Meda, S.A.; Gill, A.; Stevens, M.C.; Lorenzoni, R.P.; Glahn, D.C.; Calhoun, V.D.; Sweeney, J.A.; Tamminga, C.A.; Keshavan, M.S.; Thaker, G.; et al. Differences in resting-state functional magnetic resonance imaging functional network connectivity between schizophrenia and psychotic bipolar probands and their unaffected first-degree relatives. Biol. Psychiatry 2012, 71, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Mamah, D.; Barch, D.M.; Repovs, G. Resting state functional connectivity of five neural networks in bipolar disorder and schizophrenia. J. Affect. Disord. 2013, 150, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Argyelan, M.; Ikuta, T.; DeRosse, P.; Braga, R.J.; Burdick, K.E.; John, M.; Kingsley, P.B.; Malhotra, A.K.; Szeszko, P.R. Resting-state fMRI connectivity impairment in schizophrenia and bipolar disorder. Schizophr. Bull. 2014, 40, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Schwenzer, N.F.; Stegger, L.; Bisdas, S.; Schraml, C.; Kolb, A.; Boss, A.; Muller, M.; Reimold, M.; Ernemann, U.; Claussen, C.D.; et al. Simultaneous PET/MR imaging in a human brain PET/MR system in 50 patients—Current state of image quality. Eur. J. Radiol. 2012, 81, 3472–3478. [Google Scholar] [CrossRef] [PubMed]
- Dyson, F.J. History of science. Is science mostly driven by ideas or by tools? Science 2012, 338, 1426–1427. [Google Scholar] [CrossRef] [PubMed]
- Anazodo, U.C.; Thiessen, J.D.; Ssali, T.; Mandel, J.; Gunther, M.; Butler, J.; Pavlosky, W.; Prato, F.S.; Thompson, R.T.; St Lawrence, K.S. Feasibility of simultaneous whole-brain imaging on an integrated PET-MRI system using an enhanced 2-point dixon attenuation correction method. Front. Neurosci. 2014, 8, 434. [Google Scholar] [CrossRef] [PubMed]
- Mach, R.H. New targets for the development of PET tracers for imaging neurodegeneration in Alzheimer’s disease. J. Nucl. Med. 2014, 55, 1221–1224. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Jiang, M.; Luo, J. An improved auto-window algorithm for MR image. Zhongguo Yi Liao Qi Xie Za Zhi 2011, 35, 253–255. (in Chinese). [Google Scholar] [PubMed]
- Vallabhajosula, S. Molecular Imaging. Radiopharmaceuticals for PET and SPECT; Springer: Berlin, Germany; Heidelberg, Germany, 2009. [Google Scholar]
- Lohoff, F.W. Overview of the genetics of major depressive disorder. Curr. Psychiatry Rep. 2010, 12, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, Y.; Zhou, L.; Yuan, H.; Shen, D.; Alzheimer’s Disease Neuroimaging Initiative. Multimodal classification of Alzheimer’s disease and mild cognitive impairment. NeuroImage 2011, 55, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Fodero-Tavoletti, M.T.; Masters, C.L.; Rowe, C.C. Tau imaging: Early progress and future directions. Lancet Neurol. 2015, 14, 114–124. [Google Scholar] [CrossRef]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Update on hypothetical model of Alzheimer’s disease biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef]
- Shah, Y.B.; Marsden, C.A. The application of functional magnetic resonance imaging to neuropharmacology. Curr. Opin. Pharmacol. 2004, 4, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, N.C.; Nopoulos, P.; Magnotta, V.; Pierson, R.; Ziebell, S.; Ho, B.-C. Progressive brain change in schizophrenia: A prospective longitudinal study of first-episode schizophrenia. Biol. Psychiatry 2011, 70, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Parsey, R.V. Serotonin receptor imaging: Clinically useful? J. Nucl. Med. 2010, 51, 1495–1498. [Google Scholar] [CrossRef] [PubMed]
- Hiemke, C. Therapeutic drug monitoring in neuropsychopharmacology: Does it hold its promises? Eur. Arch. Psychiatry Clin. Neurosci. 2008, 258 (Suppl. 1), S21–S27. [Google Scholar] [CrossRef] [PubMed]
- Dazzan, P.; Morgan, K.D.; Orr, K.; Hutchinson, G.; Chitnis, X.; Suckling, J.; Fearon, P.; McGuire, P.K.; Mallett, R.M.; Jones, P.B.; et al. Different effects of typical and atypical antipsychotics on grey matter in first episode psychosis: The aesop study. Neuropsychopharmacology 2005, 30, 765–774. [Google Scholar] [CrossRef]
- Crow, T.J. The emperors of the schizophrenia polygene have no clothes. Psychol. Med. 2008, 38, 1681–1685. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.S.; Scherer, S.W.; Brzustowicz, L.M. Copy number variations in schizophrenia: Critical review and new perspectives on concepts of genetics and disease. Am. J. Psychiatry 2010, 167, 899–914. [Google Scholar] [CrossRef] [PubMed]
- Kambeitz, J.P.; Howes, O.D. The serotonin transporter in depression: Meta-analysis of in vivo and post mortem findings and implications for understanding and treating depression. J. Affect. Disord. 2015, 186, 358–366. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burhan, A.M.; Marlatt, N.M.; Palaniyappan, L.; Anazodo, U.C.; Prato, F.S. Role of Hybrid Brain Imaging in Neuropsychiatric Disorders. Diagnostics 2015, 5, 577-614. https://doi.org/10.3390/diagnostics5040577
Burhan AM, Marlatt NM, Palaniyappan L, Anazodo UC, Prato FS. Role of Hybrid Brain Imaging in Neuropsychiatric Disorders. Diagnostics. 2015; 5(4):577-614. https://doi.org/10.3390/diagnostics5040577
Chicago/Turabian StyleBurhan, Amer M., Nicole M. Marlatt, Lena Palaniyappan, Udunna C. Anazodo, and Frank S. Prato. 2015. "Role of Hybrid Brain Imaging in Neuropsychiatric Disorders" Diagnostics 5, no. 4: 577-614. https://doi.org/10.3390/diagnostics5040577