

Congenital High Airway Obstructive Syndrome (CHAOS) Survival of a Newborn with Laryngeal Atresia

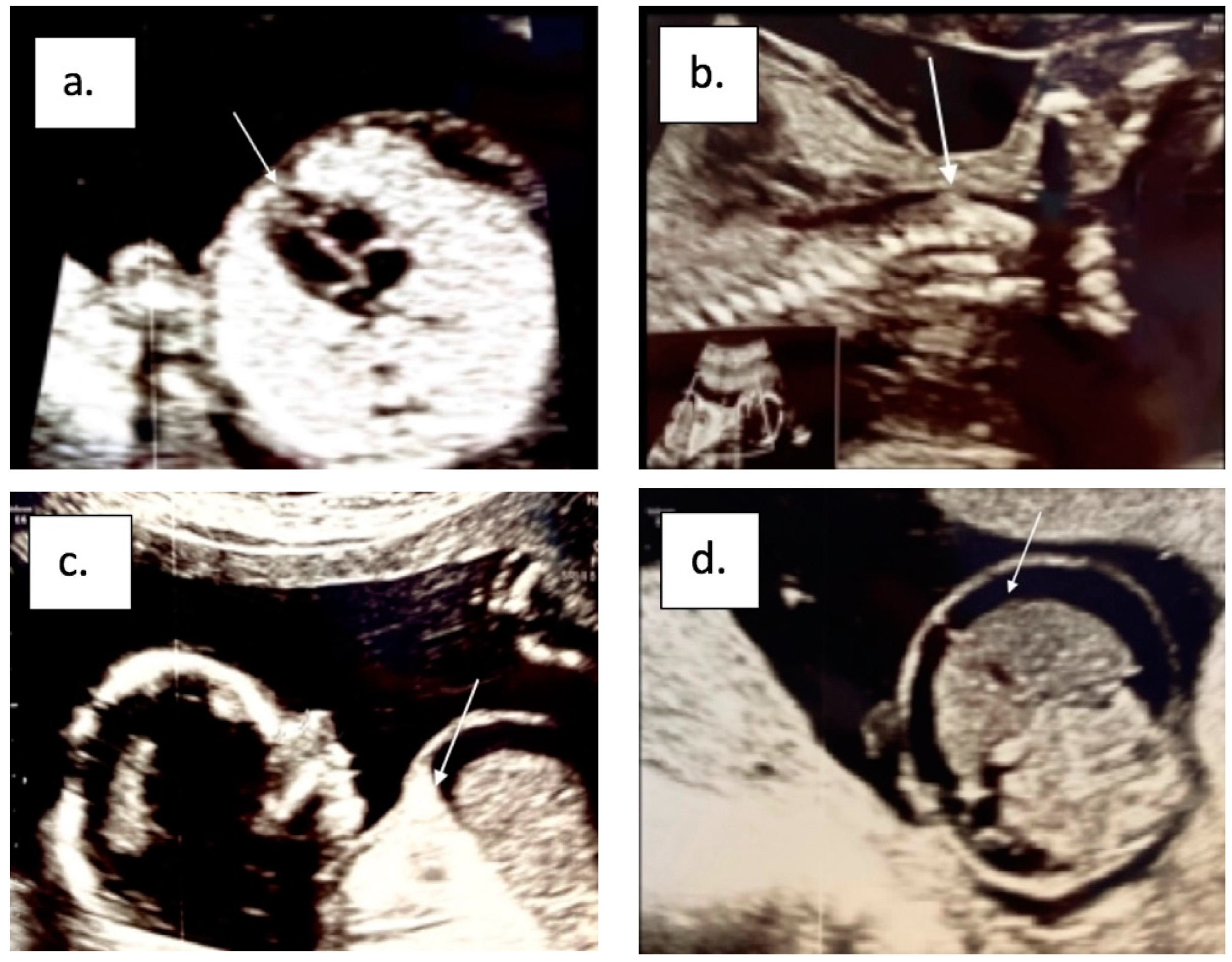{kind=link}
{kind=link}
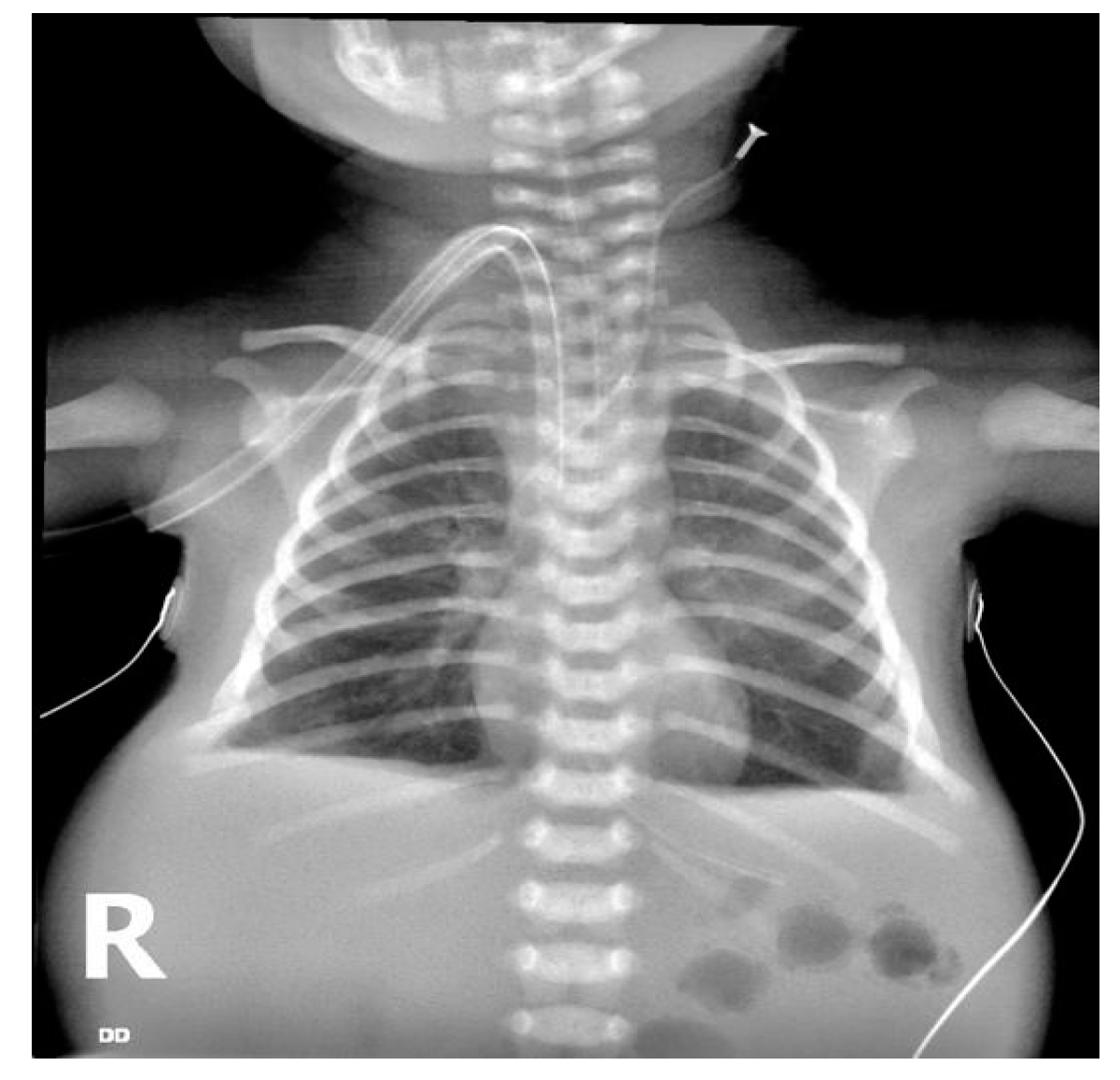{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
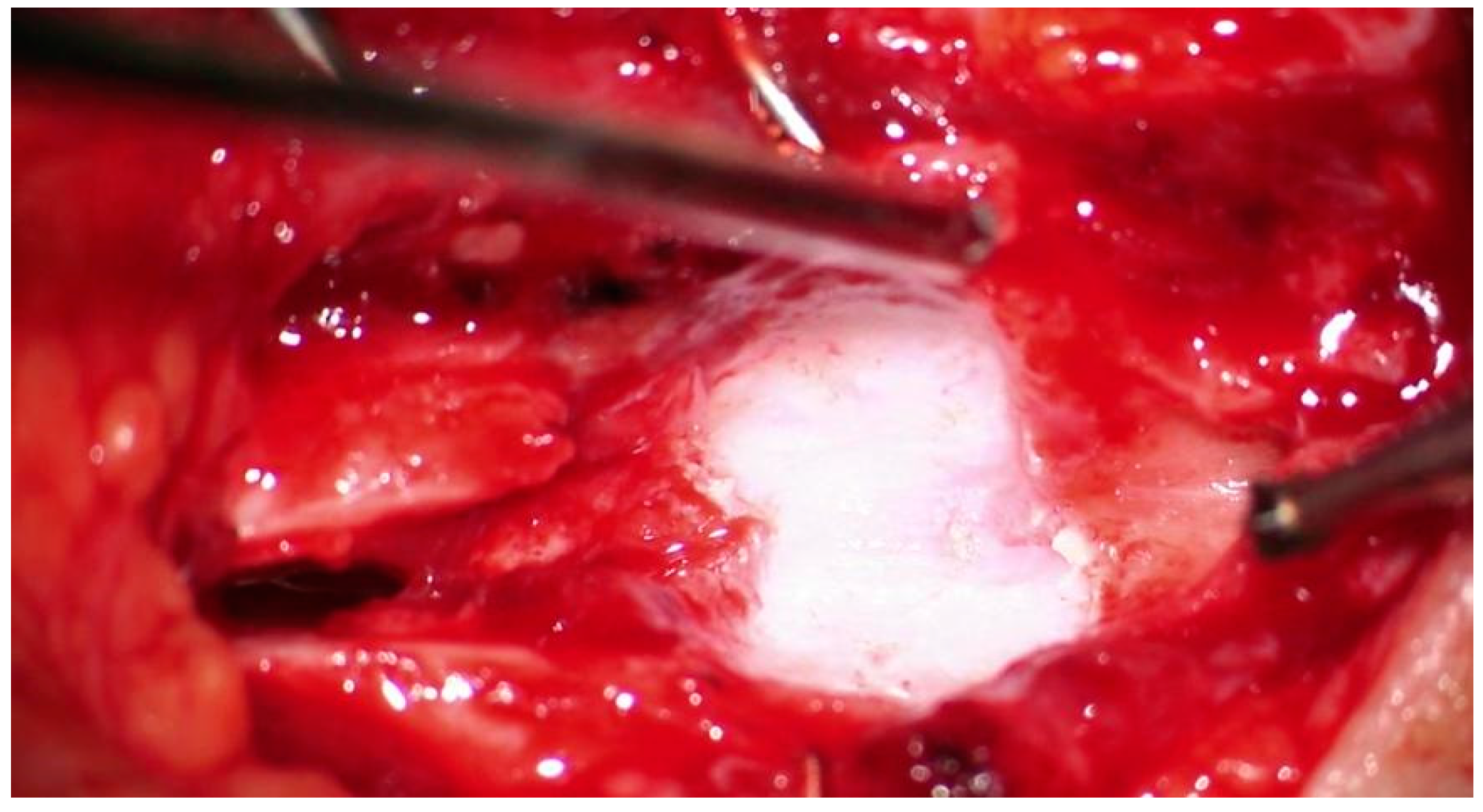{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aslan, H.; Ekiz, A.; Acar, D.K.; Aydiner, B.; Kaya, B.; Sezer, S. Prenatal diagnosis of congenital high airway obstruction syndrome (CHAOS). Five case report. Med. Ultrason. 2015, 17, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Roybal, J.L.; Liechty, K.W.; Hedrick, H.L.; Bebbington, M.W.; Johnson, M.P.; Coleman, B.G.; Adzick, N.S.; Flake, A.W. Predicting the severity of congenital high airway obstruction syndrome. J. Pediatr. Surg. 2010, 45, 1633–1639. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, L.; Güneş, I.; Halis, H.; Ketenci, İ.; Baştuğ, O.; Doğan, M.S.; Akın, M.A. A case of laryngeal atresia accompanied by persistent pharyngotracheal ductus. Turk. Arch. Pediatr. 2019, 54, 57–60. [Google Scholar] [CrossRef] [PubMed]
- de Groot-van der Mooren, M.D.; Haak, M.C.; Lakeman, P.; Cohen-Overbeek, T.E.; van der Voorn, J.P.; Bretschneider, J.H.; van Elburg, R.M. Tracheal agenesis: Approach towards this severe diagnosis. Case report and review of the literature. Eur. J. Pediatr. 2012, 171, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Lago Leal, V.; Cortés, L.M.; Seco Del Cacho, C. Prenatal diagnosis of congenital high airway obstruction syndrome. Indian J. Radiol. Imaging 2018, 28, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.K. Case report: Antenatal diagnosis of congenital high airway obstruction syndrome -laryngeal atresia. Indian J. Radiol. Imaging 2008, 18, 350–351. [Google Scholar] [CrossRef] [PubMed]
- Ryan, G.; Somme, S.; Crombleholme, T.M. Airway compromise in the fetus and neonate: Prenatal assessment and perinatal management. Semin. Fetal Neonatal Med. 2016, 21, 230–239. [Google Scholar] [CrossRef]
- Ravikanth, R.; Khanapure, V.P. Prenatal Diagnosis of Congenital High Airway Obstruction Syndrome due to Laryngeal Atresia. J. Med. Ultrasound 2021, 29, 298–299. [Google Scholar] [CrossRef]
- Gupta, A.; Yadav, C.; Dhruw, S.; Mishra, D.; Taori, A. CHAOS. J. Obstet. Gynecol. India 2016, 66, 202–208. [Google Scholar] [CrossRef]
- Joshi, P.; Satija, L.; George, R.; Chatterjee, S.; D’Souza, J.; Raheem, A. Congenital high airway obstruction syndrome-antenatal diagnosis of a rare case of airway obstruction using multimodality imaging. Med. J. Armed Forces India 2012, 68, 78–80. [Google Scholar] [CrossRef]
- Lim, F.Y.; Crombleholme, T.M.; Hedrick, H.L.; Flake, A.W.; Johnson, M.P.; Howell, L.J.; Adzick, N.S. Congenital high airway obstruction syndrome: Natural history and management. J. Pediatr. Surg. 2003, 38, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.; Cassey, J.; O’callaghan, S. Management of antenatally detected fetal airway obstruction. Int. J. Pediatr. Otorhinolaryngol. 2005, 69, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Nolan, H.R.; Gurria, J.; Peiro, J.L.; Tabbah, S.; Diaz-Primera, R.; Polzin, W.; Habli, M.; Lim, F.Y. Congenital high airway obstruction syndrome (CHAOS): Natural history, prenatal management strategies, and outcomes at a single comprehensive fetal center. J. Pediatr. Surg. 2019, 54, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Hartnick, C.J.; Rutter, M.; Lang, F.; Willging, J.P.; Cotton, R.T. Congenital high airway obstruction syndrome and airway reconstruction: An evolving paradigm. Arch. Otolaryngol. Head Neck Surg. 2002, 128, 567–570. [Google Scholar] [CrossRef]
- Smith, I.I.; Bain, A.D. Congenital atresia of the larynx. A report of nine cases. Ann. Otol. Rhinol. Laryngol. 1965, 74, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Hoka, S.; Sato, M.; Yoshitake, J.; Kukita, J. Management of a newborn infant with congenital laryngeal atresia. Obstet. Anesthesia Dig. 1989, 69, 535–536. [Google Scholar] [CrossRef]
- Miller, R.H.; Cagle, P.T.; Pitcock, J.K.; McGavran, M. Laryngeal atresia: A detailed histologic study. Int. J. Pediatr. Otorhinolaryngol. 1984, 7, 273–280. [Google Scholar] [CrossRef]
- Chaemsaithong, P.; Chansoon, T.; Chanrachakul, B.; Worawichawong, S.; Wongwaisayawan, S.; Promsonthi, P. Prenatal diagnosis and pathology of laryngeal atresia in congenital high airway obstruction syndrome. Case Rep. Radiol. 2012, 2012, 616905. [Google Scholar] [CrossRef]
- Onderoglu, L.; Saygan Karamürsel, B.; Bulun, A.; Kale, G.; Tunçbilek, E. Prenatal diagnosis of laryngeal atresia. Prenat. Diagn. 2003, 23, 277–280. [Google Scholar] [CrossRef]
- Meizner, I.; Sherizly, I.; Mashiach, R.; Shalev, J.; Kedron, D.; Ben-Rafael, Z. Prenatal sonographic diagnosis of laryngeal atresia in association with single umbilical artery. J. Clin. Ultrasound. 2000, 28, 435–438. [Google Scholar] [CrossRef]
- Kalache, K.D.; Chaoui, R.; Tennstedt, C.; Bollmann, R. Prenatal diagnosis of laryngeal atresia in two cases of congenital high airway obstruction syndrome (CHAOS). Prenat. Diagn. 1997, 17, 577–581. [Google Scholar] [CrossRef]
- Kanamori, Y.; Kitano, Y.; Hashizume, K.; Sugiyama, M.; Tomonaga, T.; Takayasu, H.; Egami, S.; Goishi, K.; Shibuya, K.; Kawana, Y.; et al. A case of laryngeal atresia (congenital high airway obstruction syndrome) with chromosome 5p deletion syndrome rescued by ex utero intrapartum treatment. J. Pediatr. Surg. 2004, 39, E25–E28. [Google Scholar] [CrossRef] [PubMed]
- Van den Boogaard, M.J.; De Pater, J.; Hennekam, R.C. A case with laryngeal atresia and partial trisomy 9 due to maternal 9;16 translocation. Genet. Couns. 1991, 2, 83–91. [Google Scholar] [PubMed]
- Fokstuen, S.; Bottani, A.; Medeiros, P.F.; Antonarakis, S.E.; Stoll, C.; Schinzel, A. Laryngeal atresia type III (glottic web) with 22q11.2 microdeletion: Report of three patients. Am. J. Med. Genet. 1997, 70, 130–133. [Google Scholar] [CrossRef]
- Kohl, T.; Hering, R.; Bauriedel, G.; Van de Vondel, P.; Heep, A.; Keiner, S.; Müller, A.; Franz, A.; Bartmann, P.; Gembruch, U. Fetoscopic and ultrasound-guided decompression of the fetal trachea in a human fetus with Fraser syndrome and congenital high airway obstruction syndrome (CHAOS) from laryngeal atresia. Ultrasound. Obstet. Gynecol. 2006, 27, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Kohl, T.; Van de Vondel, P.; Stressig, R.; Wartenberg, H.C.; Heep, A.; Keiner, S.; Müller, A.; Franz, A.; Fröhlich, S.; Willinek, W.; et al. Percutaneous fetoscopic laser decompression of congenital high airway obstruction syndrome (CHAOS) from laryngeal atresia via a single trocar--current technical constraints and potential solutions for future interventions. Fetal Diagn. Ther. 2009, 25, 67–71. [Google Scholar] [CrossRef]
- Bui, T.H.; Grunewald, C.; Frenckner, B.; Kuylenstierna, R.; Dahlgren, G.; Edner, A.; Granström, L.; Selldén, H. Successful EXIT (ex utero intrapartum treatment) procedure in a fetus diagnosed prenatally with congenital high-airway obstruction syndrome due to laryngeal atresia. Eur. J. Pediatr. Surg. 2000, 10, 328–333. [Google Scholar] [CrossRef]
- Oepkes, D.; Teunissen, A.K.; Van De Velde, M.; Devlieger, H.; Delaere, P.; Deprest, J. Congenital high airway obstruction syndrome successfully managed with ex-utero intrapartum treatment. Ultrasound Obstet. Gynecol. 2003, 22, 437–439. [Google Scholar] [CrossRef]
- van Veenendaal, M.B.; Liem, K.D.; Marres, H.A. Congenital absence of the trachea. Eur. J. Pediatr. 2000, 159, 8–13. [Google Scholar] [CrossRef]
- Zhang, P.; Herring, D.; Cook, L.; Mertz, H. Fetal laryngeal stenosis/atresia and congenital high airway obstructive syndrome (CHAOS): A case report. J. Perinatol. 2005, 25, 426–428. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heriseanu, C.; Bizubac, M.; Draghia, L.; Marcu, V.; Gheorghe, D.; Cirstoveanu, C. Congenital High Airway Obstructive Syndrome (CHAOS) Survival of a Newborn with Laryngeal Atresia. Diagnostics 2023, 13, 3658. https://doi.org/10.3390/diagnostics13243658
Heriseanu C, Bizubac M, Draghia L, Marcu V, Gheorghe D, Cirstoveanu C. Congenital High Airway Obstructive Syndrome (CHAOS) Survival of a Newborn with Laryngeal Atresia. Diagnostics. 2023; 13(24):3658. https://doi.org/10.3390/diagnostics13243658
Chicago/Turabian StyleHeriseanu, Carmen, Mihaela Bizubac, Loredana Draghia, Veronica Marcu, Dan Gheorghe, and Catalin Cirstoveanu. 2023. "Congenital High Airway Obstructive Syndrome (CHAOS) Survival of a Newborn with Laryngeal Atresia" Diagnostics 13, no. 24: 3658. https://doi.org/10.3390/diagnostics13243658