
Genomic Characterization of Rare Primary Cardiac Sarcoma Entities
 , , , , , ,
, , , , , ,
Abstract
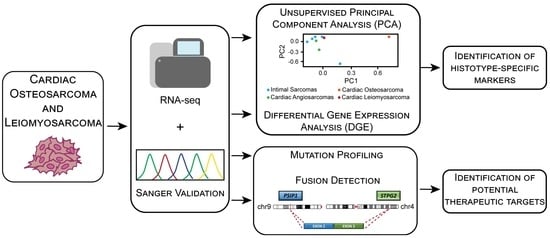
:
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Coding Transcriptome Sequencing
2.3. Bioinformatic Analysis
2.4. Sanger Sequencing
3. Results
3.1. Patients
3.2. Mutational Profile of Cardiac Osteosarcoma and Leiomyosarcoma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Isambert, N.; Ray-Coquard, I.; Italiano, A.; Rios, M.; Kerbrat, P.; Gauthier, M.; Blouet, A.; Chaigneau, L.; Duffaud, F.; Piperno-Neumann, S.; et al. Primary Cardiac Sarcomas: A Retrospective Study of the French Sarcoma Group. Eur. J. Cancer 2014, 50, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, P.; Chiara Sergi, M.; Cameli, M.; Miglioranza, M.H.; Ciccone, M.M.; Gentile, M.; Porta, C.; Tucci, M. Primary Soft Tissue Sarcoma of the Heart: An Emerging Chapter in Cardio-Oncology. Biomedicines 2021, 9, 774. [Google Scholar] [CrossRef] [PubMed]
- Siontis, B.L.; Leja, M.; Chugh, R. Current Clinical Management of Primary Cardiac Sarcoma. Expert Rev. Anticancer Ther. 2020, 20, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Moody, J.S.; Weigel, T.L.; Kozak, K.R. Primary Cardiac Sarcoma. Ann. Thorac. Surg. 2010, 90, 176–181. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.D.; Peterson, A.; Bartczak, A.; Lee, S.; Chojnowski, S.; Gajewski, P.; Loukas, M. Primary Cardiac Angiosarcoma—A Review. Med. Sci. Monit. 2014, 20, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Urbini, M.; Astolfi, A.; Indio, V.; Nannini, M.; Pizzi, C.; Paolisso, P.; Tarantino, G.; Pantaleo, M.A.; Saponara, M. Genetic Aberrations and Molecular Biology of Cardiac Sarcoma. Ther. Adv. Med. Oncol. 2020, 12, 1758835920918492. [Google Scholar] [CrossRef]
- Parissis, J.; Arvanitis, D.; Sourvinos, G.; Spandidos, D. A Primary Cardiac Leiomyosarcoma with Mutation at H-Ras Codon 12. Oncol. Rep. 1997, 4, 807–808. [Google Scholar] [CrossRef]
- Parwani, A.V.; Esposito, N.; Rao, U.N.M. Primary Cardiac Osteosarcoma with Recurrent Episodes and Unusual Patterns of Metastatic Spread. Cardiovasc. Pathol. 2008, 17, 413–417. [Google Scholar] [CrossRef]
- Saponara, M.; Indio, V.; Pizzi, C.; Serban, E.D.; Urbini, M.; Astolfi, A.; Paolisso, P.; Suarez, S.M.; Nannini, M.; Pacini, D.; et al. Successful Multidisciplinary Clinical Approach and Molecular Characterization by Whole Transcriptome Sequencing of a Cardiac Myxofibrosarcoma: A Case Report. World J. Clin. Cases 2019, 7, 3018–3026. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 30 May 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Putri, G.H.; Anders, S.; Pyl, P.T.; Pimanda, J.E.; Zanini, F. Analysing High-Throughput Sequencing Data in Python with HTSeq 2.0. Bioinformatics 2022, 38, 2943–2945. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigg, C.D.; Buhmann, J.M. Expectation-Maximization for Sparse and Non-Negative PCA. In Proceedings of the Twenty-Fifth International Conference on Machine Learning, Helsinki, Finlandia, 5–9 June 2008. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, M.; Amato, R.; Sgura, A.; Antoccia, A.; Berardinelli, F. The Multiple Facets of Atrx Protein. Cancers 2021, 13, 2211. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Garcia-Fabiani, M.B.; Carney, S.; Altshuler, D.; Núñez, F.J.; Méndez, F.M.; Núñez, F.; Lowenstein, P.R.; Castro, M.G. Mutant ATRX: Uncovering a New Therapeutic Target for Glioma. Expert Opin. Ther. Targets 2018, 22, 599–613. [Google Scholar] [CrossRef]
- Mackenzie, D.; Watters, A.K.; To, J.T.; Young, M.W.; Muratori, J.; Wilkoff, M.H.; Abraham, R.G.; Plummer, M.M.; Zhang, D. Alt Positivity in Human Cancers: Prevalence and Clinical Insights. Cancers 2021, 13, 2384. [Google Scholar] [CrossRef]
- Rickel, K.; Fang, F.; Tao, J. Molecular Genetics of Osteosarcoma. Bone 2017, 102, 69–79. [Google Scholar] [CrossRef]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent Somatic Structural Variations Contribute to Tumorigenesis in Pediatric Osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef]
- Ren, X.; Tu, C.; Tang, Z.; Ma, R.; Li, Z. Alternative Lengthening of Telomeres Phenotype and Loss of ATRX Expression in Sarcomas. Oncol. Lett. 2018, 15, 7489–7496. [Google Scholar] [CrossRef] [Green Version]
- Malik, S.S.; Mubarik, S.; Baig, M.; Masood, N.; Chaudhry, N. Genetic Polymorphism in ERCC5 and Breast Cancer Risk. Mol. Biol. Res. Commun. 2019, 8, 27–31. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, K.; Liu, X.; Jiang, G.; Liu, H. High Expression of ERCC5 Predicts a Poor Prognosis in Hepatocellular Carcinoma. Int. J. Clin. Exp. Pathol. 2018, 11, 3664–3670. [Google Scholar]
- Astolfi, A.; Nannini, M.; Indio, V.; Schipani, A.; Rizzo, A.; Perrone, A.M.; de Iaco, P.; Pirini, M.G.; de Leo, A.; Urbini, M.; et al. Genomic Database Analysis of Uterine Leiomyosarcoma Mutational Profile. Cancers 2020, 12, 2126. [Google Scholar] [CrossRef]
- Lee, P.J.; Yoo, N.S.; Hagemann, I.S.; Pfeifer, J.D.; Cottrell, C.E.; Abel, H.J.; Duncavage, E.J. Spectrum of Mutations in Leiomyosarcomas Identified by Clinical Targeted Next-Generation Sequencing. Exp. Mol. Pathol. 2017, 102, 156–161. [Google Scholar] [CrossRef]
- Ropero, S.; Setien, F.; Espada, J.; Fraga, M.F.; Herranz, M.; Asp, J.; Benassi, M.S.; Franchi, A.; Patiño, A.; Ward, L.S.; et al. Epigenetic Loss of the Familial Tumor-Suppressor Gene Exostosin-1 (EXT1) Disrupts Heparan Sulfate Synthesis in Cancer Cells. Hum. Mol. Genet. 2004, 13, 2753–2765. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhong, L.; Xu, Y.; Ding, L.; Ji, Y.; Schutz, S.; Férec, C.; Cooper, D.N.; Xu, C.; Chen, J.M.; et al. EXT1 and EXT2 Variants in 22 Chinese Families with Multiple Osteochondromas: Seven New Variants and Potentiation of Preimplantation Genetic Testing and Prenatal Diagnosis. Front. Genet. 2020, 11, 607838. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, R.; Lin, H.; Wang, H. Insights into the Molecular Regulatory Network of Pathomechanisms in Osteochondroma. J. Cell. Biochem. 2019, 120, 16362–16369. [Google Scholar] [CrossRef] [PubMed]
- D’arienzo, A.; Andreani, L.; Sacchetti, F.; Colangeli, S.; Capanna, R. Hereditary Multiple Exostoses: Current Insights. Orthop. Res. Rev. 2019, 11, 199–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeshouse, A.; Adebamowo, C.; Adebamowo, S.N.; Akbani, R.; Akeredolu, T.; Ally, A.; Anderson, M.L.; Anur, P.; Appelbaum, E.L.; Armenia, J.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, M.L.; Chavan, S.S.; Solit, D.B.; Murali, R.; Soslow, R.; Chiang, S.; Jungbluth, A.A.; Bandlamudi, C.; Srinivasan, P.; Tap, W.D.; et al. Genomic Landscape of Uterine Sarcomas Defined through Prospective Clinical Sequencing. Clin. Cancer Res. 2020, 26, 3881–3888. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Gholamalamdari, O.; Jadaliha, M.; Li, X.L.; Lin, Y.C.; Zhang, Y.; Guang, S.; Hashemikhabir, S.; Tiwari, S.; Zhu, Y.J.; et al. PSIP1/P75 Promotes Tumorigenicity in Breast Cancer Cells by Promoting the Transcription of Cell Cycle Genes. Carcinogenesis 2017, 38, 966–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liedtke, V.; Schröder, C.; Roggenbuck, D.; Weiss, R.; Stohwasser, R.; Schierack, P.; Rödiger, S.; Schenk, L. LEDGF/P75 Is Required for an Efficient Dna Damage Response. Int. J. Mol. Sci. 2021, 22, 5866. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Rojas, H.; Banerjee, H.; Cabrera, I.B.; Perez, K.Y.; de León, M.; Casiano, C.A. Expression of the Stress Response Oncoprotein LEDGF/P75 in Human Cancer: A Study of 21 Tumor Types. PLoS ONE 2012, 7, e30132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, J.D.; Johnatty, S.E.; Lu, Y.; Beesley, J.; Gao, B.; Kalimutho, M.; Henderson, M.J.; Russell, A.J.; Kar, S.; Chen, X.; et al. Germline Polymorphisms in an Enhancer of PSIP1 Are Associated with Progression-Free Survival in Epithelial Ovarian Cancer. Oncotarget 2016, 7, 6353–6368. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Xu, Y.; Zhong, J.; Wang, H.; Weng, M.; Cheng, Q.; Wu, Q.; Sun, Z.; Jiang, H.; Zhu, M.; et al. MFHAS1 Promotes Colorectal Cancer Progress by Regulating Polarization of Tumor-Associated Macrophages via STAT6 Signaling Pathway. Oncotarget 2016, 7, 78726–78735. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Jin, X.; Sun, H.; Pang, S.; Kong, X.; Bu, J.; Xu, S. Mir-139-5p Targetedly Regulates Yaf2 and Mediates the Akt/P38 Mapk Signaling Pathway to Alleviate the Metastasis of Non-Small Cell Lung Cancer Cells and Their Resistance against Cisplatin. Cancer Manag. Res. 2021, 13, 3639–3650. [Google Scholar] [CrossRef]
- Grimm, D.; Bauer, J.; Wise, P.; Krüger, M.; Simonsen, U.; Wehland, M.; Infanger, M.; Corydon, T.J. The Role of SOX Family Members in Solid Tumours and Metastasis. Semin. Cancer Biol. 2020, 67, 122–153. [Google Scholar] [CrossRef]
- Huang, S.; Zhu, Y.; Wang, C.; Li, X.; Cui, X.; Tu, S.; You, L.; Fu, J.W.; Chen, Z.; Hu, W.; et al. PAK5 Facilitates the Proliferation, Invasion and Migration in Colorectal Cancer Cells. Cancer Med. 2020, 9, 4777–4790. [Google Scholar] [CrossRef]
- Nakajima, K.; Kidani, T.; Miura, H. Molecular Profiling of Bone Remodeling Occurring in Musculoskeletal Tumors. J. Orthop. Res. 2021, 39, 1402–1410. [Google Scholar] [CrossRef]
- Baker, E.K.; Taylor, S.; Gupte, A.; Chalk, A.M.; Bhattacharya, S.; Green, A.C.; Martin, T.J.; Strbenac, D.; Robinson, M.D.; Purton, L.E.; et al. Wnt Inhibitory Factor 1 (WIF1) Is a Marker of Osteoblastic Differentiation Stage and Is Not Silenced by DNA Methylation in Osteosarcoma. Bone 2015, 73, 223–232. [Google Scholar] [CrossRef]
- Grassi, E.S.; Pietras, A. Emerging Roles of DLK1 in the Stem Cell Niche and Cancer Stemness. J. Histochem. Cytochem. 2022, 70, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Kainov, Y.; Favorskaya, I.; Delektorskaya, V.; Chemeris, G.; Komelkov, A.; Zhuravskaya, A.; Trukhanova, L.; Zueva, E.; Tavitian, B.; Dyakova, N.; et al. CRABP1 Provides High Malignancy of Transformed Mesenchymal Cells and Contributes to the Pathogenesis of Mesenchymal and Neuroendocrine Tumors. Cell Cycle 2014, 13, 1530–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, S.R.C.; Oliveira, I.D.; Okamoto, O.K.; Zago, M.A.; Alves, M.T.D.S.; Garcia Filho, R.J.; Macedo, C.R.P.D.; Petrilli, A.S. Bone Deposition, Bone Resorption, and Osteosarcoma. J. Orthop. Res. 2010, 28, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Gronchi, A.; Miah, A.B.; Dei Tos, A.P.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielack, S.; Blay, J.Y.; Bolle, S.; et al. Soft Tissue and Visceral Sarcomas: ESMO–EURACAN–GENTURIS Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2021, 32, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Strauss, S.J.; Frezza, A.M.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielack, S.; Blay, J.Y.; Bolle, S.; Bonvalot, S.; et al. Bone Sarcomas: ESMO–EURACAN–GENTURIS–ERN PaedCan Clinical Practice Guideline for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2021, 32, 1520–1536. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Gender | Age | Histotype | Disease Status Dia | Last FU 1 |
|---|---|---|---|---|---|
| CS1 | M | 50 | pulmonary artery intimal sarcoma | Advanced | DOD 2 |
| CS2 | M | 50 | pulmonary artery intimal sarcoma | Advanced | DOD 2 |
| CS3 | F | 45 | pulmonary artery intimal sarcoma | Advanced | DOD 2 |
| CS4 | M | 37 | pulmonary artery intimal sarcoma | Advanced | DOD 2 |
| CS5 | F | 69 | pulmonary artery intimal sarcoma | Advanced | DOD 2 |
| CS6 | M | 74 | angiosarcoma | Advanced | DOD 2 |
| CS7 | M | 39 | angiosarcoma | Advanced | DOD 2 |
| CS8 | M | 35 | angiosarcoma | Localized | DOD 2 |
| CS9 | M | 39 | osteosarcoma | Advanced | DOD 2 |
| CS10 | F | 73 | myxofibrosarcoma | Localized | DOD 2 |
| CS11 | F | 59 | leiomyosarcoma | NA 3 | NA 3 |
| ID | Gene | Chr | cDNA | Protein | Type | COSMIC | ClinVar | Varsome | gnomAD Frequency | Alt Allele Depth | Total Read Depth |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CS9 | ATRX | 23 | c.1283_1284ins(T) | p.(Asn428LysfsTer6) | frameshift | NA 1 | NA 1 | NA 1 | NA 1 | 32 | 39 |
| CS9 | ERCC5 | 13 | c.2636A>G | p.(Asn879Ser) | missense | pathogenic | benign | benign | 0.00933 | 19 | 38 |
| CS9 | COL1A1 | 17 | c.1461+2T>G | NA 1 | spl. don. 2 | NA 1 | lik. path. 3 | NA 1 | NA 1 | 14 | 17 |
| CS9 | USP6 | 17 | c.319G>T | p.(Gly107Cys) | missense | NA 1 | NA 1 | uncertain | 0.0011 | 29 | 62 |
| CS9 | FH | 1 | c.172G>A | p.(Gly58Ser) | missense | NA 1 | uncertain | NA 1 | NA 1 | 11 | 20 |
| CS9 | SETBP1 | 18 | c.3562G>A | p.(Glu1188Lys) | missense | NA 1 | uncertain | uncertain | 0.000079 | 41 | 65 |
| CS11 | EXT2 | 11 | c.1186G>A | p.(Val396Met) | missense | pathogenic | NA 1 | lik. Benign 4 | 0.00044 | 17 | 26 |
| CS11 | DNM2 | 19 | c.688+2T>C | NA 1 | spl. don. 2 | NA 1 | NA 1 | pathogenic | NA 1 | 13 | 16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gozzellino, L.; Nannini, M.; Pizzi, C.; Leone, O.; Corti, B.; Indio, V.; Baldovini, C.; Paolisso, P.; Foà, A.; Pacini, D.; et al. Genomic Characterization of Rare Primary Cardiac Sarcoma Entities. Diagnostics 2023, 13, 214. https://doi.org/10.3390/diagnostics13020214
Gozzellino L, Nannini M, Pizzi C, Leone O, Corti B, Indio V, Baldovini C, Paolisso P, Foà A, Pacini D, et al. Genomic Characterization of Rare Primary Cardiac Sarcoma Entities. Diagnostics. 2023; 13(2):214. https://doi.org/10.3390/diagnostics13020214
Chicago/Turabian StyleGozzellino, Livia, Margherita Nannini, Carmine Pizzi, Ornella Leone, Barbara Corti, Valentina Indio, Chiara Baldovini, Pasquale Paolisso, Alberto Foà, Davide Pacini, and et al. 2023. "Genomic Characterization of Rare Primary Cardiac Sarcoma Entities" Diagnostics 13, no. 2: 214. https://doi.org/10.3390/diagnostics13020214