
IgA Nephropathy: Current Understanding and Perspectives on Pathogenesis and Targeted Treatment
 , and
, and
Abstract
:1. Introduction
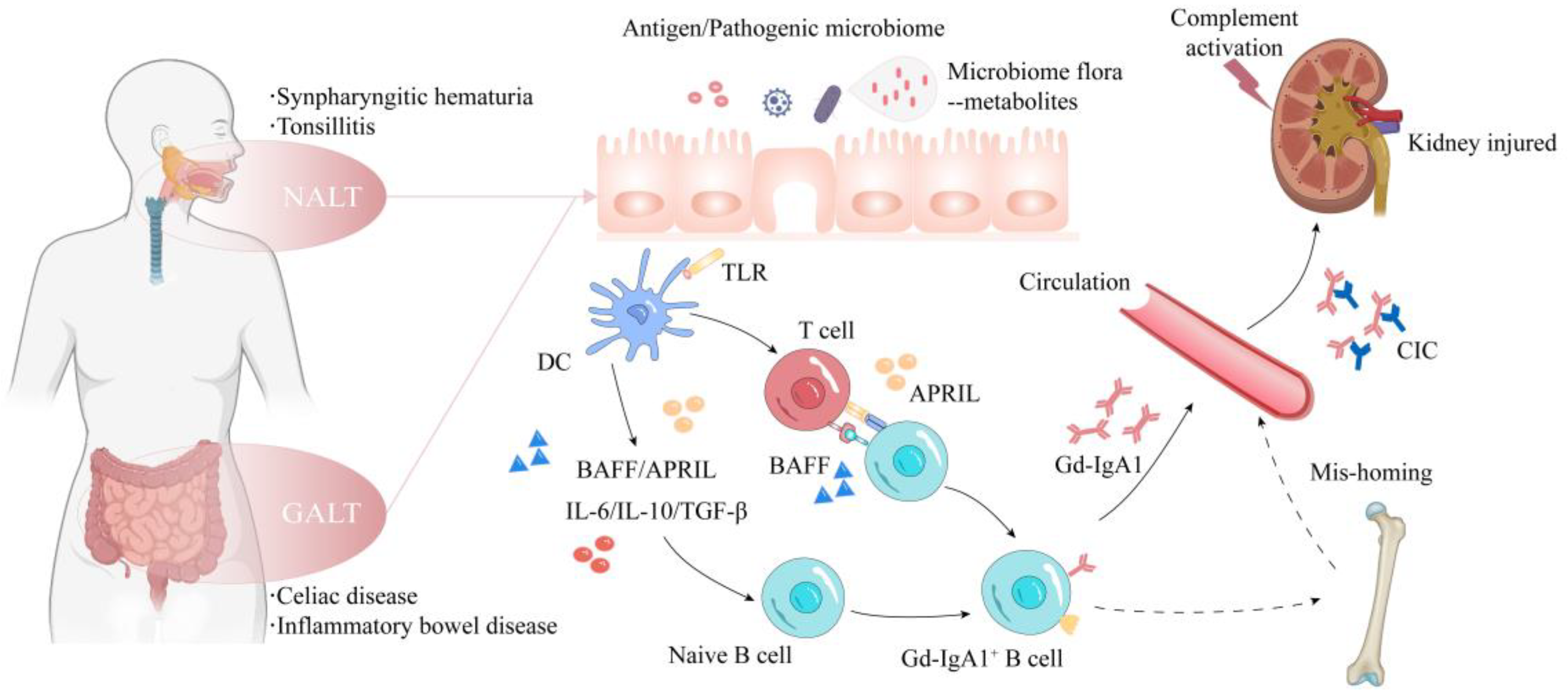
2. Mechanisms and Pathophysiology
2.1. Genetic Susceptibility
2.2. Mucosal Immunity
2.3. NALT
2.4. GALT

{kind=link}
| Mechanism | Subjects | Pathophysiologic Processes | Reference |
|---|---|---|---|
| Multi-hit Hypothesis | Gd-IgA1 | Formation of pathogenic circulating immune complexes | [13] |
| Anti-Gd-IgA1 antibodies | [14] | ||
| Soluble CD89 | [16] | ||
| Mucosal Immunity | CD40 | Promoting B-cell class switch | [10,25,26] |
| BAFF/APRIL | |||
| Transforming growth factor β, Interleukin 6, Interleukin 10 | |||
| TLRs | Activating the innate immune responses | ||
| Gut-derived uremic toxins (Indoxyl sulfate, p-cresyl sulfate, indole-3 acetic acid, trimethylamine N-oxide and phenylacetylglutamine) | Inducing intestinal inflammation and IgA overproduction | [50,51] | |
| Gut microbiota metabolites (p-tert-butyl-phenol, 4-(1,1,3,3-tetramethylbutyl) phenol, hexadecyl ester benzoic acid, methyl neopentyl phthalic acid and furanone A) | Increasing gut permeability and inducing mucosal hyper-responsivity | [40] | |
| Short-chain fatty acids | Maintaining gut barrier function and host immune system | [52,54] | |
| Bile acids | Regulation of intestinal immune responses | [53,54] | |
| Complement Pathways | C3 | Activation of alternative pathway | [60] |
| Alternative pathway proteins (Complement factor H-related protein 1 and 5) | Competitively inhibiting factor H and leading to overactivation of alternative pathway | [61,62] | |
| Lectin pathway proteins (MBL, L-ficolin, M-ficolin, MASP 1/3 and MAp19) | Promoting activation of lectin pathway | [63,64,65] |
2.5. Complement Dysregulation
3. Prediction and Prognosis of IgAN
| Subjects | Origin | Clinical Significance | Reference | |
|---|---|---|---|---|
| Tools | MEST-C score | Histopathology | Histopathological lesions associated with adverse outcomes | [5,70,71,72] |
| The International IgAN Prediction Tool | Clinical and histopathological parameters | Quantifying the risk of progression up to 6.7 years after biopsy | [73,74] | |
| Biomarkers | Proteinuria | Urine | Risk factor for disease progression of IgAN | [68] |
| Serum Gd-IgA1 | Serum | Independent risk factor for disease progression of IgAN | [79] | |
| Gd-IgA1-specific IgG and IgA autoantibodies | Serum | Association with the risk of disease progression | [80] | |
| Recombinant CD89-bound poly-IgA immune complex | Serum | Association with the severity of IgAN and with treatment response to steroids and immunosuppressants | [81] | |
| C4d | Histopathology | Association with clinical and histopathological severity of IgAN | [63,82] | |
| Laminin G-like 3 | Urine | Association with clinical severity and prognosis | [84] | |
| Free kappa light chains | Urine | |||
| Urinary Dickkopf-3 | Urine | Predicting kidney prognosis over the next 12 months | [85] | |
| miR-200 family, miR-205, miR-192 | Urine | Association with disease severity and rate of progression | [86] | |
| miR-148b | Peripheral blood mononuclear cell | The level of miR-148b positively correlated with serum level of Gd-IgA1 in IgAN patients | [87] | |
| miR-374b | Blood B-cell | The level of miR-374b in B-cells was positively related to urine protein level and pathological MEST score | [88] |
4. Current Treatment of IgAN
4.1. Supportive Therapy
4.2. Immunosuppression Therapy
4.3. Outlook of Future Alternatives to Conventional Immunosuppression
4.3.1. Regulation of Pathogenic IgA1 and CIC Production
- Inhibition of TLRs/BAFF/APRIL signaling
- Depletion of Gd-IgA1-producing plasma cells
4.3.2. Clearance of IgA Deposits
4.3.3. Modulation of Mucosal Immunity
- Modulation of NALT
- Modulation of GALT and Gut microbiota
- Gut-targeted immunosuppressionEvidence suggests that the GALT, including Peyer’s patches, may play a critical role as a potential source of Gd-IgA1 in IgAN, where antigens, microbes or products of gut microbial activity initiate mucosal pathogenic IgA synthesis [130]. Hence, immunosuppression targeted to dysregulated GALT immune responses may provide an alternative to conventional systemic immunosuppression. Tarpeyo is a distal ileum targeted-release budesonide, which has been shown to reduce the level of CICs in a dose-dependent manner [131]. Recently, tarpeyo was granted accelerated approval by the FDA since two clinical trials (NEFIGAN, NCT01738035; NefIgArd Part A, NCT03643965) preliminarily demonstrated its efficacy in reducing proteinuria and stabilizing kidney function despite optimized RAS inhibition [132]. Zhang et al. developed a novel ileocecum targeting medication based on an orange-derived and dexamethasone-encapsulated extracellular vesicle (EVs-DexP) delivery system [133]. Evs-DexP exerted its effects by reducing intestinal IgA production and kidney IgA deposition in IgAN mice. It can also suppress lymphocyte activation in vitro while decreasing the ratio of IgA+B220+ lymphocytes in Peyer’s patches.
- Gut microbiota modulationSeveral studies indicated that dysbiosis of the gut microbiota might be associated with the progression, clinical features and treatment responses of IgAN [134,135,136]. Coupled with the pathogenesis of IgAN, approaches focused on the restoration of intestinal flora homeostasis, such as regulation of microbial diversity and metabolites, would be promising adjuvant therapeutic options against IgAN. Broad-spectrum antibiotics exhibited therapeutic effects on modulating microbiota, resulting in reduced IgA1-related CICs and mesangial IgA1 deposition in humanized mouse models of IgAN [137,138]. Another potential option is regulating gut immunity by the supplementation of probiotics/prebiotics/specific microbial metabolites or by the transplantation of fecal microbiota [139,140,141]. Some of these strategies have been shown to improve pathophysiological and clinical parameters in IgAN patients.
4.3.4. Blockade of Complement Cascades
| Agent | Mechanism of Action | Registration No. | Phase | Primary Outcome Measures | Trial Results/Status | Reference |
|---|---|---|---|---|---|---|
| Hydroxychloroquine | TLR signaling inhibitor | NCT02942381 | 2 | Proteinuria (every 2 months, total 6 months) | In addition to optimized renin-angiotensin–aldosterone system inhibition, hydroxychloroquine significantly reduced proteinuria without evidence of adverse event | [102] |
| Blisibimod | Monoclonal antibody of soluble and membrane BAFF | NCT02062684 | 2/3 | Proportion of subjects achieving reduction in proteinuria from baseline (24 weeks) | The interim results of blisibimod treatment showed a reduction in the level of proteinuria, peripheral B-cells and immunoglobulins | - |
| VIS649 | Monoclonal IgG2κ antibody targeting APRIL | NCT03719443 | 1 | Number of participants with adverse events and frequency of ECG abnormalities (112 days) | VIS649 treatment reduced serum levels of APRIL, IgA and Gd-IgA1 without evidence of severe adverse event | [109,110] |
| BION-1301 | Monoclonal IgG4 antibody targeting APRIL | NCT03945318 | 1/2 | Incidence and severity of adverse events (76 weeks) | Recruiting | - |
| Atacicept | BAFF/APRIL dual inhibitor | NCT02808429 | 2 | Percentage of adverse events (96 weeks) | 1. Atacicept treatment demonstrated an acceptable safety profile 2. The interim results of atacicept treatment showed early reduction in proteinuria and dose-dependent reduction in Gd-IgA1 | [111] |
| NCT04716231 | 2 | Change from baseline in UPCR (24 weeks) | Active, not recruiting | - | ||
| Telitacicept | BAFF/APRIL dual inhibitor | NCT04291781 | 2 | Change from baseline in 24-h urine protein excretion level (24 weeks) | Results not yet available | - |
| NCT04905212 | 2 | Recruiting | - | |||
| Rituximab | Monoclonal anti-CD20 antibody | NCT00498368 | 4 | Change in proteinuria (12 months) | Rituximab treatment did not significantly improve kidney function or proteinuria and failed to reduce serum levels of Gd-IgA1 and anti-Gd-IgA1 antibodies | [113] |
| Felzartamab | Monoclonal IgG1 antibody targeting CD38 | NCT05065970 | 2 | Relative change in proteinuria value (9 months) | Recruiting | - |
| Fostamatinib | Spleen tyrosine kinase inhibitor | NCT02112838 | 2 | Mean change from baseline in proteinuria (24 weeks) | Fostamatinib treatment did not significantly improve proteinuria or eGFR | [121] |
| NEFECON (TARPEYO) | Distal ileum targeted-release budesonide formulation targeting B-cells in mucosal lymphoid tissue | NCT01738035 | 2 | Change from baseline in UPCR (9 months) | Nefecon treatment reduced proteinuria and preserved kidney function | [132] |
| NCT03643965 | 3 | Change in proteinuria (9 months) and eGFR (up to 2 years) | [131] | |||
| CCX168 | Anti-C5a receptor antagonist | NCT02384317 | 2 | The number of patients with adverse events (169 days) | CCX168 treatment improved proteinuria | [142] |
| ALXN1210 | Long-acting C5-blocking antibody | NCT04564339 | 2 | Percentage change from baseline in proteinuria (26 weeks) | Recruiting | - |
| ALN-CC5 | Small interfering RNA targeting C5 | NCT03841448 | 2 | Percentage change from baseline in UPCR (32 weeks) | Active, not recruiting | - |
| APL-2 | Cyclic peptide inhibitor of C3 | NCT03453619 | 2 | Proteinuria (48 weeks) | Active, not recruiting | - |
| LNP023 | Selective complement factor B inhibitor | NCT03373461 | 2 | Multiple comparison procedure modeling estimates of the ratio to baseline of UPCR (90 days) | LNP023 treatment led to continuous reduction in proteinuria and strong inhibition of alternative pathway activity | [143] |
| NCT04578834 | 3 | Ratio to baseline in UPCR (9 months) and annualized total eGFR slope (24 months) | Recruiting | - | ||
| IONIS-FB-LRx | Antisense inhibitor of complement factor B | NCT04014335 | 2 | Percent reduction in 24-h urine protein excretion (29 weeks) | Recruiting | - |
| OMS721 | Monoclonal antibody against mannan-associated lectin-binding serine protease-2 | NCT02682407 | 2 | Proportion of adverse events (Up to 104 weeks) and change from baseline in serum and urine complement component levels (38 weeks) | OMS721 treatment reduced proteinuria and preserved kidney function | [144] |
| NCT03608033 | 3 | Change from baseline in 24-h urine protein excretion (36 weeks) | Recruiting | - |
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lai, K.N.; Tang, S.C.; Schena, F.P.; Novak, J.; Tomino, Y.; Fogo, A.B.; Glassock, R.J. IgA nephropathy. Nat. Rev. Dis. Prim. 2016, 2, 16001. [Google Scholar] [CrossRef]
- Rauen, T.; Wied, S.; Fitzner, C.; Eitner, F.; Sommerer, C.; Zeier, M.; Otte, B.; Panzer, U.; Budde, K.; Benck, U.; et al. After ten years of follow-up, no difference between supportive care plus immunosuppression and supportive care alone in IgA nephropathy. Kidney Int. 2020, 98, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.C.; Cheung, C.K.; Barratt, J. New insights into the pathogenesis of IgA nephropathy. Pediatr. Nephrol. 2017, 33, 763–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, G. Natural history of idiopathic IgA nephropathy and factors predictive of disease outcome. Semin. Nephrol. 2004, 24, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Honda, K.; Tanabe, K.; Toma, H.; Nihei, H.; Yamaguchi, Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int. 2003, 63, 2286–2294. [Google Scholar] [CrossRef] [Green Version]
- Gharavi, A.G.; Moldoveanu, Z.; Wyatt, R.; Barker, C.V.; Woodford, S.Y.; Lifton, R.P.; Mestecky, J.; Novak, J.; Julian, B.A. Aberrant IgA1 Glycosylation Is Inherited in Familial and Sporadic IgA Nephropathy. J. Am. Soc. Nephrol. 2008, 19, 1008–1014. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, S.; Imamura, R.; Kawamura, M.; Kato, T.; Abe, T.; Namba, T.; Iwatani, H.; Yamanaka, K.; Uemura, M.; Kishikawa, H.; et al. Difference in IgA1 O-glycosylation between IgA deposition donors and IgA nephropathy recipients. Biochem. Biophys. Res. Commun. 2018, 508, 1106–1112. [Google Scholar] [CrossRef]
- Gaber, L.W.; Khan, F.N.; Graviss, E.A.; Nguyen, D.T.; Moore, L.W.; Truong, L.D.; Barrios, R.J.; Suki, W.N. Prevalence, Characteristics, and Outcomes of Incidental IgA Glomerular Deposits in Donor Kidneys. Kidney Int. Rep. 2020, 5, 1914–1924. [Google Scholar] [CrossRef]
- Gesualdo, L.; Di Leo, V.; Coppo, R. The mucosal immune system and IgA nephropathy. Semin. Immunopathol. 2021, 43, 657–668. [Google Scholar] [CrossRef]
- He, J.-W.; Zhou, X.-J.; Lv, J.-C.; Zhang, H. Perspectives on how mucosal immune responses, infections and gut microbiome shape IgA nephropathy and future therapies. Theranostics 2020, 10, 11462–11478. [Google Scholar] [CrossRef]
- Medjeral-Thomas, N.R.; Cook, H.T.; Pickering, M.C. Complement activation in IgA nephropathy. Semin. Immunopathol. 2021, 43, 679–690. [Google Scholar] [CrossRef]
- Schena, F.P.; Nistor, I. Epidemiology of IgA Nephropathy: A Global Perspective. Semin. Nephrol. 2018, 38, 435–442. [Google Scholar] [CrossRef]
- Suzuki, H.; Kiryluk, K.; Novak, J.; Moldoveanu, Z.; Herr, A.B.; Renfrow, M.B.; Wyatt, R.J.; Scolari, F.; Mestecky, J.; Gharavi, A.G.; et al. The Pathophysiology of IgA Nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H. Biomarkers for IgA nephropathy on the basis of multi-hit pathogenesis. Clin. Exp. Nephrol. 2018, 23, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Gao, L.; Liu, P.; Lv, J.; Lu, W.-H.; Zhang, H.; Jin, J. Propensity of IgA to self-aggregate via tailpiece cysteine-471 and treatment of IgA nephropathy using cysteamine. J. Clin. Investig. 2021, 6, e150551. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.C. Recent advances in the physiopathology of IgA nephropathy. Néphrol. Thér. 2018, 14 (Suppl. S1), S1–S8. [Google Scholar] [CrossRef]
- Kiryluk, K.; Li, Y.; Sanna-Cherchi, S.; Rohanizadegan, M.; Suzuki, H.; Eitner, F.; Snyder, H.J.; Choi, M.; Hou, P.; Scolari, F.; et al. Geographic Differences in Genetic Susceptibility to IgA Nephropathy: GWAS Replication Study and Geospatial Risk Analysis. PLOS Genet. 2012, 8, e1002765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Mei, Y.; Mao, Z.; Long, L.; Han, Q.; You, Y.; Zhu, H. Association of Immune and Inflammatory Gene Polymorphism With the Risk of IgA Nephropathy: A Systematic Review and Meta-Analysis of 45 Studies. Front. Immunol. 2021, 12, 683913. [Google Scholar] [CrossRef]
- Zhou, X.-J.; Tsoi, L.C.; Hu, Y.; Patrick, M.T.; He, K.; Berthier, C.C.; Li, Y.; Wang, Y.-N.; Qi, Y.-Y.; Zhang, Y.-M.; et al. Exome Chip Analyses and Genetic Risk for IgA Nephropathy among Han Chinese. Clin. J. Am. Soc. Nephrol. 2021, 16, 213–224. [Google Scholar] [CrossRef]
- Knoppova, B.; Reily, C.; Maillard, N.; Rizk, D.V.; Moldoveanu, Z.; Mestecky, J.; Raska, M.; Renfrow, M.B.; Julian, B.A.; Novak, J. The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy. Front. Immunol. 2016, 7, 117. [Google Scholar] [CrossRef]
- Gormley, P.D.; Powell-Richards, A.O.; Azuara-Blanco, A.; Donoso, L.A.; Dua, H.S. Lymphocyte subsets in conjunctival mucosa-associated-lymphoid-tissue after exposure to retinal-S-antigen. Int. Ophthalmol. 1998, 22, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Kiyono, H.; Fukuyama, S. NALT-versus Peyer’s-patch-mediated mucosal immunity. Nat. Rev. Immunol. 2004, 4, 699–710. [Google Scholar] [CrossRef]
- Kano, T.; Suzuki, H.; Makita, Y.; Fukao, Y.; Suzuki, Y. Nasal-associated lymphoid tissue is the major induction site for nephritogenic IgA in murine IgA nephropathy. Kidney Int. 2021, 100, 364–376. [Google Scholar] [CrossRef]
- Barratt, J.; Rovin, B.H.; Cattran, D.; Floege, J.; Lafayette, R.; Tesar, V.; Trimarchi, H.; Zhang, H.; NefIgArd Study Steering Committee. Why Target the Gut to Treat IgA Nephropathy? Kidney Int. Rep. 2020, 5, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, A. The regulation of IgA class switching. Nat. Rev. Immunol. 2008, 8, 421–434. [Google Scholar] [CrossRef]
- Nakawesi, J.; This, S.; Hütter, J.; Boucard-Jourdin, M.; Barateau, V.; Muleta, K.G.; Gooday, L.J.; Thomsen, K.F.; López, A.G.; Ulmert, I.; et al. αvβ8 integrin-expression by BATF3-dependent dendritic cells facilitates early IgA responses to Rotavirus. Mucosal Immunol. 2020, 14, 53–67. [Google Scholar] [CrossRef]
- Suzuki, H.; Suzuki, Y.; Narita, I.; Aizawa, M.; Kihara, M.; Yamanaka, T.; Kanou, T.; Tsukaguchi, H.; Novak, J.; Horikoshi, S.; et al. Toll-Like Receptor 9 Affects Severity of IgA Nephropathy. J. Am. Soc. Nephrol. 2008, 19, 2384–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, A.; Kraft, M.; Roth, K.; Riedel, R.; Lammerding, D.; E Hauser, A. Long-lived plasma cells are generated in mucosal immune responses and contribute to the bone marrow plasma cell pool in mice. Mucosal Immunol. 2016, 9, 83–97. [Google Scholar] [CrossRef] [Green Version]
- Coppo, R. Treatment of IgA nephropathy: Recent advances and prospects. Néphrol. Thér. 2018, 14 (Suppl. 1), S13–S21. [Google Scholar] [CrossRef]
- Inoue, T.; Sugiyama, H.; Kitagawa, M.; Takiue, K.; Morinaga, H.; Kikumoto, Y.; Maeshima, Y.; Fukushima, K.; Nishizaki, K.; Akagi, H.; et al. Abnormalities of Glycogenes in Tonsillar Lymphocytes in IgA Nephropathy. Adv. Otorhinolaryngol. 2011, 72, 71–74. [Google Scholar] [CrossRef]
- Suzuki, Y.; Suzuki, H.; Nakata, J.; Sato, D.; Kajiyama, T.; Watanabe, T.; Tomino, Y. Pathological Role of Tonsillar B Cells in IgA Nephropathy. J. Immunol. Res. 2011, 2011, 639074. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, T.; Yoshimura, M.; Miyazaki, Y.; Okamoto, H.; Kimura, K.; Hirano, K.; Matsushima, M.; Utsunomiya, Y.; Ogura, M.; Yokoo, T.; et al. A multicenter randomized controlled trial of tonsillectomy combined with steroid pulse therapy in patients with immunoglobulin A nephropathy. Nephrol. Dial. Transplant. 2014, 29, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, M.; Yamamoto, I.; Yamakawa, T.; Katsumata, H.; Isaka, N.; Katsuma, A.; Nakada, Y.; Kobayashi, A.; Koike, K.; Ueda, H.; et al. Association Between Galactose-Deficient IgA1 Derived From the Tonsils and Recurrence of IgA Nephropathy in Patients Who Underwent Kidney Transplantation. Front. Immunol. 2020, 11, 2068. [Google Scholar] [CrossRef] [PubMed]
- Feehally, J.; Coppo, R.; Troyanov, S.; Bellur, S.S.; Cattran, D.; Cook, T.; Roberts, I.S.; Verhave, J.C.; Camilla, R.; Vergano, L.; et al. Tonsillectomy in a European Cohort of 1,147 Patients with IgA Nephropathy. Nephron 2015, 132, 15–24. [Google Scholar] [CrossRef]
- Zachova, K.; Jemelkova, J.; Kosztyu, P.; Ohyama, Y.; Takahashi, K.; Zadrazil, J.; Orsag, J.; Matousovic, K.; Galuszkova, D.; Petejova, N.; et al. Galactose-Deficient IgA1 B cells in the Circulation of IgA Nephropathy Patients Carry Preferentially Lambda Light Chains and Mucosal Homing Receptors. J. Am. Soc. Nephrol. 2022, 33, 908–917. [Google Scholar] [CrossRef]
- Park, J.I.; Kim, T.-Y.; Oh, B.; Cho, H.; Kim, J.E.; Yoo, S.H.; Lee, J.P.; Kim, Y.S.; Chun, J.; Kim, B.-S.; et al. Comparative analysis of the tonsillar microbiota in IgA nephropathy and other glomerular diseases. Sci. Rep. 2020, 10, 16206. [Google Scholar] [CrossRef]
- Fujieda, S.; Suzuki, S.; Sunaga, H.; Yamamoto, H.; Seki, M.; Sugimoto, H.; Saito, H. Induction of IgA against Haemophilus parainfluenzae antigens in tonsillar mononuclear cells from patients with IgA nephropathy. Clin. Immunol. 2000, 95, 235–243. [Google Scholar] [CrossRef]
- Ito, S.; Misaki, T.; Naka, S.; Wato, K.; Nagasawa, Y.; Nomura, R.; Otsugu, M.; Matsumoto-Nakano, M.; Nakano, K.; Kumagai, H.; et al. Specific strains of Streptococcus mutans, a pathogen of dental caries, in the tonsils, are associated with IgA nephropathy. Sci. Rep. 2019, 9, 20130. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Goto, S.; Mori, H.; Higashi, K.; Hosomichi, K.; Aizawa, N.; Takahashi, N.; Tsuchida, M.; Suzuki, Y.; Yamada, T.; et al. Comprehensive microbiome analysis of tonsillar crypts in IgA nephropathy. Nephrol. Dial. Transplant. 2016, 32, 2072–2079. [Google Scholar] [CrossRef]
- Nyangale, E.P.; Mottram, D.S.; Gibson, G.R. Gut Microbial Activity, Implications for Health and Disease: The Potential Role of Metabolite Analysis. J. Proteome Res. 2012, 11, 5573–5585. [Google Scholar] [CrossRef]
- Coppo, R. The intestine-renal connection in IgA nephropathy. Nephrol. Dial. Transplant. 2014, 30, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, R.C.; Rafeh, D.; Gleeson, P.J. Is There a Role for Gut Microbiome Dysbiosis in IgA Nephropathy? Microorganisms 2022, 10, 683. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.; Montemurno, E.; Piccolo, M.; Vannini, L.; Lauriero, G.; Maranzano, V.; Gozzi, G.; Serrazanetti, D.; Dalfino, G.; Gobbetti, M.; et al. Microbiota and metabolome associated with immunoglobulin A nephropathy (IgAN). PLoS ONE 2014, 9, e99006. [Google Scholar] [CrossRef] [Green Version]
- Dong, R.; Bai, M.; Zhao, J.; Wang, D.; Ning, X.; Sun, S. A Comparative Study of the Gut Microbiota Associated With Immunoglobulin a Nephropathy and Membranous Nephropathy. Front. Cell. Infect. Microbiol. 2020, 10, 557368. [Google Scholar] [CrossRef]
- Satoh-Takayama, N.; Kato, T.; Motomura, Y.; Kageyama, T.; Taguchi-Atarashi, N.; Kinoshita-Daitoku, R.; Kuroda, E.; Di Santo, J.P.; Mimuro, H.; Moro, K.; et al. Bacteria-Induced Group 2 Innate Lymphoid Cells in the Stomach Provide Immune Protection through Induction of IgA. Immunity 2020, 52, 635–649.e4. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, M.; Cerdà-Cuéllar, M.; Martínez, V. Antibiotic-induced dysbiosis alters host-bacterial interactions and leads to colonic sensory and motor changes in mice. Gut Microbes 2015, 6, 10–23. [Google Scholar] [CrossRef] [Green Version]
- Obata, T.; Goto, Y.; Kunisawa, J.; Sato, S.; Sakamoto, M.; Setoyama, H.; Matsuki, T.; Nonaka, K.; Shibata, N.; Gohda, M.; et al. Indigenous opportunistic bacteria inhabit mammalian gut-associated lymphoid tissues and share a mucosal antibody-mediated symbiosis. Proc. Natl. Acad. Sci. USA 2010, 107, 7419–7424. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Mogno, I.; Contijoch, E.J.; Borgerding, J.N.; Aggarwala, V.; Li, Z.; Siu, S.; Grasset, E.K.; Helmus, D.S.; Dubinsky, M.C.; et al. Fecal IgA Levels Are Determined by Strain-Level Differences in Bacteroides ovatus and Are Modifiable by Gut Microbiota Manipulation. Cell Host Microbe 2020, 27, 467–475.e6. [Google Scholar] [CrossRef] [Green Version]
- Yiu, J.H.C.; Dorweiler, B.; Woo, C.W. Interaction between gut microbiota and toll-like receptor: From immunity to metabolism. J. Mol. Med. 2016, 95, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-Y.; Chen, D.-Q.; Chen, L.; Liu, J.-R.; Vaziri, N.D.; Guo, Y.; Zhao, Y.-Y. Microbiome–metabolome reveals the contribution of gut–kidney axis on kidney disease. J. Transl. Med. 2019, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhou, J.; Wang, S.; Xiong, J.; Chen, Y.; Liu, Y.; Xiao, T.; Li, Y.; He, T.; Li, Y.; et al. Indoxyl sulfate induces intestinal barrier injury through IRF1-DRP1 axis-mediated mitophagy impairment. Theranostics 2020, 10, 7384–7400. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Savoj, J.; Nakata, M.B.; Vaziri, N.D. Altered microbiome in chronic kidney disease: Systemic effects of gut-derived uremic toxins. Clin. Sci. 2018, 132, 509–522. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Pearson, J.A.; Peng, J.; Hu, Y.; Sha, S.; Xing, Y.; Huang, G.; Li, X.; Hu, F.; Xie, Z.; et al. Gut microbial metabolites alter IgA immunity in type 1 diabetes. J. Clin. Investig. 2020, 5, e135718. [Google Scholar] [CrossRef] [PubMed]
- Luck, H.; Khan, S.; Kim, J.H.; Copeland, J.K.; Revelo, X.S.; Tsai, S.; Chakraborty, M.; Cheng, K.; Chan, Y.T.; Nøhr, M.K.; et al. Gut-associated IgA+ immune cells regulate obesity-related insulin resistance. Nat. Commun. 2019, 10, 3650. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.A.; Velazquez, K.T.; Herbert, K.M. Influence of high-fat diet on gut microbiota: A driving force for chronic disease risk. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 515–520. [Google Scholar] [CrossRef]
- Papista, C.; Lechner, S.; Ben Mkaddem, S.; LeStang, M.-B.; Abbad, L.; Bex-Coudrat, J.; Pillebout, E.; Chemouny, J.M.; Jablonski, M.; Flamant, M.; et al. Gluten exacerbates IgA nephropathy in humanized mice through gliadin–CD89 interaction. Kidney Int. 2015, 88, 276–285. [Google Scholar] [CrossRef]
- Coppo, R.; Roccatello, D.; Amore, A.; Quattrocchio, G.; Molino, A.; Gianoglio, B.; Amoroso, A.; Bajardi, P.; Piccoli, G. Effects of a gluten-free diet in primary IgA nephropathy. Clin. Nephrol. 1990, 33, 72–86. [Google Scholar]
- Yap, H.K.; Sakai, R.S.; Woo, K.T.; Lim, C.H.; Jordan, S.C. Detection of bovine serum albumin in the circulating IgA immune complexes of patients with IgA nephropathy. Clin. Immunol. Immunopathol. 1987, 43, 395–402. [Google Scholar] [CrossRef]
- Barbour, T.D.; Pickering, M.C.; Cook, H.T. Recent insights into C3 glomerulopathy. Nephrol. Dial. Transplant. 2013, 28, 1685–1693. [Google Scholar] [CrossRef] [Green Version]
- Medjeral-Thomas, N.R.; Lomax-Browne, H.J.; Beckwith, H.; Willicombe, M.; McLean, A.G.; Brookes, P.; Pusey, C.D.; Falchi, M.; Cook, H.T.; Pickering, M.C. Circulating complement factor H–related proteins 1 and 5 correlate with disease activity in IgA nephropathy. Kidney Int. 2017, 92, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Tortajada, A.; Gutiérrez, E.; de Jorge, E.G.; Anter, J.; Segarra, A.; Espinosa, M.; Blasco, M.; Roman, E.; Marco, H.; Quintana, L.F.; et al. Elevated factor H–related protein 1 and factor H pathogenic variants decrease complement regulation in IgA nephropathy. Kidney Int. 2017, 92, 953–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa, M.; Ortega, R.; Sánchez, M.; Segarra, A.; Salcedo, M.T.; González, F.; Camacho, R.; Valdivia, M.A.; Cabrera, R.; López, K.; et al. Association of C4d Deposition with Clinical Outcomes in IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2014, 9, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Roos, A.; Rastaldi, M.P.; Calvaresi, N.; Oortwijn, B.D.; Schlagwein, N.; Van Gijlswijk-Janssen, D.J.; Stahl, G.; Matsushita, M.; Fujita, T.; van Kooten, C.; et al. Glomerular Activation of the Lectin Pathway of Complement in IgA Nephropathy Is Associated with More Severe Renal Disease. J. Am. Soc. Nephrol. 2006, 17, 1724–1734. [Google Scholar] [CrossRef] [Green Version]
- Medjeral-Thomas, N.R.; Troldborg, A.; Constantinou, N.; Lomax-Browne, H.J.; Hansen, A.G.; Willicombe, M.; Pusey, C.D.; Cook, H.T.; Thiel, S.; Pickering, M.C. Progressive IgA Nephropathy Is Associated With Low Circulating Mannan-Binding Lectin–Associated Serine Protease-3 (MASP-3) and Increased Glomerular Factor H–Related Protein-5 (FHR5) Deposition. Kidney Int. Rep. 2017, 3, 426–438. [Google Scholar] [CrossRef] [Green Version]
- Stangou, M.; Alexopoulos, E.; Pantzaki, A.; Leonstini, M.; Memmos, D. C5b-9 glomerular deposition and tubular alpha3beta1-integrin expression are implicated in the development of chronic lesions and predict renal function outcome in immunoglobulin A nephropathy. Scand. J. Urol. Nephrol. 2008, 42, 373–380. [Google Scholar] [CrossRef]
- Ouyang, Y.; Zhu, L.; Shi, M.; Yu, S.; Jin, Y.; Wang, Z.; Ma, J.; Yang, M.; Zhang, X.; Pan, X.; et al. A Rare Genetic Defect of MBL2 Increased the Risk for Progression of IgA Nephropathy. Front. Immunol. 2019, 10, 537. [Google Scholar] [CrossRef]
- Barbour, S.J.; Reich, H.N. Risk Stratification of Patients With IgA Nephropathy. Am. J. Kidney Dis. 2012, 59, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Cattran, D.C.; Feehally, J.; Cook, H.T.; Liu, Z.H.; Fervenza, F.C.; Mezzano, S.A.; Floege, J.; Nachman, P.H.; Gipson, D.S.; Praga, M.; et al. Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int. 2021, 100, S1–S276. [Google Scholar]
- Roberts, I.S.; A Working Group of the International IgA Nephropathy Network and the Renal Pathology Society; Cook, H.T.; Troyanov, S.; Alpers, C.E.; Amore, A.; Barratt, J.; Berthoux, F.; Bonsib, S.; Bruijn, J.A.; et al. The Oxford classification of IgA nephropathy: Pathology definitions, correlations, and reproducibility. Kidney Int. 2009, 76, 546–556. [Google Scholar] [CrossRef] [Green Version]
- A Working Group of the International IgA Nephropathy Network and the Renal Pathology Society; Cattran, D.C.; Coppo, R.; Cook, H.T.; Feehally, J.; Roberts, I.S.; Troyanov, S.; Alpers, C.E.; Amore, A.; Barratt, J.; et al. The Oxford classification of IgA nephropathy: Rationale, clinicopathological correlations, and classification. Kidney Int. 2009, 76, 534–545. [Google Scholar] [CrossRef] [Green Version]
- Haas, M.; Verhave, J.C.; Liu, Z.-H.; Alpers, C.E.; Barratt, J.; Becker, J.U.; Cattran, D.; Cook, H.T.; Coppo, R.; Feehally, J.; et al. A Multicenter Study of the Predictive Value of Crescents in IgA Nephropathy. J. Am. Soc. Nephrol. 2016, 28, 691–701. [Google Scholar] [CrossRef]
- Barbour, S.J.; Coppo, R.; Zhang, H.; Liu, Z.-H.; Suzuki, Y.; Matsuzaki, K.; Katafuchi, R.; Er, L.; Espino-Hernandez, G.; Kim, S.J.; et al. Evaluating a New International Risk-Prediction Tool in IgA Nephropathy. JAMA Intern. Med. 2019, 179, 942–952. [Google Scholar] [CrossRef]
- Barbour, S.J.; Coppo, R.; Zhang, H.; Liu, Z.-H.; Suzuki, Y.; Matsuzaki, K.; Er, L.; Reich, H.N.; Barratt, J.; Cattran, D.C.; et al. Application of the International IgA Nephropathy Prediction Tool one or two years post-biopsy. Kidney Int. 2022, 102, 160–172. [Google Scholar] [CrossRef]
- Shimozato, S.; Hiki, Y.; Odani, H.; Takahashi, K.; Yamamoto, K.; Sugiyama, S. Serum under-galactosylated IgA1 is increased in Japanese patients with IgA nephropathy. Nephrol. Dial. Transplant. 2008, 23, 1931–1939. [Google Scholar] [CrossRef] [Green Version]
- Camilla, R.; Suzuki, H.; Daprà, V.; Loiacono, E.; Peruzzi, L.; Amore, A.; Ghiggeri, G.M.; Mazzucco, G.; Scolari, F.; Gharavi, A.G.; et al. Oxidative Stress and Galactose-Deficient IgA1 as Markers of Progression in IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2011, 6, 1903–1911. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Li, Q.; Zhang, Y.; Shang, W.; Wei, L.; Li, H.; Gao, S.; Yan, T.; Jia, J.; Liu, Y.; et al. Clinical Significance of Galactose-Deficient IgA1 by KM55 in Patients with IgA Nephropathy. Kidney Blood Press. Res. 2019, 44, 1196–1206. [Google Scholar] [CrossRef]
- Yanagawa, H.; Suzuki, H.; Suzuki, Y.; Kiryluk, K.; Gharavi, A.G.; Matsuoka, K.; Makita, Y.; Julian, B.A.; Novak, J.; Tomino, Y. A Panel of Serum Biomarkers Differentiates IgA Nephropathy from Other Renal Diseases. PLoS ONE 2014, 9, e98081. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Hou, P.; Lv, J.; Moldoveanu, Z.; Li, Y.; Kiryluk, K.; Gharavi, A.G.; Novak, J.; Zhang, H. The level of galactose-deficient IgA1 in the sera of patients with IgA nephropathy is associated with disease progression. Kidney Int. 2012, 82, 790–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthoux, F.; Suzuki, H.; Thibaudin, L.; Yanagawa, H.; Maillard, N.; Mariat, C.; Tomino, Y.; Julian, B.A.; Novak, J. Autoantibodies Targeting Galactose-Deficient IgA1 Associate with Progression of IgA Nephropathy. J. Am. Soc. Nephrol. 2012, 23, 1579–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Lv, J.; Liu, P.; Xie, X.; Wang, M.; Liu, D.; Zhang, H.; Jin, J. Poly-IgA Complexes and Disease Severity in IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2021, 16, 1652–1664. [Google Scholar] [CrossRef] [PubMed]
- Baek, H.S.; Han, M.H.; Kim, Y.J.; Cho, M.H. Clinical Relevance of C4d Deposition in Pediatric Immunoglobulin A Nephropathy. Fetal Pediatr. Pathol. 2018, 37, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Marek-Bukowiec, K.; Konieczny, A.; Ratajczyk, K.; Witkiewicz, W. Candidate Urine Peptide Biomarkers for IgA Nephropathy: Where Are We Now? Dis. Mark. 2018, 2018, 5205831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocchetti, M.T.; Papale, M.; D’Apollo, A.M.; Suriano, I.V.; Di Palma, A.M.; Vocino, G.; Montemurno, E.; Varraso, L.; Grandaliano, G.; Di Paolo, S.; et al. Association of Urinary Laminin G-Like 3 and Free K Light Chains with Disease Activity and Histological Injury in IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2013, 8, 1115–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zewinger, S.; Rauen, T.; Rudnicki, M.; Federico, G.; Wagner, M.; Triem, S.; Schunk, S.J.; Petrakis, I.; Schmit, D.; Wagenpfeil, S.; et al. Dickkopf-3 (DKK3) in Urine Identifies Patients with Short-Term Risk of eGFR Loss. J. Am. Soc. Nephrol. 2018, 29, 2722–2733. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Kwan, B.C.-H.; Lai, F.M.-M.; Chow, K.-M.; Kam-Tao Li, P.; Szeto, C.-C. Expression of microRNAs in the urinary sediment of patients with IgA nephropathy. Dis. Mark. 2010, 28, 79–86. [Google Scholar] [CrossRef]
- Serino, G.; Sallustio, F.; Cox, S.N.; Pesce, F.; Schena, F.P. Abnormal miR-148b Expression Promotes Aberrant Glycosylation of IgA1 in IgA Nephropathy. J. Am. Soc. Nephrol. 2012, 23, 814–824. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Bao, H.; Xu, X.; Zhou, X.; Qin, W.; Zeng, C.; Liu, Z. Increased miR-374b promotes cell proliferation and the production of aberrant glycosylated IgA1 in B cells of IgA nephropathy. FEBS Lett. 2015, 589, 4019–4025. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Shi, S.; Lv, J.; Liu, L.; Zhou, X.; Zhu, L.; Chen, P.; Yang, H.; Wang, Z.; Wang, S.; et al. Rapidly progressive IgA nephropathy: Clinicopathological characteristics and outcomes assessed according to the revised definition of the KDIGO 2021 Guideline. Nephrol. Dial. Transplant. 2022, 37, 2429–2437. [Google Scholar] [CrossRef]
- Floege, J.; Feehally, J. Treatment of IgA nephropathy and Henoch-Schönlein nephritis. Nat. Rev. Nephrol. 2013, 9, 320–327. [Google Scholar] [CrossRef]
- Lennartz, D.P.; Seikrit, C.; Wied, S.; Fitzner, C.; Eitner, F.; Hilgers, R.-D.; Rauen, T.; Floege, J. Single versus dual blockade of the renin-angiotensin system in patients with IgA nephropathy. J. Nephrol. 2020, 33, 1231–1239. [Google Scholar] [CrossRef]
- Wheeler, D.C.; Toto, R.D.; Stefánsson, B.V.; Jongs, N.; Chertow, G.M.; Greene, T.; Hou, F.F.; McMurray, J.J.; Pecoits-Filho, R.; Correa-Rotter, R.; et al. A pre-specified analysis of the DAPA-CKD trial demonstrates the effects of dapagliflozin on major adverse kidney events in patients with IgA nephropathy. Kidney Int. 2021, 100, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Rauen, T.; Eitner, F.; Fitzner, C.; Sommerer, C.; Zeier, M.; Otte, B.; Panzer, U.; Peters, H.; Benck, U.; Mertens, P.R.; et al. Intensive Supportive Care plus Immunosuppression in IgA Nephropathy. N. Engl. J. Med. 2015, 373, 2225–2236. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, H.; Wong, M.G.; Jardine, M.J.; Hladunewich, M.; Jha, V.; Monaghan, H.; Zhao, M.; Barbour, S.; Reich, H.; et al. Effect of Oral Methylprednisolone on Clinical Outcomes in Patients With IgA Nephropathy: The TESTING Randomized Clinical Trial. JAMA 2017, 318, 432–442. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.G.; Lv, J.; Hladunewich, M.A.; Jha, V.; Hooi, L.S.; Monaghan, H.; Zhao, M.; Barbour, S.; Reich, H.N.; Cattran, D.; et al. The Therapeutic Evaluation of Steroids in IgA Nephropathy Global (TESTING) Study: Trial Design and Baseline Characteristics. Am. J. Nephrol. 2021, 52, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhang, H.; Wong, M.G.; Jardine, M.J.; Hladunewich, M.; Jha, V.; Monaghan, H.; Zhao, M.; Barbour, S.; Reich, H.; et al. Effect of Oral Methylprednisolone on Decline in Kidney Function or Kidney Failure in Patients With IgA Nephropathy: The TESTING Randomized Clinical Trial. JAMA 2022, 327, 1888–1898. [Google Scholar] [CrossRef]
- Tang, S.; Leung, J.C.; Chan, L.Y.; Lui, Y.H.; Tang, C.S.; Kan, C.H.; Ho, Y.W.; Lai, K.N. Mycophenolate mofetil alleviates persistent proteinuria in IgA nephropathy. Kidney Int. 2005, 68, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.C.; Tang, A.W.; Wong, S.S.; Leung, J.C.; Ho, Y.W.; Lai, K.N. Long-term study of mycophenolate mofetil treatment in IgA nephropathy. Kidney Int. 2010, 77, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Floege, J.; Rauen, T.; Tang, S.C.W. Current treatment of IgA nephropathy. Semin. Immunopathol. 2021, 43, 717–728. [Google Scholar] [CrossRef]
- Makita, Y.; Suzuki, H.; Kano, T.; Takahata, A.; Julian, B.A.; Novak, J.; Suzuki, Y. TLR9 activation induces aberrant IgA glycosylation via APRIL- and IL-6–mediated pathways in IgA nephropathy. Kidney Int. 2019, 97, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Torigoe, M.; Sakata, K.; Ishii, A.; Iwata, S.; Nakayamada, S.; Tanaka, Y. Hydroxychloroquine efficiently suppresses inflammatory responses of human class-switched memory B cells via Toll-like receptor 9 inhibition. Clin. Immunol. 2018, 195, 1–7. [Google Scholar] [CrossRef]
- Liu, L.-J.; Yang, Y.-Z.; Shi, S.-F.; Bao, Y.-F.; Yang, C.; Zhu, S.-N.; Sui, G.-L.; Chen, Y.-Q.; Lv, J.-C.; Zhang, H. Effects of Hydroxychloroquine on Proteinuria in IgA Nephropathy: A Randomized Controlled Trial. Am. J. Kidney Dis. 2019, 74, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Wu, W.; Wen, Y.; Li, X. Hydroxychloroquine alleviates persistent proteinuria in IgA nephropathy. Int. Urol. Nephrol. 2017, 49, 1233–1241. [Google Scholar] [CrossRef]
- Tang, C.; Lv, J.-C.; Shi, S.-F.; Chen, Y.-Q.; Liu, L.-J.; Zhang, H. Long-term safety and efficacy of hydroxychloroquine in patients with IgA nephropathy: A single-center experience. J. Nephrol. 2021, 35, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Stefan, G.; Mircescu, G. Hydroxychloroquine in IgA nephropathy: A systematic review. Ren. Fail. 2021, 43, 1520–1527. [Google Scholar] [CrossRef]
- Rollino, C.; Vischini, G.; Coppo, R. IgA nephropathy and infections. J. Nephrol. 2016, 29, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Takahara, M.; Nagato, T.; Nozaki, Y.; Kumai, T.; Katada, A.; Hayashi, T.; Harabuchi, Y. A proliferation-inducing ligand (APRIL) induced hyper-production of IgA from tonsillar mononuclear cells in patients with IgA nephropathy. Cell. Immunol. 2019, 341, 103925. [Google Scholar] [CrossRef]
- Zheng, N.; Xie, K.; Ye, H.; Dong, Y.; Wang, B.; Luo, N.; Fan, J.; Tan, J.; Chen, W.; Yu, X. TLR7 in B cells promotes renal inflammation and Gd-IgA1 synthesis in IgA nephropathy. J. Clin. Investig. 2020, 5, e136965. [Google Scholar] [CrossRef]
- Myette, J.R.; Kano, T.; Suzuki, H.; Sloan, S.E.; Szretter, K.J.; Ramakrishnan, B.; Adari, H.; Deotale, K.D.; Engler, F.; Shriver, Z.; et al. A Proliferation Inducing Ligand (APRIL) targeted antibody is a safe and effective treatment of murine IgA nephropathy. Kidney Int. 2019, 96, 104–116. [Google Scholar] [CrossRef]
- Mathur, M.; Barratt, J.; Suzuki, Y.; Engler, F.; Pasetti, M.F.; Yarbrough, J.; Sloan, S.; Oldach, D. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of VIS649 (Sibeprenlimab), an APRIL-Neutralizing IgG2 Monoclonal Antibody, in Healthy Volunteers. Kidney Int. Rep. 2022, 7, 993–1003. [Google Scholar] [CrossRef]
- Barratt, J.; Tumlin, J.; Suzuki, Y.; Kao, A.; Aydemir, A.; Pudota, K.; Jin, H.; Gühring, H.; Appel, G. Randomized Phase II JANUS Study of Atacicept in Patients With IgA Nephropathy and Persistent Proteinuria. Kidney Int. Rep. 2022, 7, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Maixnerova, D.; El Mehdi, D.; Rizk, D.V.; Zhang, H.; Tesar, V. New Treatment Strategies for IgA Nephropathy: Targeting Plasma Cells as the Main Source of Pathogenic Antibodies. J. Clin. Med. 2022, 11, 2810. [Google Scholar] [CrossRef] [PubMed]
- Lafayette, R.A.; Canetta, P.; Rovin, B.H.; Appel, G.B.; Novak, J.; Nath, K.A.; Sethi, S.; Tumlin, J.A.; Mehta, K.; Hogan, M.; et al. A Randomized, Controlled Trial of Rituximab in IgA Nephropathy with Proteinuria and Renal Dysfunction. J. Am. Soc. Nephrol. 2016, 28, 1306–1313. [Google Scholar] [CrossRef] [Green Version]
- Lechner, S.M.; Papista, C.; Chemouny, J.M.; Berthelot, L.; Monteiro, R.C. Role of IgA receptors in the pathogenesis of IgA nephropathy. J. Nephrol. 2015, 29, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Jhee, J.H.; Nam, B.Y.; Park, J.T.; Kim, H.W.; Chang, T.I.; Kang, E.W.; Lim, B.J.; Yoo, T.-H.; Kang, S.-W.; Jeong, H.J.; et al. CD71 mesangial IgA1 receptor and the progression of IgA nephropathy. Transl. Res. 2020, 230, 34–43. [Google Scholar] [CrossRef]
- Moresco, R.N.; Speeckaert, M.M.; Zmonarski, S.C.; Krajewska, M.; Komuda-Leszek, E.; Perkowska-Ptasinska, A.; Gesualdo, L.; Rocchetti, M.T.; Delanghe, S.E.; Vanholder, R.; et al. Urinary myeloid IgA Fc alpha receptor (CD89) and transglutaminase-2 as new biomarkers for active IgA nephropathy and henoch-Schönlein purpura nephritis. BBA Clin. 2016, 5, 79–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamm, M.E.; Emancipator, S.N.; Robinson, J.K.; Yamashita, M.; Fujioka, H.; Qiu, J.; Plaut, A.G. Microbial IgA Protease Removes IgA Immune Complexes from Mouse Glomeruli In Vivo: Potential Therapy for IgA Nephropathy. Am. J. Pathol. 2008, 172, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, X.; Shen, H.; Mao, N.; Wang, H.; Cui, L.; Cheng, Y.; Fan, J. Bacterial IgA protease-mediated degradation of agIgA1 and agIgA1 immune complexes as a potential therapy for IgA Nephropathy. Sci. Rep. 2016, 6, 30964. [Google Scholar] [CrossRef] [Green Version]
- Lechner, S.M.; Abbad, L.; Boedec, E.; Papista, C.; Le Stang, M.-B.; Moal, C.; Maillard, J.; Jamin, A.; Bex-Coudrat, J.; Wang, Y.; et al. IgA1 Protease Treatment Reverses Mesangial Deposits and Hematuria in a Model of IgA Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2622–2629. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Li, J.; Liu, P.; Wang, M.; Gao, L.; Wan, F.; Lv, J.; Zhang, H.; Jin, J. Chimeric Fusion between Clostridium Ramosum IgA Protease and IgG Fc Provides Long-Lasting Clearance of IgA Deposits in Mouse Models of IgA Nephropathy. J. Am. Soc. Nephrol. 2022, 33, 918–935. [Google Scholar] [CrossRef]
- Tam, F.; Tumlin, J.; Barratt, J.; Rovin, B.; Roberts, I.; Roufosse, C.; Cook, H.; Tong, S.; Magilavy, D. Lafayette. Sun-036 Spleen Tyrosine Kinase (Syk) Inhibition in Iga Nephropathy: A Global, Phase Ii, Randomised Placebo-Controlled Trial of Fostamatinib. Kidney Int. Rep. 2019, 4, S168. [Google Scholar] [CrossRef] [Green Version]
- McAdoo, S.; Tam, F.W. Role of the Spleen Tyrosine Kinase Pathway in Driving Inflammation in IgA Nephropathy. Semin. Nephrol. 2018, 38, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Yiu, W.H.; Chan, K.W.; Chan, L.Y.Y.; Leung, J.C.K.; Lai, K.N.; Tang, S.C.W. Spleen Tyrosine Kinase Inhibition Ameliorates Tubular Inflammation in IgA Nephropathy. Front. Physiol. 2021, 12, 650888. [Google Scholar] [CrossRef]
- Nakata, J.; Suzuki, Y.; Suzuki, H.; Sato, D.; Kano, T.; Yanagawa, H.; Matsuzaki, K.; Horikoshi, S.; Novak, J.; Tomino, Y. Changes in Nephritogenic Serum Galactose-Deficient IgA1 in IgA Nephropathy following Tonsillectomy and Steroid Therapy. PLoS ONE 2014, 9, e89707. [Google Scholar] [CrossRef]
- Moriyama, T.; Karasawa, K.; Miyabe, Y.; Akiyama, K.; Iwabuchi, Y.; Ogura, S.; Takabe, T.; Sugiura, N.; Seki, M.; Hanafusa, N.; et al. Long-Term Beneficial Effects of Tonsillectomy on Patients with Immunoglobulin A Nephropathy. Kidney360 2020, 1, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Matsuzaki, K.; Yasuda, T.; Nishikawa, M.; Yasuda, Y.; Koike, K.; Maruyama, S.; Yokoo, T.; Matsuo, S.; Kawamura, T.; et al. Association Between Tonsillectomy and Outcomes in Patients With Immunoglobulin A Nephropathy. JAMA Netw. Open 2019, 2, e194772. [Google Scholar] [CrossRef] [Green Version]
- Bager, P.; Gørtz, S.; Feenstra, B.; Andersen, N.N.; Jess, T.; Frisch, M.; Melbye, M. Increased Risk of Inflammatory Bowel Disease in Families with Tonsillectomy: A Danish National Cohort Study. Epidemiology 2019, 30, 256–262. [Google Scholar] [CrossRef]
- Wu, M.-C.; Ma, K.S.-K.; Wang, Y.-H.; Wei, J.C.-C. Impact of tonsillectomy on irritable bowel syndrome: A nationwide population-based cohort study. PLoS ONE 2020, 15, e0238242. [Google Scholar] [CrossRef]
- Lee, M.; Suzuki, H.; Kato, R.; Nakayama, M.; Fukao, Y.; Kano, T.; Makita, Y.; Kobayashi, T.; Sato, D.; Kihara, M.; et al. Study Protocol for Validation of the Safety and Efficacy of Topical Steroid Therapy Targeting Nasal-Associated Lymphoid Tissue in IgA Nephropathy: A Single-Centre, Open-Label, Historical Controlled Study. Research Square. Available online: https://doi.org/10.21203/rs.3.rs-1699588/v1 (accessed on 1 December 2022).
- Coppo, R. The Gut-Renal Connection in IgA Nephropathy. Semin. Nephrol. 2018, 38, 504–512. [Google Scholar] [CrossRef]
- Barratt, J.; Lafayette, R.; Kristensen, J.; Stone, A.; Cattran, D.; Floege, J.; Tesar, V.; Trimarchi, H.; Zhang, H.; Eren, N.; et al. Results from part A of the multi-center, double-blind, randomized, placebo-controlled NefIgArd trial, which evaluated targeted-release formulation of budesonide for the treatment of primary immunoglobulin A nephropathy. Kidney Int. 2022. [Google Scholar] [CrossRef]
- Fellström, B.C.; Barratt, J.; Cook, H.; Coppo, R.; Feehally, J.; de Fijter, J.W.; Floege, J.; Hetzel, G.; Jardine, A.G.; Locatelli, F.; et al. Targeted-release budesonide versus placebo in patients with IgA nephropathy (NEFIGAN): A double-blind, randomised, placebo-controlled phase 2b trial. Lancet 2017, 389, 2117–2127. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Yuan, Y.; Li, X.; Luo, J.; Zhou, Z.; Yu, L.; Wang, G. Orange-derived and dexamethasone-encapsulated extracellular vesicles reduced proteinuria and alleviated pathological lesions in IgA nephropathy by targeting intestinal lymphocytes. Front. Immunol. 2022, 13, 900963. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Tan, J.; Tan, L.; Tang, Y.; Qiu, Z.; Pei, G.; Qin, W. Modifications of gut microbiota are associated with the severity of IgA nephropathy in the Chinese population. Int. Immunopharmacol. 2020, 89 (Pt B), 107085. [Google Scholar] [CrossRef]
- He, J.-W.; Zhou, X.-J.; Li, Y.-F.; Wang, Y.-N.; Liu, L.-J.; Shi, S.-F.; Xin, X.-H.; Li, R.-S.; Falchi, M.; Lv, J.-C.; et al. Associations of Genetic Variants Contributing to Gut Microbiota Composition in Immunoglobin A Nephropathy. Msystems 2021, 6, e00819-20. [Google Scholar] [CrossRef]
- Zhao, J.; Bai, M.; Ning, X.; Qin, Y.; Wang, Y.; Yu, Z.; Dong, R.; Zhang, Y.; Sun, S. Expansion of Escherichia-Shigella in Gut Is Associated with the Onset and Response to Immunosuppressive Therapy of IgA Nephropathy. J. Am. Soc. Nephrol. 2022, 33, 2276–2292. [Google Scholar] [CrossRef]
- Chemouny, J.M.; Gleeson, P.J.; Abbad, L.; Lauriero, G.; Boedec, E.; Le Roux, K.; Monot, C.; Bredel, M.; Bex-Coudrat, J.; Sannier, A.; et al. Modulation of the microbiota by oral antibiotics treats immunoglobulin A nephropathy in humanized mice. Nephrol. Dial. Transplant. 2018, 34, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Di Leo, V.; Gleeson, P.J.; Sallustio, F.; Bounaix, C.; Da Silva, J.; Loreto, G.; Ben Mkaddem, S.; Monteiro, R.C. Rifaximin as a Potential Treatment for IgA Nephropathy in a Humanized Mice Model. J. Pers. Med. 2021, 11, 309. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Dong, L.; Jiang, Z.; Tan, L.; Luo, X.; Pei, G.; Qin, A.; Zhong, Z.; Liu, X.; Tang, Y.; et al. Probiotics ameliorate IgA nephropathy by improving gut dysbiosis and blunting NLRP3 signaling. J. Transl. Med. 2022, 20, 382. [Google Scholar] [CrossRef]
- Chai, L.; Luo, Q.; Cai, K.; Wang, K.; Xu, B. Reduced fecal short-chain fatty acids levels and the relationship with gut microbiota in IgA nephropathy. BMC Nephrol. 2021, 22, 209. [Google Scholar] [CrossRef]
- Lauriero, G.; Abbad, L.; Vacca, M.; Celano, G.; Chemouny, J.M.; Calasso, M.; Berthelot, L.; Gesualdo, L.; De Angelis, M.; Monteiro, R.C. Fecal Microbiota Transplantation Modulates Renal Phenotype in the Humanized Mouse Model of IgA Nephropathy. Front. Immunol. 2021, 12, 694787. [Google Scholar] [CrossRef]
- Bruchfeld, A.; Magin, H.; Nachman, P.; Parikh, S.; Lafayette, R.; Potarca, A.; Miao, S.; Bekker, P. C5a receptor inhibitor avacopan in immunoglobulin A nephropathy—An open-label pilot study. Clin. Kidney J. 2022, 15, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Barratt, J.; Rovin, B.; Zhang, H.; Kashihara, N.; Maes, B.; Rizk, D.; Trimarchi, H.; Sprangers, B.; Meier, M.; Kollins, D.; et al. Pos-546 Efficacy and Safety of Iptacopan in Iga Nephropathy: Results of a Randomized Double-Blind Placebo-Controlled Phase 2 Study at 6 Months. Kidney Int. Rep. 2022, 7, S236. [Google Scholar] [CrossRef]
- Lafayette, R.A.; Rovin, B.H.; Reich, H.N.; Tumlin, J.A.; Floege, J.; Barratt, J. Safety, Tolerability and Efficacy of Narsoplimab, a Novel MASP-2 Inhibitor for the Treatment of IgA Nephropathy. Kidney Int. Rep. 2020, 5, 2032–2041. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Y.; Cheng, T.; Liu, C.; Zhu, T.; Guo, C.; Li, S.; Rao, X.; Li, J. IgA Nephropathy: Current Understanding and Perspectives on Pathogenesis and Targeted Treatment. Diagnostics 2023, 13, 303. https://doi.org/10.3390/diagnostics13020303
Du Y, Cheng T, Liu C, Zhu T, Guo C, Li S, Rao X, Li J. IgA Nephropathy: Current Understanding and Perspectives on Pathogenesis and Targeted Treatment. Diagnostics. 2023; 13(2):303. https://doi.org/10.3390/diagnostics13020303
Chicago/Turabian StyleDu, Yating, Tingzhu Cheng, Chenxuan Liu, Tingting Zhu, Chuan Guo, Shen Li, Xiangrong Rao, and Jinpu Li. 2023. "IgA Nephropathy: Current Understanding and Perspectives on Pathogenesis and Targeted Treatment" Diagnostics 13, no. 2: 303. https://doi.org/10.3390/diagnostics13020303