
Proteomics in Childhood Acute Lymphoblastic Leukemia: Challenges and Opportunities


Abstract
:1. Introduction
2. Molecular Basis of Acute Lymphoblastic Leukemia
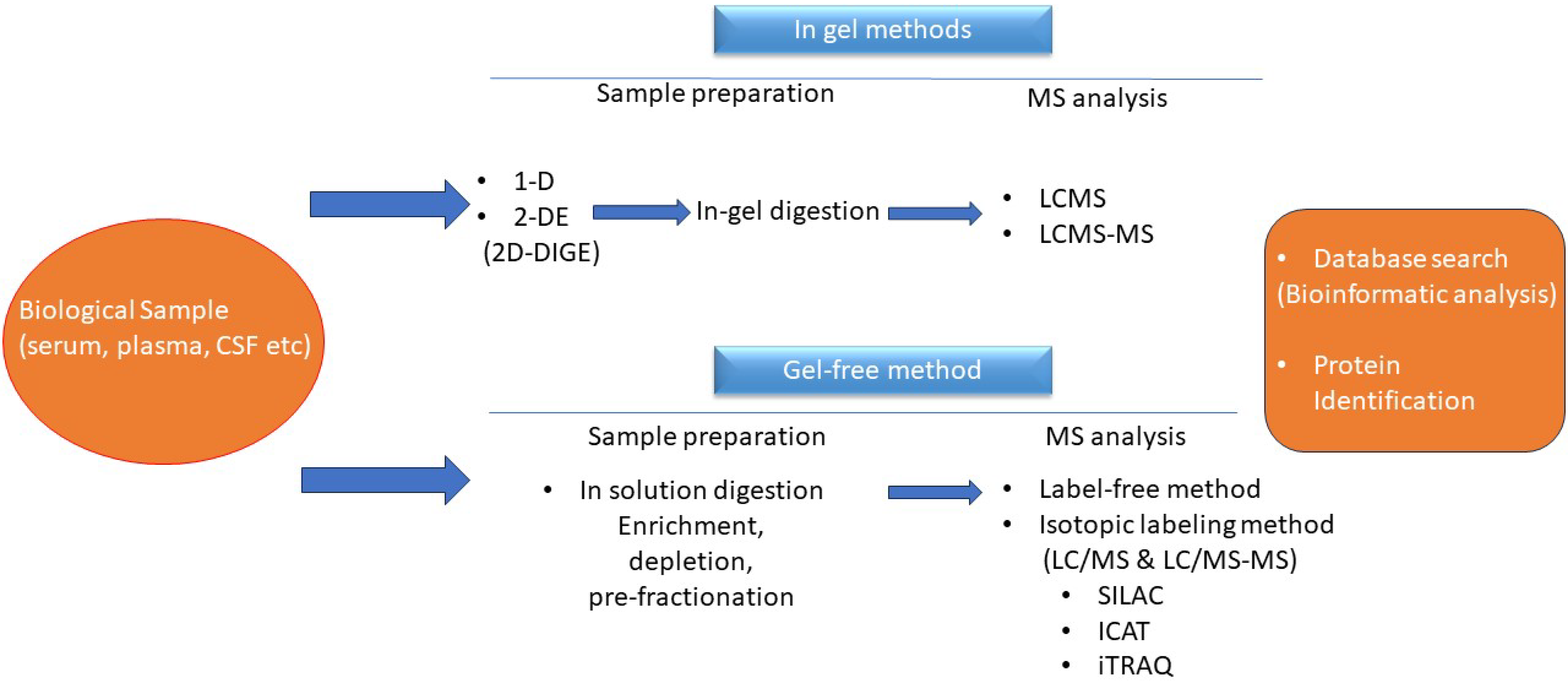
3. The Era of Proteomics
4. Discussion—Application of Proteomics in Childhood ALL
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Inaba, H.; Mullighan, C.G. Pediatric Acute Lymphoblastic Leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Nichols, K.E.; Yang, J.J. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 2019, 16, 227–240. [Google Scholar] [CrossRef]
- Jeha, S.; Pei, D.; Choi, J.; Cheng, C.; Sandlund, J.T.; Coustan-Smith, E.; Campana, D.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia without Cranial Irradiation: St Jude Total Therapy Study 16. J. Clin. Oncol. 2019, 37, 3377–3391. [Google Scholar] [CrossRef] [PubMed]
- Borowitz, M.J.; Wood, B.L.; Devidas, M.; Loh, M.L.; Raetz, E.A.; Salzer, W.L.; Nachman, J.B.; Carroll, A.J.; Heerema, N.A.; Gastier-Foster, J.M.; et al. Prognostic significance of minimal residual disease in high risk B-ALL: A report from Children’s Oncology Group study AALL0232. Blood 2015, 126, 964–971. [Google Scholar] [CrossRef]
- Wood, B.; Wu, D.; Crossley, B.; Dai, Y.; Williamson, D.; Gawad, C.; Borowitz, M.J.; Devidas, M.; Maloney, K.W.; Larsen, E.; et al. Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood 2018, 131, 1350–1359. [Google Scholar] [CrossRef]
- Smith, M.A.; Seibel, N.L.; Altekruse, S.F.; Ries, L.A.; Melbert, D.L.; O’Leary, M.; Smith, F.O.; Reaman, G.H. Outcomes for children and adolescents with cancer: Challenges for the twenty-first century. J. Clin. Oncol. 2010, 28, 2625–2634. [Google Scholar] [CrossRef]
- Kwon, Y.W.; Jo, H.S.; Bae, S.; Seo, Y.; Song, P.; Song, M.; Yoon, J.H. Application of Proteomics in Cancer: Recent Trends and Approaches for Biomarkers Discovery. Front. Med. 2021, 8, 747333. [Google Scholar] [CrossRef]
- Noone, A.M.; Cronin, K.A.; Altekruse, S.F.; Howlader, N.; Lewis, D.R.; Petkov, V.I.; Penberthy, L. Cancer Incidence and Survival Trends by Subtype Using Data from the Surveillance Epidemiology and End Results Program, 1992–2013. Cancer Epidemiol. Biomark. Prev. 2017, 26, 632–641. [Google Scholar] [CrossRef]
- National Cancer Institute. Age-Adjusted and Age-Specific SEER Cancer Incidence Rates, 2014–2018. Available online: https://seer.cancer.gov/csr/1975_2018/results_merged/sect_02_childhood_cancer_iccc.pdf (accessed on 18 April 2021).
- Lim, J.Y.; Bhatia, S.; Robison, L.L.; Yang, J.J. Genomics of racial and ethnic disparities in childhood acute lymphoblastic leukemia. Cancer 2014, 120, 955–962. [Google Scholar] [CrossRef]
- Wiemels, J. Perspectives on the causes of childhood leukemia. Chem. Biol. Interact. 2012, 196, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Stieglitz, E.; Loh, M.L. Genetic predispositions to childhood leukemia. Ther. Adv. Hematol. 2013, 4, 270–290. [Google Scholar] [CrossRef] [PubMed]
- Buitenkamp, T.D.; Izraeli, S.; Zimmermann, M.; Forestier, E.; Heerema, N.A.; van den Heuvel-Eibrink, M.M.; Pieters, R.; Korbijn, C.M.; Silverman, L.B.; Schmiegelow, K.; et al. Acute lymphoblastic leukemia in children with Down syndrome: A retrospective analysis from the Ponte di Legno study group. Blood 2014, 123, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Collins-Underwood, J.R.; Phillips, L.A.; Loudin, M.G.; Liu, W.; Zhang, J.; Ma, J.; Coustan-Smith, E.; Harvey, R.C.; Willman, C.L.; et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat. Genet. 2009, 41, 1243–1246. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Fioretos, T. New oncogenic subtypes in pediatric B-cell precursor acute lymphoblastic leukemia. Blood 2017, 130, 1395–1401. [Google Scholar] [CrossRef]
- Bastian, L.; Schroeder, M.P.; Eckert, C.; Schlee, C.; Tanchez, J.O.; Kämpf, S.; Wagner, D.L.; Schulze, V.; Isaakidis, K.; Lázaro-Navarro, J.; et al. PAX5 biallelic genomic alterations define a novel subgroup of B-cell precursor acute lymphoblastic leukemia. Leukemia 2019, 33, 1895–1909. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.; Roberts, K.; Li, Y.; Liu, Y.; Harvey, R.C.; McCastlain, K.; Reshmi, S.C.; Payne-Turner, D.; Iacobucci, I.; et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 13331. [Google Scholar] [CrossRef]
- Paulsson, K.; Lilljebjörn, H.; Biloglav, A.; Olsson, L.; Rissler, M.; Castor, A.; Barbany, G.; Fogelstrand, L.; Nordgren, A.; Sjögren, H.; et al. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nat. Genet. 2015, 47, 672–676. [Google Scholar] [CrossRef]
- Vora, A.; Goulden, N.; Wade, R.; Mitchell, C.; Hancock, J.; Hough, R.; Rowntree, C.; Richards, S. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): A randomised controlled trial. Lancet Oncol. 2013, 14, 199–209. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef]
- Pui, C.H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J. Clin. Oncol. 2015, 3, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Holmfeldt, L.; Wei, L.; Diaz-Flores, E.; Walsh, M.; Zhang, J.; Ding, L.; Payne-Turner, D.; Churchman, M.; Andersson, A.; Chen, S.C.; et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Safavi, S.; Olsson, L.; Biloglav, A.; Veerla, S.; Blendberg, M.; Tayebwa, J.; Behrendtz, M.; Castor, A.; Hansson, M.; Johansson, B.; et al. Genetic and epigenetic characterization of hypodiploid acute lymphoblastic leukemia. Oncotarget 2015, 6, 42793–42802. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J. Cytogenetics of paediatric and adolescent acute lymphoblastic leukaemia. Br. J. Haematol. 2009, 144, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M. Darwin and evolutionary tales in leukemia. The Ham-Wasserman Lecture. Hematol. Am. Soc. Hematol. Educ. Program. 2009, 2009, 3–12. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 11790. [Google Scholar] [CrossRef]
- Felice, M.S.; Gallego, M.S.; Alonso, C.N.; Alfaro, E.M.; Guitter, M.R.; Bernasconi, A.R.; Rubio, P.L.; Zubizarreta, P.A.; Rossi, J.G. Prognostic impact of t(1;19)/ TCF3-PBX1 in childhood acute lymphoblastic leukemia in the context of Berlin-Frankfurt-Münster-based protocols. Leuk. Lymphoma 2011, 52, 1215–1221. [Google Scholar] [CrossRef]
- Jeha, S.; Pei, D.; Raimondi, S.C.; Onciu, M.; Campana, D.; Cheng, C.; Sandlund, J.T.; Ribeiro, R.C.; Rubnitz, J.E.; Howard, S.C.; et al. Increased risk for CNS relapse in pre-B cell leukemia with the t(1;19)/TCF3-PBX1. Leukemia 2009, 23, 1406–1409. [Google Scholar] [CrossRef]
- Mullighan, C.G. Molecular genetics of B-precursor acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3407–3415. [Google Scholar] [CrossRef]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef]
- Brown, P.; Levis, M.; Shurtleff, S.; Campana, D.; Downing, J.; Small, D. FLT3 inhibition selectively kills childhood acute lymphoblastic leukemia cells with high levels of FLT3 expression. Blood 2005, 105, 812–820. [Google Scholar] [CrossRef]
- Brown, P.A.; Kairalla, J.A.; Hilden, J.M.; Dreyer, Z.E.; Carroll, A.J.; Heerema, N.A.; Wang, C.; Devidas, M.; Gore, L.; Salzer, W.L.; et al. FLT3 inhibitor lestaurtinib plus chemotherapy for newly diagnosed KMT2A-rearranged infant acute lymphoblastic leukemia: Children’s Oncology Group trial AALL0631. Leukemia 2021, 35, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.J.; Liu, Y.F.; Xu, L.Z.; Long, Z.J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.X.; Pan, Y.J.; Yan, J.S.; et al. The Philadelphia chromosome in leukemogenesis. Chin. J. Cancer 2016, 35, 48. [Google Scholar] [CrossRef] [PubMed]
- Biondi, A.; Gandemer, V.; De Lorenzo, P.; Cario, G.; Campbell, M.; Castor, A.; Pieters, R.; Baruchel, A.; Vora, A.; Leoni, V.; et al. Imatinib treatment of paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (EsPhALL2010): A prospective, intergroup, open-label, single-arm clinical trial. Lancet Haematol. 2018, 5, e641–e652. [Google Scholar] [CrossRef]
- Schultz, K.R.; Carroll, A.; Heerema, N.A.; Bowman, W.P.; Aledo, A.; Slayton, W.B.; Sather, H.; Devidas, M.; Zheng, H.W.; Davies, S.M.; et al. Children’s Oncology Group. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia 2014, 28, 1467–1471. [Google Scholar] [CrossRef]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef]
- Ding, Y.Y.; Stern, J.W.; Jubelirer, T.F.; Wertheim, G.B.; Lin, F.; Chang, F.; Gu, Z.; Mullighan, C.G.; Li, Y.; Harvey, R.C.; et al. Clinical efficacy of ruxolitinib and chemotherapy in a child with Philadelphia chromosome-like acute lymphoblastic leukemia with GOLGA5-JAK2 fusion and induction failure. Haematologica 2018, 103, e427–e431. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Sig. Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Dillon, M.; Lopez, A.; Lin, E.; Sales, D.; Perets, R.; Jain, P. Progress on Ras/MAPK Signaling Research and Targeting in Blood and Solid Cancers. Cancers 2021, 13, 5059. [Google Scholar] [CrossRef]
- Stivala, S.; Codilupi, T.; Brkic, S.; Baerenwaldt, A.; Ghosh, N.; Hao-Shen, H.; Dirnhofer, S.; Dettmer, M.S.; Simillion, C.; Kaufmann, B.A.; et al. Targeting compensatory MEK/ERK activation increases JAK inhibitor efficacy in myeloproliferative neoplasms. J. Clin. Investig. 2019, 129, 1596–1611. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- D’Angiò, M.; Valsecchi, M.G.; Testi, A.M.; Conter, V.; Nunes, V.; Parasole, R.; Colombini, A.; Santoro, N.; Varotto, S.; Caniglia, M.; et al. Clinical features and outcome of SIL/TAL1-positive T-cell acute lymphoblastic leukemia in children and adolescents: A 10-year experience of the AIEOP group. Haematologica 2015, 100, e10–e13. [Google Scholar] [CrossRef] [PubMed]
- Girardi, T.; Vicente, C.; Cools, J.; De Keersmaecker, K. The genetics and molecular biology of T-ALL. Blood 2017, 129, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Dolai, S.; Delgado-Martin, C.; Vincent, T.; Robbins, A.; Selvanathan, A.; Ryan, T.; Hall, J.; Wood, A.C.; Tasian, S.K.; et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood 2015, 125, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Kathpalia, M.; Mishra, P.; Bajpai, R.; Bhurani, D.; Agarwal, N. Efficacy and safety of nelarabine in patients with relapsed or refractory T-cell acute lymphoblastic leukemia: A systematic review and meta-analysis. Ann. Hematol. 2022, 101, 1655–1666. [Google Scholar] [CrossRef]
- Teachey, D.T.; Devidas, M.; Wood, B.L.; Chen, Z.; Hayashi, R.J.; Hermiston, M.L.; Annett, R.D.; Archer, J.H.; Asselin, B.L.; August, K.J.; et al. Children’s Oncology Group Trial AALL1231: A Phase III Clinical Trial Testing Bortezomib in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia and Lymphoma. J. Clin. Oncol. 2022, 40, 2106–2118. [Google Scholar] [CrossRef]
- Patrick, K.; Vora, A. Update on biology and treatment of T-cell acute lymphoblastic leukaemia. Curr. Opin. Pediatr. 2015, 27, 44–49. [Google Scholar] [CrossRef]
- Waanders, E.; Gu, Z.; Dobson, S.M.; Antić, Ž.; Crawford, J.C.; Ma, X.; Edmonson, M.N.; Payne-Turner, D.; van de Vorst, M.; Jongmans, M.C.J.; et al. Mutational landscape and patterns of clonal evolution in relapsed pediatric acute lymphoblastic leukemia. Blood Cancer Discov. 2020, 1, 96–111. [Google Scholar] [CrossRef]
- Burke, M.J.; Kostadinov, R.; Sposto, R.; Gore, L.; Kelley, S.M.; Rabik, C.; Trepel, J.B.; Lee, M.J.; Yuno, A.; Lee, S.; et al. Decitabine and Vorinostat with Chemotherapy in Relapsed Pediatric Acute Lymphoblastic Leukemia: A TACL Pilot Study. Clin. Cancer Res. 2020, 26, 2297–2307. [Google Scholar] [CrossRef]
- Place, A.E.; Pikman, Y.; Stevenson, K.E.; Harris, M.H.; Pauly, M.; Sulis, M.L.; Hijiya, N.; Gore, L.; Cooper, T.M.; Loh, M.L.; et al. Phase I trial of the mTOR inhibitor everolimus in combination with multi-agent chemotherapy in relapsed childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2018, 65, e27062. [Google Scholar] [CrossRef]
- Hiramatsu, H. Current status of CAR-T cell therapy for pediatric hematologic malignancies. Int. J. Clin. Oncol. 2023, 28, 729–735. [Google Scholar] [CrossRef]
- Rafei, H.; Kantarjian, H.M.; Jabbour, E.J. Targeted therapy paves the way for the cure of acute lymphoblastic leukaemia. Br. J. Haematol. 2020, 188, 207–223. [Google Scholar] [CrossRef]
- Altelaar, A.F.; Munoz, J.; Heck, A.J. Next-generation proteomics: Towards an integrative view of proteome dynamics. Nat. Rev. Genet. 2013, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Macklin, A.; Khan, S.; Kislinger, T. Recent advances in mass spectrometry based clinical proteomics: Applications to cancer research. Clin. Proteom. 2020, 24, 17. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef]
- Melani, R.D.; Gerbasi, V.R.; Anderson, L.C.; Sikora, J.W.; Toby, T.K.; Hutton, J.E.; Butcher, D.S.; Negrão, F.; Seckler, H.S.; Srzentić, K.; et al. The Blood Proteoform Atlas: A reference map of proteoforms in human hematopoietic cells. Science 2022, 28, 411–418. [Google Scholar] [CrossRef]
- Ercan, H.; Resch, U.; Hsu, F.; Mitulovic, G.; Bileck, A.; Gerner, C.; Yang, J.-W.; Geiger, M.; Miller, I.; Zellner, M. A Practical and Analytical Comparative Study of Gel-Based Top-Down and Gel-Free Bottom-Up Proteomics Including Unbiased Proteoform Detection. Cells 2023, 12, 747. [Google Scholar] [CrossRef]
- Cunningham, R.; Ma, D.; Li, L. Mass Spectrometry-based Proteomics and Peptidomics for Systems Biology and Biomarker Discovery. Front. Biol. 2012, 1, 313–335. [Google Scholar] [CrossRef]
- Pursiheimo, A.; Vehmas, A.P.; Afzal, S.; Suomi, T.; Chand, T.; Strauss, L.; Poutanen, M.; Rokka, A.; Corthals, G.L.; Elo, L.L. Optimization of Statistical Methods Impact on Quantitative Proteomics Data. J. Proteome Res. 2015, 14, 4118–4126. [Google Scholar] [CrossRef]
- Pietrowska, M.; Wlosowicz, A.; Gawin, M.; Widlak, P. MS-Based Proteomic Analysis of Serum and Plasma: Problem of High Abundant Components and Lights and Shadows of Albumin Removal. Adv. Exp. Med. Biol. 2019, 1073, 57–76. [Google Scholar] [PubMed]
- Dunphy, K.; O’Mahoney, K.; Dowling, P.; O’Gorman, P.; Bazou, D. Clinical Proteomics of Biofluids in Haematological Malignancies. Int. J. Mol. Sci. 2021, 22, 8021. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Conrads, T.P.; Lucas, D.A.; Janini, G.M.; Schaefer, C.F.; Buetow, K.H.; Issaq, H.J.; Veenstra, T.D. Direct ampholyte-free liquid-phase isoelectric peptide focusing: Application to the human serum proteome. Electrophoresis 2004, 25, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Jiang, X.; Mao, B.; Li, Q.X. The design, analysis and application of mouse clinical trials in oncology drug development. BMC Cancer 2019, 19, 718. [Google Scholar] [CrossRef] [PubMed]
- Georges, L.M.C.; De Wever, O.; Galván, J.A.; Dawson, H.; Lugli, A.; Demetter, P.; Zlobec, I. Cell Line Derived Xenograft Mouse Models Are a Suitable in vivo Model for Studying Tumor Budding in Colorectal Cancer. Front. Med. 2019, 27, 139. [Google Scholar] [CrossRef]
- Collins, A.T.; Lang, S.H. A systematic review of the validity of patient derived xenograft (PDX) models: The implications for translational research and personalised medicine. PeerJ 2018, 6, e5981. [Google Scholar] [CrossRef] [PubMed]
- Zecha, J.; Gabriel, W.; Spallek, R.; Chang, Y.C.; Mergner, J.; Wilhelm, M.; Bassermann, F.; Kuster, B. Linking post-translational modifications and protein turnover by site-resolved protein turnover profiling. Nat. Commun. 2022, 13, 165. [Google Scholar] [CrossRef]
- Bantscheff, M.; Schirle, M.; Sweetman, G.; Rick, J.; Kuster, B. Quantitative mass spectrometry in proteomics: A critical review. Anal. Bioanal. Chem. 2007, 389, 1017–1031. [Google Scholar] [CrossRef]
- Zhu, W.; Smith, J.W.; Huang, C.M. Mass spectrometry-based label-free quantitative proteomics. J. Biomed. Biotechnol. 2010, 2010, 840518. [Google Scholar] [CrossRef]
- Yu, Q.; Zhong, X.; Chen, B.; Feng, Y.; Ma, M.; Diamond, C.A.; Voeller, J.S.; Kim, M.; DeSantes, K.B.; Capitini, C.M.; et al. Isobaric Labeling Strategy Utilizing 4-Plex N,N-Dimethyl Leucine (DiLeu) Tags Reveals Proteomic Changes Induced by Chemotherapy in Cerebrospinal Fluid of Children with B-Cell Acute Lymphoblastic Leukemia. J. Proteome Res. 2020, 19, 2606–2616. [Google Scholar] [CrossRef]
- Marcus, K.; Lelong, C.; Rabilloud, T. What Room for Two-Dimensional Gel-Based Proteomics in a Shotgun Proteomics World? Proteomes 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Rodríguez, S.I.; Sanabria-Salas, M.C.; Umaña-Perez, A. A comparative proteomic study of plasma in Colombian childhood acute lymphoblastic leukemia. PLoS ONE 2019, 14, e0221509. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lv, Y.Q.; Liu, Y.F.; Du, X.J.; Li, B. Differential protein analysis of lymphocytes between children with acute lymphoblastic leukemia and healthy children. Leuk. Lymphoma 2013, 54, 381–386. [Google Scholar] [CrossRef]
- Cavalcante Mde, S.; Torres-Romero, J.C.; Lobo, M.D.; Moreno, F.B.; Bezerra, L.P.; Lima, D.S.; Matos, J.C.; Moreira Rde, A.; Monteiro-Moreira, A.C. A panel of glycoproteins as candidate biomarkers for early diagnosis and treatment evaluation of B-cell acute lymphoblastic leukemia. Biomark. Res. 2016, 27, 4. [Google Scholar] [CrossRef]
- Yu, R.; Yang, S.; Liu, Y.; Zhu, Z. Identification and validation of serum autoantibodies in children with B-cell acute lymphoblastic leukemia by serological proteome analysis. Proteome Sci. 2022, 20, 3. [Google Scholar] [CrossRef] [PubMed]
- Braoudaki, M.; Lambrou, G.I.; Vougas, K.; Karamolegou, K.; Tsangaris, G.T.; Tzortzatou-Stathopoulou, F. Protein biomarkers distinguish between high- and low-risk pediatric acute lymphoblastic leukemia in a tissue specific manner. J. Hematol. Oncol. 2013, 6, 52. [Google Scholar] [CrossRef]
- Xu, G.; Li, Z.; Wang, L.; Chen, F.; Chi, Z.; Gu, M.; Li, S.; Wu, D.; Miao, J.; Zhang, Y.; et al. Label-free quantitative proteomics reveals differentially expressed proteins in high risk childhood acute lymphoblastic leukemia. J. Proteom. 2017, 150, 1–8. [Google Scholar] [CrossRef]
- Jiang, N.; Kham, S.K.; Koh, G.S.; Suang Lim, J.Y.; Ariffin, H.; Chew, F.T.; Yeoh, A.E. Identification of prognostic protein biomarkers in childhood acute lymphoblastic leukemia (ALL). J. Proteom. 2011, 74, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Lin, M.; Liu, T.; Li, J.; Chen, B.; Chen, Y. DIGE-based proteomic analysis identifies nucleophosmin/B23 and nucleolin C23 as over-expressed proteins in relapsed/refractory acute leukemia. Leuk. Res. 2011, 35, 1087–1092. [Google Scholar] [CrossRef]
- Guzmán-Ortiz, A.L.; Aparicio-Ozores, G.; Valle-Rios, R.; Medina-Contreras, O.; Patiño-López, G.; Quezada, H. Proteomic changes in a childhood acute lymphoblastic leukemia cell line during the adaptation to vincristine. Bol. Med. Hosp. Infant. Mex. 2017, 74, 181–192. [Google Scholar]
- Risinger, A.L.; Giles, F.J.; Mooberry, S.L. Microtubule dynamics as a target in oncology. Cancer Treat. Rev. 2009, 35, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Verrills, N.M.; Liem, N.L.; Liaw, T.Y.; Hood, B.D.; Lock, R.B.; Kavallaris, M. Proteomic analysis reveals a novel role for the actin cytoskeleton in vincristine resistant childhood leukemia an in vivo study. Proteomics 2006, 6, 1681–1694. [Google Scholar] [CrossRef] [PubMed]
- Kemper, E.K.; Zhang, Y.; Dix, M.M.; Cravatt, B.F. Global profiling of phosphorylation-dependent changes in cysteine reactivity. Nat. Methods 2022, 19, 341–352. [Google Scholar] [CrossRef]
- Floyd, B.M.; Drew, K.; Marcotte, E.M. Systematic Identification of Protein Phosphorylation-Mediated Interactions. J. Proteome Res. 2021, 20, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- de Smith, A.J.; Ojha, J.; Francis, S.S.; Sanders, E.; Endicott, A.A.; Hansen, H.M.; Smirnov, I.; Termuhlen, A.M.; Walsh, K.M.; Metayer, C.; et al. Clonal and microclonal mutational heterogeneity in high hyperdiploid acute lymphoblastic leukemia. Oncotarget 2016, 7, 72733–72745. [Google Scholar] [CrossRef]
- Malinowska-Ozdowy, K.; Frech, C.; Schönegger, A.; Eckert, C.; Cazzaniga, G.; Stanulla, M.; zur Stadt, U.; Mecklenbräuker, A.; Schuster, M.; Kneidinger, D.; et al. KRAS and CREBBP mutations: A relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia 2015, 29, 1656–1667. [Google Scholar] [CrossRef]
- Siekmann, I.K.; Dierck, K.; Prall, S.; Klokow, M.; Strauss, J.; Buhs, S.; Wrzeszcz, A.; Bockmayr, M.; Beck, F.; Trochimiuk, M.; et al. Combined inhibition of receptor tyrosine and p21-activated kinases as a therapeutic strategy in childhood ALL. Blood Adv. 2018, 2, 2554–2567. [Google Scholar] [CrossRef]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef]
- Liu, X.; Qu, C.K. Protein Tyrosine Phosphatase SHP-2 (PTPN11) in Hematopoiesis and Leukemogenesis. J. Signal Transduct. 2011, 2011, 195239. [Google Scholar] [CrossRef]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch signaling pathway: Architecture, disease, and therapeutics. Signal Transduct Target Ther. 2022, 7, 95. [Google Scholar] [CrossRef]
- Yuan, X.; Wu, H.; Xu, H.; Xiong, H.; Chu, Q.; Yu, S.; Wu, G.S.; Wu, K. Notch signaling: An emerging therapeutic target for cancer treatment. Cancer Lett. 2015, 369, 20–27. [Google Scholar] [CrossRef] [PubMed]
- López-Nieva, P.; González-Sánchez, L.; Cobos-Fernández, M.Á.; Córdoba, R.; Santos, J.; Fernández-Piqueras, J. More Insights on the Use of γ-Secretase Inhibitors in Cancer Treatment. Oncologist 2021, 26, e298–e305. [Google Scholar] [CrossRef]
- Simioni, C.; Martelli, A.M.; Zauli, G.; Melloni, E.; Neri, L.M. Targeting mTOR in Acute Lymphoblastic Leukemia. Cells 2019, 8, 190. [Google Scholar] [CrossRef] [PubMed]
- Pinchinat, A.; Raetz, E. How do mTOR inhibitors fit in the landscape of treatment for relapsed acute lymphoblastic leukemia? Haematologica 2022, 107, 2292–2294. [Google Scholar] [CrossRef] [PubMed]
- Uzozie, A.C.; Ergin, E.K.; Rolf, N.; Tsui, J.; Lorentzian, A.; Weng, S.S.H.; Nierves, L.; Smith, T.G.; Lim, C.J.; Maxwell, C.A.; et al. PDX models reflect the proteome landscape of pediatric acute lymphoblastic leukemia but divert in select pathways. J. Exp. Clin. Cancer Res. 2021, 40, 96. [Google Scholar] [CrossRef]
- Yang, M.; Vesterlund, M.; Siavelis, I.; Moura-Castro, L.H.; Castor, A.; Fioretos, T.; Jafari, R.; Lilljebjörn, H.; Odom, D.T.; Olsson, L.; et al. Proteogenomics and Hi-C reveal transcriptional dysregulation in high hyperdiploid childhood acute lymphoblastic leukemia. Nat. Commun. 2019, 10, 1519. [Google Scholar] [CrossRef] [PubMed]
- Costa, O.; Schneider, P.; Coquet, L.; Chan, P.; Penther, D.; Legrand, E.; Jouenne, T.; Vasse, M.; Vannier, J.P. Proteomic profile of pre- B2 lymphoblasts from children with acute lymphoblastic leukemia (ALL) in relation with the translocation (12; 21). Clin. Proteom. 2014, 11, 31. [Google Scholar] [CrossRef]
- Serafin, V.; Capuzzo, G.; Milani, G.; Minuzzo, S.A.; Pinazza, M.; Bortolozzi, R.; Bresolin, S.; Porcù, E.; Frasson, C.; Indraccolo, S.; et al. Glucocorticoid resistance is reverted by LCK inhibition in pediatric T-cell acute lymphoblastic leukemia. Blood 2017, 130, 2750–2761. [Google Scholar] [CrossRef]
- Cordo’, V.; Meijer, M.T.; Hagelaar, R.; de Goeij-de Haas, R.R.; Poort, V.M.; Henneman, A.A.; Piersma, S.R.; Pham, T.V.; Oshima, K.; Ferrando, A.A.; et al. Phosphoproteomic profiling of T cell acute lymphoblastic leukemia reveals targetable kinases and combination treatment strategies. Nat. Commun. 2022, 13, 1048. [Google Scholar] [CrossRef]
- Beekhof, R.; van Alphen, C.; Henneman, A.A.; Knol, J.C.; Pham, T.V.; Rolfs, F.; Labots, M.; Henneberry, E.; Le Large, T.Y.; de Haas, R.R.; et al. INKA, an integrative data analysis pipeline for phosphoproteomic inference of active kinases. Mol. Syst. Biol. 2019, 15, e8250. [Google Scholar] [CrossRef]
- Lawton, M.L.; Emili, A. Mass Spectrometry-Based Phosphoproteomics and Systems Biology: Approaches to Study T Lymphocyte Activation and Exhaustion. J. Mol. Biol. 2021, 433, 167318. [Google Scholar] [CrossRef] [PubMed]
- Broto, G.E.; Corrêa, S.; Trigo, F.C.; Dos Santos, E.C.; Tomiotto-Pelissier, F.; Pavanelli, W.R.; Silveira, G.F.; Abdelhay, E.; Panis, C. Comparative Analysis of Systemic and Tumor Microenvironment Proteomes from Children with B-Cell Acute Lymphocytic Leukemia at Diagnosis and after Induction Treatment. Front. Oncol. 2020, 10, 550213. [Google Scholar] [CrossRef] [PubMed]
- Leo, I.R.; Aswad, L.; Stahl, M.; Kunold, E.; Post, F.; Erkers, T.; Struyf, N.; Mermelekas, G.; Joshi, R.N.; Gracia-Villacampa, E.; et al. Integrative multi-omics and drug response profiling of childhood acute lymphoblastic leukemia cell lines. Nat. Commun. 2022, 13, 1691. [Google Scholar] [CrossRef] [PubMed]
- Baytan, B.; Evim, M.S.; Güler, S.; Güneş, A.M.; Okan, M. Acute Central Nervous System Complications in Pediatric Acute Lymphoblastic Leukemia. Pediatr. Neurol. 2015, 53, 312–318. [Google Scholar] [CrossRef]
- Manley, S.; Keenan, R.; Campbell, H.; Caswell, M.; Pizer, B. No evidence for routine cerebrospinal fluid cytology in detecting asymptomatic central nervous system relapse in children with acute lymphoblastic leukaemia: 20 years’ experience of a UK primary treatment centre. Br. J. Haematol. 2014, 164, 462–464. [Google Scholar] [CrossRef]
- Thastrup, M.; Duguid, A.; Mirian, C.; Schmiegelow, K.; Halsey, C. Central nervous system involvement in childhood acute lymphoblastic leukemia: Challenges and solutions. Leukemia 2022, 36, 2751–2768. [Google Scholar] [CrossRef]
- Galicia, N.; Díez, P.; Dégano, R.M.; Guest, P.C.; Ibarrola, N.; Fuentes, M. Proteomic Biomarker Identification in Cerebrospinal Fluid for Leptomeningeal Metastases with Neurological Complications. Adv. Exp. Med. Biol. 2017, 974, 85–96. [Google Scholar]
- Roy, S.; Josephson, S.A.; Fridlyand, J.; Karch, J.; Kadoch, C.; Karrim, J.; Damon, L.; Treseler, P.; Kunwar, S.; Shuman, M.A.; et al. Protein biomarker identification in the CSF of patients with CNS lymphoma. J. Clin. Oncol. 2008, 26, 96–105. [Google Scholar] [CrossRef]
- Carbonara, K.; Andonovski, M.; Coorssen, J.R. Proteomes Are of Proteoforms: Embracing the Complexity. Proteomes 2021, 9, 38. [Google Scholar] [CrossRef]
- Priola, G.M.; Foster, M.W.; Deal, A.M.; Richardson, B.M.; Thompson, J.W.; Blatt, J. Cerebrospinal fluid proteomics in children during induction for acute lymphoblastic leukemia: A pilot study. Pediatr. Blood Cancer 2015, 62, 1190–1194. [Google Scholar] [CrossRef]
- Guo, L.; Ren, H.; Zeng, H.; Gong, Y.; Ma, X. Proteomic analysis of cerebrospinal fluid in pediatric acute lymphoblastic leukemia patients: A pilot study. OncoTargets Ther. 2019, 12, 3859–3868. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Ma, X.; Liu, X.; Zhou, R.; Zhao, Y.; Zhou, H. Altered CSF Proteomic Profiling of Paediatric Acute Lymphocytic Leukemia Patients with CNS Infiltration. J. Oncol. 2019, 2019, 3283629. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Author/Year/Country | Objective | Sample | Proteomic Approach | Main Results |
|---|---|---|---|---|
| Jiang N et al. [79] 2011 Singapore | Identify potential prognostic protein biomarkers. Discover promising regulators of PRED-induced apoptosis | Cell Line Bone marrow | 2-DE & MALDI-TOF | PCNA was highly predictive of PRED response in patients independent of molecular subtype. |
| Braoudaki M et al. [77] 2013 Greece | Evaluate differential expression in proteomic profiles of low risk- and high risk-ALL. Characterize candidate biomarkers related to diagnosis, prognosis and patient targeted therapy | Plasma Bone marrow | 2-DE & MALDI-TOF | CLUS, CERU, APOE, APOA4, APOA1, GELS, S10A9, AMBP, ACTB, CATA and AFAM proteins play a role in leukemia prognosis Vitronectin and plasminogen: contribute to leukemogenesis. Bicaudal D-related protein 1: biomarker for pediatric ALL therapeutics. |
| Wang D et al. [74] 2013 China | Compare differential proteins in lymphocytes between children with c-ALL and healthy children. Explore the mechanisms of c-ALL. Find new diagnostic and therapeutic strategies for c-ALL. | Bone marrow | SDS-PAGE MALDI-TOF | 15 proteins differentially expressed. 2 high expressions (Glutathione S-transferase P, PHB) 6 low expressions (PRDX4, 60s acidic ribosomal protein P0, Cytoplasmic actin,pyridoxine-5′-phosphate oxidase, Triosephosphate isomerase 1, FLJ26567) |
| Costa O et al. [98] 2014 France | Compare the lymphoblastes proteome in c-ALL in accordance with the presence of t(12;21) | Bone marrow | 2-DE nanoLC Ion trap MS/MS | Over-expression of CNN2, MAT-2β, hnRNPA2, PITPβ, PSMB2, HSPC263 (OTUB1). Under-expression: BUB3, hnRNPE2, PSMB6, CK2a |
| Cavalcante MS et al. [75] 2016 Brazil | Perform proteomic analysis of serum from pediatric patients with B-ALL Identify candidate biomarker proteins, for use in early diagnosis and evaluation of treatment. | Serum | FPLC nanoUPLC-ESI-MS/MS | Upregulation and candidate biomarkers for early diagnosis: Leucine-rich alpha-2-glycoprotein 1 (LRG1), Clusterin (CLU), thrombin (F2), heparin cofactor II (SERPIND1), alpha-2-macroglobulin (A2M), alpha-2-antiplasmin (SERPINF2), Alpha-1 antitrypsin (SERPINA1), Complement factor B (CFB) and Complement C3 (C3). |
| Xu G et al. [78] 2017 China | Figure out the critical altered proteins which can indicate the risk rank | Bone marrow | LC-ESI-MS/MS | 86 differently expressed proteins in the high-risk B-ALL 35 proteins have directive interactions |
| Guzmán-Ortiz AL et al. [81] 2017 Mexico | Identify changes in the proteome after adaptation to vincristine. | B-ALL cell line | MS ESI-MS/MS | 135 proteins exclusively expressed in the presence of vincristine—represent potential therapeutic targets (Toll receptor signaling pathway, Ras Pathway, B-T cell activation, CCKR signaling map cytokine-mediated signaling pathway, oxidative phosphorylation.) |
| Serafin V et al. [99] 2017 Italy | Identification of deregulated signaling pathways to point out new targeted approaches. | Bone marrow Cell lines Primary xenograft (PDX) cells | Reverse-phase protein arrays (RPPA) | Lymphocyte cell-specific protein-tyrosine kinase (LCK): aberrantly activated in PPR patients. Resistance to glucocorticoid treatment in pediatric T-ALL can be reversed by LCK inhibitors in vitro and in vivo. IL-4 overexpression contributes to LCK-induced glucocorticoid resistance. |
| Siekmann IK et al. [88] 2018 Germany Canada UK | Decipher signaling circuits that link RTK activity with biological output in vivo | Patient-derived xenograft ALL (PDX-ALL) models with dependencies on fms-like tyrosine kinase 3 (FLT3) and platelet-derived growth factor receptor b (PDGFRB) | Phosphoproteomics iTRAQ nano-LC MS/MS | Group I PAKs act as signaling hubs in RTK-dependent pathways in ALL. Inhibition of group I PAKs by FRAX486 augments the antileukemic efficacy of midostaurin in FLT3-driven ALL. |
| Calderon-Rodrıguez SI et al. [73] 2019 Colombia | Study of the plasma proteome of Colombian children diagnosed with B-cell ALL (B-ALL) to determine potential disease markers that reflect processes altered by the presence of the disease or in response to it. | Plasma | Nano-LC-MS/MS | 25 proteins were differentially expressed in B-ALL Potential biomarkers that could be used to differentiate unhealthy patients from healthy individuals |
| Yang M et al. [97] 2019 Sweden Germany UK | Proteogenomic analysis of a series of pediatric BCP-ALL, including high hyperdiploid and diploid/near-diploid ETV6/RUNX1-positive cases, aiming to determine the effects of aneuploidy. | Bone marrow | HiRIEF LC-MS/MS | Proteins differentially expressed between hyperdiploid and ETV6/RUNX1-positive leukemia had higher mRNA-protein correlations. CTCF and cohesin: low expression in hyperdiploid ALL |
| Broto GE et al. [103] 2020 Brazil | Comparing bone marrow and peripheral blood profiles, before chemotherapy—at diagnosis, and the cumulative effects found after the induction treatment | Bone marrow Peripheral blood B-ALL | Nano-LC-MS/MS | D0, PB characterized as a pro-inflammatory environment, with the involvement of several down-regulated. Coagulation proteins as KNG, plasmin, and plasminogen. D28 characterized by immune response-related processes and the super expression of the transcription factor IRF3 and transthyretin. RUNX1 found in both D0 and D28. IRF3-IFN γ axis induction as a possible mechanism enrolled in disease resolution, transthyretin as an up-regulated protein induced by the induction phase of chemotherapy. |
| Uzozie A et al. [96] 2021 Canada | Reveal proteome signatures characteristic of leukemic subtypes. Explore how effectively PDX models replicate the primary leukemic proteome. | Cell lines Bone marrow Peripheral blood PDXs | LC-MSMS | Proteome differentiates pediatric B-ALL and T-ALL from non-leukemic cells. The proteome of xenograft ALL closely resemble matched patient proteome. Relapse specific changes in patients are retained in PDXs. Proliferation and immune response processes differ between PDXs and patients. Xenografts recapitulate proteome response to structural genomic changes in patients. |
| Cordo V et al. [100] 2022 Netherlands | Provide information on pathway activation and signaling networks that offer opportunities for targeted therapy. | T-cell ALL lines PDXs | Reverse phase protein array (RPPA) | 21,000 phosphosites on 4896 phosphoproteins, including 217 kinases. Validate putative targets for therapy ex vivo and identify potential combination treatments, such as the inhibition of the INSR/IGF-1R axis to increase the sensitivity to dasatinib treatment. |
| Yu R et al. [76] 2022 China | Screen serum autoantibodies of pediatric B-ALL, aiming to contribute to the early detection of B-ALL in children. | Pooled B-ALL cell lines (NALM-6, REH and BALL-1 cells) | Serological proteome analysis (SERPA) | α-enolase and VDAC1 were identified as candidate autoantigens in children with B-ALL and potential serological markers. |
| Leo IR et al. [104] 2022 Sweden | Perform comprehensive multi-omic analyses using proteomics, transcriptomics, and pharmacoproteomic characterization | Childhood ALL cell lines | LC-MS | In-depth proteomic analysis of childhood ALL cell lines covering numerous cytogenetic subtypes quantifying more than 12,000 proteins and 19,000 protein-coding transcripts as well as sensitivity to 528 oncology and investigational drugs. Identify the diacylglycerol-analog bryostatin-1 as a therapeutic candidate in the MEF2D-HNRNPUL1 fusion high-risk subtype ALL |
| Author/Year/Country | Proteomic Approach | Main Results |
|---|---|---|
| Priola GM et al. [111] 2015 USA | LC-MS/MS | Protein C Inhibitor (SERPINA5) and heparin cofactor II (SERPIND1) changed over the course of therapy. Antithrombin III (ATIII) and plasminogen (PLMN) levels decreased expression over time in one child with CNS thrombosis. |
| Guo L et al. [112] 2019 China | LC-MS/MS | 51 proteins identified, 32 up-regulated and 19 down-regulated including TIMP1, LGALS3BP, A2M, AHSG, FN1, HRG,ITIH4, CF I, C2, C4A. |
| Mo F et al. [113] 2019 China | LC-MS/MS | 428 unique proteins identified, 10 altered proteins during treatment |
| Yu Q et al. [71] 2020 USA | 4-plex N,N dimethyl leucine (DiLeu) isobaric labeling strategy and MS | 63 proteins were significantly altered |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kourti, M.; Aivaliotis, M.; Hatzipantelis, E. Proteomics in Childhood Acute Lymphoblastic Leukemia: Challenges and Opportunities. Diagnostics 2023, 13, 2748. https://doi.org/10.3390/diagnostics13172748
Kourti M, Aivaliotis M, Hatzipantelis E. Proteomics in Childhood Acute Lymphoblastic Leukemia: Challenges and Opportunities. Diagnostics. 2023; 13(17):2748. https://doi.org/10.3390/diagnostics13172748
Chicago/Turabian StyleKourti, Maria, Michalis Aivaliotis, and Emmanouel Hatzipantelis. 2023. "Proteomics in Childhood Acute Lymphoblastic Leukemia: Challenges and Opportunities" Diagnostics 13, no. 17: 2748. https://doi.org/10.3390/diagnostics13172748