
Hodgkin Lymphoma: Biology and Differential Diagnostic Problem


Abstract
:1. Introduction
2. Classical Hodgkin Lymphoma
2.1. Epidemiology
2.2. Clinical Features
2.3. Histological Feature
2.3.1. NSCHL
2.3.2. MCCHL
2.3.3. LDCHL
2.3.4. LRCHL
2.4. Immunophenotype
2.5. Cellular Origin of Hodgkin Lymphoma
2.6. Genetic Alterations
2.7. EBV Infection
2.8. Microenvironment
3. Nodular Lymphocyte-Predominant Hodgkin Lymphoma (NLPHL)
3.1. Epidemiology and Clinical Features
3.2. Histology and Immunophenotype
3.3. Cellular Origin and Molecular Biology
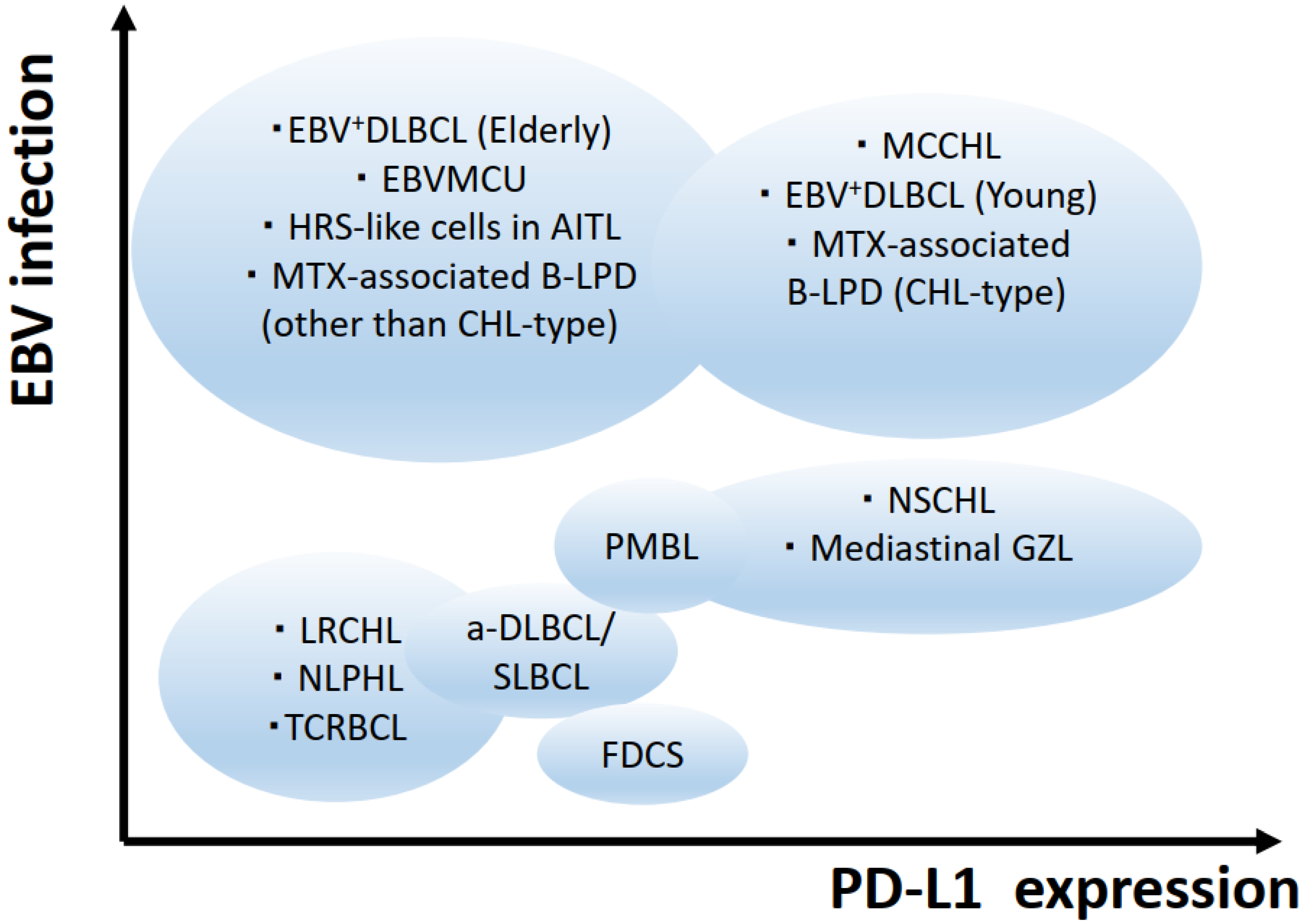
4. Differential Diagnosis of Hodgkin Lymphomas
4.1. Differential Diagnosis of CHLs
4.1.1. Primary Mediastinal Large B-Cell Lymphoma and B-Cell Lymphoma Unclassifiable, with Features Intermediate between DLBCL and CHL (Gray Zone Lymphoma)
4.1.2. DLBCL and Other B-Cell LPD with “HRS-like” Cells
4.1.3. Anaplastic Large Cell Lymphoma, ALK-Positive and -Negative
4.1.4. T-Cell Lymphomas with “HRS-like” Cells
4.1.5. Dendritic Cell Neoplasms
4.2. Differential Diagnosis of NLPHL
5. Future Perspectives
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hodgkin, T. On some Morbid Appearances of the Absorbent Glands and Spleen. Med. Chir. Trans. 1832, 17, 68–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, N.L.; Jaffe, E.S.; Stein, H.; Banks, P.M.; Chan, J.K.; Cleary, M.L.; Delsol, G.; De Wolf-Peeters, C.; Falini, B.; Gatter, K.C.; et al. A revised European-American classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood 1994, 84, 1361–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A Report from the Clinical Advisory Committee. Blood 2022. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 2. [Google Scholar]
- Saito, E.; Matsuoka, J. International comparison of Hodgkin and non-Hodgkin lymphoma incidence. Jpn. J. Clin. Oncol. 2020, 50, 96–97. [Google Scholar] [CrossRef]
- National Cancer Institute Surveillance Epidemiology and End Results Program. Cancer Stat Facts: Hodgkin Lymphoma. In SEER; 2019. Available online: https://seer.cancer.gov/statfacts/html/hodg.html (accessed on 19 June 2022).
- Connors, J.M.; Cozen, W.; Steidl, C.; Carbone, A.; Hoppe, R.T.; Flechtner, H.H.; Bartlett, N.L. Hodgkin lymphoma. Nat. Rev. Dis. Primers 2020, 6, 61. [Google Scholar] [CrossRef]
- Cozen, W.; Katz, J.; Mack, T.M. Risk patterns of Hodgkin’s disease in Los Angeles vary by cell type. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 1992, 1, 261–268. [Google Scholar]
- Correa, P.; O’Conor, G.T. Epidemiologic patterns of Hodgkin’s disease. Int. J. Cancer 1971, 8, 192–201. [Google Scholar] [CrossRef]
- Cozen, W.; Hamilton, A.S.; Zhao, P.; Salam, M.T.; Deapen, D.M.; Nathwani, B.N.; Weiss, L.M.; Mack, T.M. A protective role for early oral exposures in the etiology of young adult Hodgkin lymphoma. Blood 2009, 114, 4014–4020. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, M.; Hayward, A.; Warren-Gash, C.; Denaxas, S.; Gonzalez-Izquierdo, A.; Lyratzopoulos, G.; Thomas, S. Allergic disease, corticosteroid use, and risk of Hodgkin lymphoma: A United Kingdom nationwide case-control study. J. Allergy Clin. Immunol. 2020, 145, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Campos, A.; Moreira, A.; Ribeiro, K.B.; Paes, R.P.; Zerbini, M.C.; Aldred, V.; de Souza, C.A.; Neto, C.S.; Soares, F.A.; Vassallo, J. Frequency of EBV associated classical Hodgkin lymphoma decreases over a 54-year period in a Brazilian population. Sci. Rep. 2018, 8, 1849. [Google Scholar] [CrossRef] [Green Version]
- Shimabukuro-Vornhagen, A.; Haverkamp, H.; Engert, A.; Balleisen, L.; Majunke, P.; Heil, G.; Eich, H.T.; Stein, H.; Diehl, V.; Josting, A. Lymphocyte-rich classical Hodgkin’s lymphoma: Clinical presentation and treatment outcome in 100 patients treated within German Hodgkin’s Study Group trials. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 5739–5745. [Google Scholar] [CrossRef] [PubMed]
- Smithers, D.W.; Lillicrap, S.C.; Barnes, A. Patterns of lymph node involvement in relation to hypotheses about the modes of spread of Hodgkin’s disease. Cancer 1974, 34, 1779–1786. [Google Scholar] [CrossRef]
- Klimm, B.; Franklin, J.; Stein, H.; Eichenauer, D.A.; Haverkamp, H.; Diehl, V.; Fuchs, M.; Borchmann, P.; Engert, A. Lymphocyte-depleted classical Hodgkin’s lymphoma: A comprehensive analysis from the German Hodgkin study group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 3914–3920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.; Olszewski, A.J. Disparate survival and risk of secondary non-Hodgkin lymphoma in histologic subtypes of Hodgkin lymphoma: A population-based study. Leuk. Lymphoma 2014, 55, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Parente, P.; Zanelli, M.; Sanguedolce, F.; Mastracci, L.; Graziano, P. Hodgkin Reed-Sternberg-Like Cells in Non-Hodgkin Lymphoma. Diagnostics 2020, 10, 1019. [Google Scholar] [CrossRef]
- von Wasielewski, S.; Franklin, J.; Fischer, R.; Hubner, K.; Hansmann, M.L.; Diehl, V.; Georgii, A.; von Wasielewski, R. Nodular sclerosing Hodgkin disease: New grading predicts prognosis in intermediate and advanced stages. Blood 2003, 101, 4063–4069. [Google Scholar] [CrossRef] [Green Version]
- Drakos, E.; Rassidakis, G.Z.; Leventaki, V.; Cotta, C.V.; Vega, F.; Medeiros, L.J. Nodular lymphocyte predominant Hodgkin lymphoma with clusters of LP Cells, acute inflammation, and fibrosis: A syncytial variant. Am. J. Surg. Pathol. 2009, 33, 1725–1731. [Google Scholar] [CrossRef]
- Zhang, Q.; Kim, D.H.; Xu, Y.; Wang, W.; Medeiros, L.J. Clinicopathological features of syncytial variant nodular sclerosis Hodgkin lymphoma. Hum. Pathol. 2022, 119, 105–113. [Google Scholar] [CrossRef]
- Haybittle, J.L.; Hayhoe, F.G.; Easterling, M.J.; Jelliffe, A.M.; Bennett, M.H.; Vaughan Hudson, G.; Vaughan Hudson, B.; MacLennan, K.A. Review of British National Lymphoma Investigation studies of Hodgkin’s disease and development of prognostic index. Lancet 1985, 1, 967–972. [Google Scholar] [CrossRef]
- MacLennan, K.A.; Bennett, M.H.; Tu, A.; Hudson, B.V.; Easterling, M.J.; Hudson, G.V.; Jelliffe, A.M. Relationship of histopathologic features to survival and relapse in nodular sclerosing Hodgkin’s disease. A study of 1659 patients. Cancer 1989, 64, 1686–1693. [Google Scholar] [CrossRef]
- Delsol, G.; Brousset, P.; Chittal, S.; Rigal-Huguet, F. Correlation of the expression of Epstein–Barr virus latent membrane protein and in situ hybridization with biotinylated BamHI-W probes in Hodgkin’s disease. Am. J. Pathol. 1992, 140, 247–253. [Google Scholar] [PubMed]
- Mani, H.; Jaffe, E.S. Hodgkin lymphoma: An update on its biology with new insights into classification. Clin. Lymphoma Myeloma 2009, 9, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.P.; Byrne, G.E.; Jones, S.E. Mistaken clinical and pathologic diagnoses of Hodgkin’s disease: A Southwest oncology group study. Cancer Treat. Rep. 1982, 66, 645–651. [Google Scholar] [PubMed]
- Lukes, R.J.; Butler, J.J. The pathology and nomenclature of Hodgkin’s disease. Cancer Res. 1966, 26, 1063–1083. [Google Scholar]
- Slack, G.W.; Ferry, J.A.; Hasserjian, R.P.; Sohani, A.R.; Longtine, J.A.; Harris, N.L.; Zukerberg, L.R. Lymphocyte depleted Hodgkin lymphoma: An evaluation with immunophenotyping and genetic analysis. Leuk. Lymphoma 2009, 50, 937–943. [Google Scholar] [CrossRef]
- Diehl, V.; Sextro, M.; Franklin, J.; Hansmann, M.L.; Harris, N.; Jaffe, E.; Poppema, S.; Harris, M.; Franssila, K.; van Krieken, J.; et al. Clinical presentation, course, and prognostic factors in lymphocyte-predominant Hodgkin’s disease and lymphocyte-rich classical Hodgkin’s disease: Report from the European Task Force on Lymphoma Project on Lymphocyte-Predominant Hodgkin’s Disease. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1999, 17, 776–783. [Google Scholar] [CrossRef]
- Anagnostopoulos, I.; Hansmann, M.L.; Franssila, K.; Harris, M.; Harris, N.L.; Jaffe, E.S.; Han, J.; van Krieken, J.M.; Poppema, S.; Marafioti, T.; et al. European Task Force on Lymphoma project on lymphocyte predominance Hodgkin disease: Histologic and immunohistologic analysis of submitted cases reveals 2 types of Hodgkin disease with a nodular growth pattern and abundant lymphocytes. Blood 2000, 96, 1889–1899. [Google Scholar]
- Durkop, H.; Latza, U.; Hummel, M.; Eitelbach, F.; Seed, B.; Stein, H. Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin’s disease. Cell 1992, 68, 421–427. [Google Scholar] [CrossRef]
- Kuze, T.; Nakamura, N.; Hashimoto, Y.; Sasaki, Y.; Abe, M. The characteristics of Epstein–Barr virus (EBV)-positive diffuse large B-cell lymphoma: Comparison between EBV(+) and EBV(−) cases in Japanese population. Jpn. J. Cancer Res. Gann 2000, 91, 1233–1240. [Google Scholar] [CrossRef]
- Nakamura, S.; Takagi, N.; Kojima, M.; Motoori, T.; Kitoh, K.; Osada, H.; Suzuki, H.; Ogura, M.; Kurita, S.; Oyama, A.; et al. Clinicopathologic study of large cell anaplastic lymphoma (Ki-1-positive large cell lymphoma) among the Japanese. Cancer 1991, 68, 118–129. [Google Scholar] [CrossRef]
- Hu, S.; Xu-Monette, Z.Y.; Balasubramanyam, A.; Manyam, G.C.; Visco, C.; Tzankov, A.; Liu, W.M.; Miranda, R.N.; Zhang, L.; Montes-Moreno, S.; et al. CD30 expression defines a novel subgroup of diffuse large B-cell lymphoma with favorable prognosis and distinct gene expression signature: A report from the International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2013, 121, 2715–2724. [Google Scholar] [CrossRef] [PubMed]
- von Wasielewski, R.; Mengel, M.; Fischer, R.; Hansmann, M.L.; Hubner, K.; Franklin, J.; Tesch, H.; Paulus, U.; Werner, M.; Diehl, V.; et al. Classical Hodgkin’s disease. Clinical impact of the immunophenotype. Am. J. Pathol. 1997, 151, 1123–1130. [Google Scholar] [PubMed]
- Carbone, A.; Gloghini, A.; Carlo-Stella, C. Are EBV-related and EBV-unrelated Hodgkin lymphomas different with regard to susceptibility to checkpoint blockade? Blood 2018, 132, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Panjwani, P.K.; Charu, V.; DeLisser, M.; Molina-Kirsch, H.; Natkunam, Y.; Zhao, S. Programmed death-1 ligands PD-L1 and PD-L2 show distinctive and restricted patterns of expression in lymphoma subtypes. Hum. Pathol. 2018, 71, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [Green Version]
- Gerhard-Hartmann, E.; Goergen, H.; Brockelmann, P.J.; Mottok, A.; Steinmuller, T.; Grund, J.; Zamo, A.; Ben-Neriah, S.; Sasse, S.; Borchmann, S.; et al. 9p24.1 alterations and programmed cell death 1 ligand 1 expression in early stage unfavourable classical Hodgkin lymphoma: An analysis from the German Hodgkin Study Group NIVAHL trial. Br. J. Haematol. 2022, 196, 116–126. [Google Scholar] [CrossRef]
- Volaric, A.; Bacchi, C.E.; Gru, A.A. PD-1 and PD-L1 Immunohistochemistry as a Diagnostic Tool for Classic Hodgkin Lymphoma in Small-volume Biopsies. Am. J. Surg. Pathol. 2020, 44, 1353–1366. [Google Scholar] [CrossRef]
- Venkataraman, G.; Song, J.Y.; Tzankov, A.; Dirnhofer, S.; Heinze, G.; Kohl, M.; Traverse-Glehen, A.; Eberle, F.C.; Hanson, J.C.; Raffeld, M.A.; et al. Aberrant T-cell antigen expression in classical Hodgkin lymphoma is associated with decreased event-free survival and overall survival. Blood 2013, 121, 1795–1804. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar] [CrossRef]
- Sakakibara, A.; Kohno, K.; Eladl, A.E.; Klaisuwan, T.; Ishikawa, E.; Suzuki, Y.; Shimada, S.; Nakaguro, M.; Shimoyama, Y.; Takahara, T.; et al. Immunohistochemical assessment of the diagnostic utility of PD-L1: A preliminary analysis of anti-PD-L1 antibody (SP142) for lymphoproliferative diseases with tumour and non-malignant Hodgkin-Reed-Sternberg (HRS)-like cells. Histopathology 2018, 72, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Marshall, A.; Chavez, E.A.; Miyata-Takata, T.; Ben-Neriah, S.; Unrau, D.; Telenius, A.; et al. Single-cell profiling reveals the importance of CXCL13/CXCR5 axis biology in lymphocyte-rich classic Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2021, 118, e2105822118. [Google Scholar] [CrossRef] [PubMed]
- Schwering, I.; Brauninger, A.; Klein, U.; Jungnickel, B.; Tinguely, M.; Diehl, V.; Hansmann, M.L.; Dalla-Favera, R.; Rajewsky, K.; Kuppers, R. Loss of the B-lineage-specific gene expression program in Hodgkin and Reed-Sternberg cells of Hodgkin lymphoma. Blood 2003, 101, 1505–1512. [Google Scholar] [CrossRef]
- Hertel, C.B.; Zhou, X.G.; Hamilton-Dutoit, S.J.; Junker, S. Loss of B cell identity correlates with loss of B cell-specific transcription factors in Hodgkin/Reed-Sternberg cells of classical Hodgkin lymphoma. Oncogene 2002, 21, 4908–4920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, K.C.; Higgins, J.P.; Montgomery, K.; Kaygusuz, G.; van de Rijn, M.; Natkunam, Y. The utility of PAX5 immunohistochemistry in the diagnosis of undifferentiated malignant neoplasms. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2007, 20, 871–877. [Google Scholar] [CrossRef]
- Elsayed, A.A.; Asano, N.; Ohshima, K.; Izutsu, K.; Kinoshita, T.; Nakamura, S. Prognostic significance of CD20 expression and Epstein–Barr virus (EBV) association in classical Hodgkin lymphoma in Japan: A clinicopathologic study. Pathol. Int. 2014, 64, 336–345. [Google Scholar] [CrossRef]
- Tzankov, A.; Zimpfer, A.; Pehrs, A.C.; Lugli, A.; Went, P.; Maurer, R.; Pileri, S.; Dirnhofer, S. Expression of B-cell markers in classical hodgkin lymphoma: A tissue microarray analysis of 330 cases. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2003, 16, 1141–1147. [Google Scholar] [CrossRef] [Green Version]
- Vassallo, J.; Metze, K.; Traina, F.; de Souza, C.A.; Lorand-Metze, I. Further remarks on the expression of CD20 in classical Hodgkin’s lymphomas. Haematologica 2002, 87, ELT17. [Google Scholar]
- Carbone, A.; Gloghini, A.; Aldinucci, D.; Gattei, V.; Dalla-Favera, R.; Gaidano, G. Expression pattern of MUM1/IRF4 in the spectrum of pathology of Hodgkin’s disease. Br. J. Haematol. 2002, 117, 366–372. [Google Scholar] [CrossRef]
- Bai, M.; Panoulas, V.; Papoudou-Bai, A.; Horianopoulos, N.; Kitsoulis, P.; Stefanaki, K.; Rontogianni, D.; Agnantis, N.J.; Kanavaros, P. B-cell differentiation immunophenotypes in classical Hodgkin lymphomas. Leuk. Lymphoma 2006, 47, 495–501. [Google Scholar] [CrossRef]
- Marafioti, T.; Hummel, M.; Foss, H.D.; Laumen, H.; Korbjuhn, P.; Anagnostopoulos, I.; Lammert, H.; Demel, G.; Theil, J.; Wirth, T.; et al. Hodgkin and reed-sternberg cells represent an expansion of a single clone originating from a germinal center B-cell with functional immunoglobulin gene rearrangements but defective immunoglobulin transcription. Blood 2000, 95, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Kanzler, H.; Kuppers, R.; Hansmann, M.L.; Rajewsky, K. Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J. Exp. Med. 1996, 184, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Xu, Y.; Puthalakath, H.; Light, A.; Willis, S.N.; Strasser, A.; Tarlinton, D.M. CD45 links the B cell receptor with cell survival and is required for the persistence of germinal centers. Nat. Immunol. 2006, 7, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Kinoshita, T.; Tamaru, J.; Ohshima, K.; Yoshino, T.; Niitsu, N.; Tsukamoto, N.; Hirabayashi, K.; Izutsu, K.; Taniwaki, M.; et al. Cytotoxic molecule-positive classical Hodgkin’s lymphoma: A clinicopathological comparison with cytotoxic molecule-positive peripheral T-cell lymphoma of not otherwise specified type. Haematologica 2011, 96, 1636–1643. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [Green Version]
- Van Roosbroeck, K.; Ferreiro, J.F.; Tousseyn, T.; van der Krogt, J.A.; Michaux, L.; Pienkowska-Grela, B.; Theate, I.; De Paepe, P.; Dierickx, D.; Doyen, C.; et al. Genomic alterations of the JAK2 and PDL loci occur in a broad spectrum of lymphoid malignancies. Genes Chromosomes Cancer 2016, 55, 428–441. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Abdul Razak, F.R.; Terpstra, M.; Chan, F.C.; Saber, A.; Nijland, M.; van Imhoff, G.; Visser, L.; Gascoyne, R.; Steidl, C.; et al. The mutational landscape of Hodgkin lymphoma cell lines determined by whole-exome sequencing. Leukemia 2014, 28, 2248–2251. [Google Scholar] [CrossRef]
- Wienand, K.; Chapuy, B.; Stewart, C.; Dunford, A.J.; Wu, D.; Kim, J.; Kamburov, A.; Wood, T.R.; Cader, F.Z.; Ducar, M.D.; et al. Genomic analyses of flow-sorted Hodgkin Reed-Sternberg cells reveal complementary mechanisms of immune evasion. Blood Adv. 2019, 3, 4065–4080. [Google Scholar] [CrossRef] [Green Version]
- Tiacci, E.; Ladewig, E.; Schiavoni, G.; Penson, A.; Fortini, E.; Pettirossi, V.; Wang, Y.; Rosseto, A.; Venanzi, A.; Vlasevska, S.; et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood 2018, 131, 2454–2465. [Google Scholar] [CrossRef] [Green Version]
- Joos, S.; Menz, C.K.; Wrobel, G.; Siebert, R.; Gesk, S.; Ohl, S.; Mechtersheimer, G.; Trumper, L.; Moller, P.; Lichter, P.; et al. Classical Hodgkin lymphoma is characterized by recurrent copy number gains of the short arm of chromosome 2. Blood 2002, 99, 1381–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, R.; Hansmann, M.L.; Bohle, V.; Martin-Subero, J.I.; Hartmann, S.; Mechtersheimer, G.; Klapper, W.; Vater, I.; Giefing, M.; Gesk, S.; et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J. Exp. Med. 2009, 206, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desch, A.K.; Hartung, K.; Botzen, A.; Brobeil, A.; Rummel, M.; Kurch, L.; Georgi, T.; Jox, T.; Bielack, S.; Burdach, S.; et al. Genotyping circulating tumor DNA of pediatric Hodgkin lymphoma. Leukemia 2020, 34, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Havranek, O.; Xu, J.; Kohrer, S.; Wang, Z.; Becker, L.; Comer, J.M.; Henderson, J.; Ma, W.; Man Chun Ma, J.; Westin, J.R.; et al. Tonic B-cell receptor signaling in diffuse large B-cell lymphoma. Blood 2017, 130, 995–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechtel, D.; Kurth, J.; Unkel, C.; Kuppers, R. Transformation of BCR-deficient germinal-center B cells by EBV supports a major role of the virus in the pathogenesis of Hodgkin and posttransplantation lymphomas. Blood 2005, 106, 4345–4350. [Google Scholar] [CrossRef]
- Weniger, M.A.; Melzner, I.; Menz, C.K.; Wegener, S.; Bucur, A.J.; Dorsch, K.; Mattfeldt, T.; Barth, T.F.; Moller, P. Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho-STAT5 accumulation. Oncogene 2006, 25, 2679–2684. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, Y.; Yamamoto, N.; Dewan, M.Z.; Sugimoto, H.; Martinez Bruyn, V.J.; Iwasaki, Y.; Matsubara, K.; Qi, X.; Saitoh, T.; Imoto, I.; et al. Overexpressed NF-kappaB-inducing kinase contributes to the tumorigenesis of adult T-cell leukemia and Hodgkin Reed-Sternberg cells. Blood 2008, 111, 5118–5129. [Google Scholar] [CrossRef] [Green Version]
- Otto, C.; Giefing, M.; Massow, A.; Vater, I.; Gesk, S.; Schlesner, M.; Richter, J.; Klapper, W.; Hansmann, M.L.; Siebert, R.; et al. Genetic lesions of the TRAF3 and MAP3K14 genes in classical Hodgkin lymphoma. Br. J. Haematol. 2012, 157, 702–708. [Google Scholar] [CrossRef]
- Martin-Subero, J.I.; Wlodarska, I.; Bastard, C.; Picquenot, J.M.; Hoppner, J.; Giefing, M.; Klapper, W.; Siebert, R. Chromosomal rearrangements involving the BCL3 locus are recurrent in classical Hodgkin and peripheral T-cell lymphoma. Blood 2006, 108, 401–402. [Google Scholar] [CrossRef]
- Hartmann, S.; Schuhmacher, B.; Rausch, T.; Fuller, L.; Doring, C.; Weniger, M.; Lollies, A.; Weiser, C.; Thurner, L.; Rengstl, B.; et al. Highly recurrent mutations of SGK1, DUSP2 and JUNB in nodular lymphocyte predominant Hodgkin lymphoma. Leukemia 2016, 30, 844–853. [Google Scholar] [CrossRef]
- Wlodarska, I.; Stul, M.; De Wolf-Peeters, C.; Hagemeijer, A. Heterogeneity of BCL6 rearrangements in nodular lymphocyte predominant Hodgkin’s lymphoma. Haematologica 2004, 89, 965–972. [Google Scholar] [PubMed]
- Wlodarska, I.; Nooyen, P.; Maes, B.; Martin-Subero, J.I.; Siebert, R.; Pauwels, P.; De Wolf-Peeters, C.; Hagemeijer, A. Frequent occurrence of BCL6 rearrangements in nodular lymphocyte predominance Hodgkin lymphoma but not in classical Hodgkin lymphoma. Blood 2003, 101, 706–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, C.; Hoeller, S.; Bourgau, C.; Hirschmann, P.; Schwaller, J.; Went, P.; Pileri, S.A.; Reiter, A.; Dirnhofer, S.; Tzankov, A. Recurrent numerical aberrations of JAK2 and deregulation of the JAK2-STAT cascade in lymphomas. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2009, 22, 476–487. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.C.; Khen, N.T.; Jones, D.; Su, I.J. Epstein–Barr virus is associated with all histological subtypes of Hodgkin lymphoma in Vietnamese children with special emphasis on the entity of lymphocyte predominance subtype. Hum. Pathol. 2005, 36, 747–755. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, Y.; Choi, J.W.; Kim, Y.S. Prevalence and prognostic significance of Epstein–Barr virus infection in classical Hodgkin’s lymphoma: A meta-analysis. Arch. Med. Res. 2014, 45, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Asano, N. Clinicopathological Features of Nodular Sclerosis-Type Classical Hodgkin Lymphoma In the Elderly: Multicenter Study of Hodgkin Lymphoma In Japan. Blood 2010, 116, 2677. [Google Scholar] [CrossRef]
- Huen, D.S.; Henderson, S.A.; Croom-Carter, D.; Rowe, M. The Epstein–Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene 1995, 10, 549–560. [Google Scholar]
- Mancao, C.; Hammerschmidt, W. Epstein–Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood 2007, 110, 3715–3721. [Google Scholar] [CrossRef] [Green Version]
- Vockerodt, M.; Wei, W.; Nagy, E.; Prouzova, Z.; Schrader, A.; Kube, D.; Rowe, M.; Woodman, C.B.; Murray, P.G. Suppression of the LMP2A target gene, EGR-1, protects Hodgkin’s lymphoma cells from entry to the EBV lytic cycle. J. Pathol. 2013, 230, 399–409. [Google Scholar] [CrossRef]
- Vrzalikova, K.; Ibrahim, M.; Nagy, E.; Vockerodt, M.; Perry, T.; Wei, W.; Woodman, C.; Murray, P. Co-Expression of the Epstein–Barr Virus-Encoded Latent Membrane Proteins and the Pathogenesis of Classic Hodgkin Lymphoma. Cancers 2018, 10, 285. [Google Scholar] [CrossRef] [Green Version]
- Cader, F.Z.; Schackmann, R.C.J.; Hu, X.; Wienand, K.; Redd, R.; Chapuy, B.; Ouyang, J.; Paul, N.; Gjini, E.; Lipschitz, M.; et al. Mass cytometry of Hodgkin lymphoma reveals a CD4(+) regulatory T-cell-rich and exhausted T-effector microenvironment. Blood 2018, 132, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.B.; Challoner, P.B.; Neiman, P.E.; Groudine, M. Expression of the c-myb proto-oncogene during cellular proliferation. Nature 1986, 319, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Nishikori, M.; Kitawaki, T.; Sakai, T.; Hishizawa, M.; Tashima, M.; Kondo, T.; Ohmori, K.; Kurata, M.; Hayashi, T.; et al. PD-1-PD-1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood 2008, 111, 3220–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wein, F.; Kuppers, R. The role of T cells in the microenvironment of Hodgkin lymphoma. J. Leukoc. Biol. 2016, 99, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldman, J.; Visser, L.; Huberts-Kregel, M.; Muller, N.; Hepkema, B.; van den Berg, A.; Diepstra, A. Rosetting T cells in Hodgkin lymphoma are activated by immunological synapse components HLA class II and CD58. Blood 2020, 136, 2437–2441. [Google Scholar] [CrossRef]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Hav, M.; Colombo, A.; Chavez, E.A.; Nissen, M.; Wang, X.; Miyata-Takata, T.; et al. Single-Cell Transcriptome Analysis Reveals Disease-Defining T-cell Subsets in the Tumor Microenvironment of Classic Hodgkin Lymphoma. Cancer Discov. 2020, 10, 406–421. [Google Scholar] [CrossRef] [Green Version]
- Le, K.S.; Ame-Thomas, P.; Tarte, K.; Gondois-Rey, F.; Granjeaud, S.; Orlanducci, F.; Foucher, E.D.; Broussais, F.; Bouabdallah, R.; Fest, T.; et al. CXCR5 and ICOS expression identifies a CD8 T-cell subset with TFH features in Hodgkin lymphomas. Blood Adv. 2018, 2, 1889–1900. [Google Scholar] [CrossRef]
- Menendez, V.; Solorzano, J.L.; Fernandez, S.; Montalban, C.; Garcia, J.F. The Hodgkin Lymphoma Immune Microenvironment: Turning Bad News into Good. Cancers 2022, 14, 1360. [Google Scholar] [CrossRef]
- Carey, C.D.; Gusenleitner, D.; Lipschitz, M.; Roemer, M.G.M.; Stack, E.C.; Gjini, E.; Hu, X.; Redd, R.; Freeman, G.J.; Neuberg, D.; et al. Topological analysis reveals a PD-L1-associated microenvironmental niche for Reed-Sternberg cells in Hodgkin lymphoma. Blood 2017, 130, 2420–2430. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrarini, I.; Rigo, A.; Visco, C.; Krampera, M.; Vinante, F. The Evolving Knowledge on T and NK Cells in Classic Hodgkin Lymphoma: Insights into Novel Subsets Populating the Immune Microenvironment. Cancers 2020, 12, 3757. [Google Scholar] [CrossRef] [PubMed]
- Morton, L.M.; Wang, S.S.; Devesa, S.S.; Hartge, P.; Weisenburger, D.D.; Linet, M.S. Lymphoma incidence patterns by WHO subtype in the United States, 1992–2001. Blood 2006, 107, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Nogova, L.; Reineke, T.; Josting, A.; Muller-Hermelink, H.K.; Eich, H.T.; Behringer, K.; Muller, R.P.; Diehl, V.; Engert, A. Lymphocyte-predominant and classical Hodgkin’s lymphoma--comparison of outcomes. Eur. J. Haematology. Suppl. 2005, 75, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Lazarovici, J.; Dartigues, P.; Brice, P.; Oberic, L.; Gaillard, I.; Hunault-Berger, M.; Broussais-Guillaumot, F.; Gyan, E.; Bologna, S.; Nicolas-Virelizier, E.; et al. Nodular lymphocyte predominant Hodgkin lymphoma: A Lymphoma Study Association retrospective study. Haematologica 2015, 100, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Agbay, R.; Loghavi, S.; Zuo, Z.; Fayad, L.; Dabaja, B.; Medeiros, L.J.; Khoury, J.D. Bone Marrow Involvement in Patients with Nodular Lymphocyte Predominant Hodgkin Lymphoma. Am. J. Surg. Pathol. 2018, 42, 492–499. [Google Scholar] [CrossRef]
- Eichenauer, D.A.; Plutschow, A.; Fuchs, M.; von Tresckow, B.; Boll, B.; Behringer, K.; Diehl, V.; Eich, H.T.; Borchmann, P.; Engert, A. Long-Term Course of Patients with Stage IA Nodular Lymphocyte-Predominant Hodgkin Lymphoma: A Report From the German Hodgkin Study Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 2857–2862. [Google Scholar] [CrossRef]
- Song, J.Y.; Eberle, F.C.; Xi, L.; Raffeld, M.; Rahma, O.; Wilson, W.H.; Dunleavy, K.; Pittaluga, S.; Jaffe, E.S. Coexisting and clonally identical classic hodgkin lymphoma and nodular lymphocyte predominant hodgkin lymphoma. Am. J. Surg. Pathol. 2011, 35, 767–772. [Google Scholar] [CrossRef] [Green Version]
- Karube, K.; Takatori, M.; Sakihama, S.; Tsuruta, Y.; Miyagi, T.; Morichika, K.; Kitamura, S.; Nakada, N.; Hayashi, M.; Tomori, S.; et al. Clinicopathological features of adult T-cell leukemia/lymphoma with HTLV-1-infected Hodgkin and Reed-Sternberg-like cells. Blood Adv. 2021, 5, 198–206. [Google Scholar] [CrossRef]
- Wang, H.W.; Balakrishna, J.P.; Pittaluga, S.; Jaffe, E.S. Diagnosis of Hodgkin lymphoma in the modern era. Br. J. Haematol. 2019, 184, 45–59. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Natkunam, Y.; Bair, E.; Tibshirani, R.; Warnke, R.A. Characterization of variant patterns of nodular lymphocyte predominant hodgkin lymphoma with immunohistologic and clinical correlation. Am. J. Surg. Pathol. 2003, 27, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Boudova, L.; Torlakovic, E.; Delabie, J.; Reimer, P.; Pfistner, B.; Wiedenmann, S.; Diehl, V.; Muller-Hermelink, H.K.; Rudiger, T. Nodular lymphocyte-predominant Hodgkin lymphoma with nodules resembling T-cell/histiocyte-rich B-cell lymphoma: Differential diagnosis between nodular lymphocyte-predominant Hodgkin lymphoma and T-cell/histiocyte-rich B-cell lymphoma. Blood 2003, 102, 3753–3758. [Google Scholar] [CrossRef] [PubMed]
- Browne, P.; Petrosyan, K.; Hernandez, A.; Chan, J.A. The B-cell transcription factors BSAP, Oct-2, and BOB.1 and the pan-B-cell markers CD20, CD22, and CD79a are useful in the differential diagnosis of classic Hodgkin lymphoma. Am. J. Clin. Pathol. 2003, 120, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Uherova, P.; Valdez, R.; Ross, C.W.; Schnitzer, B.; Finn, W.G. Nodular lymphocyte predominant Hodgkin lymphoma. An immunophenotypic reappraisal based on a single-institution experience. Am. J. Clin. Pathol. 2003, 119, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, G.; Raffeld, M.; Pittaluga, S.; Jaffe, E.S. CD15-expressing nodular lymphocyte-predominant Hodgkin lymphoma. Histopathology 2011, 58, 803–805. [Google Scholar] [CrossRef]
- Nam-Cha, S.H.; Montes-Moreno, S.; Salcedo, M.T.; Sanjuan, J.; Garcia, J.F.; Piris, M.A. Lymphocyte-rich classical Hodgkin’s lymphoma: Distinctive tumor and microenvironment markers. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc 2009, 22, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- Huppmann, A.R.; Nicolae, A.; Slack, G.W.; Pittaluga, S.; Davies-Hill, T.; Ferry, J.A.; Harris, N.L.; Jaffe, E.S.; Hasserjian, R.P. EBV may be expressed in the LP cells of nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) in both children and adults. Am. J. Surg. Pathol. 2014, 38, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Menter, T.; Bodmer-Haecki, A.; Dirnhofer, S.; Tzankov, A. Evaluation of the diagnostic and prognostic value of PDL1 expression in Hodgkin and B-cell lymphomas. Hum. Pathol. 2016, 54, 17–24. [Google Scholar] [CrossRef]
- Eladl, A.E.; Satou, A.; Elsayed, A.A.; Suzuki, Y.; Shimizu-Kohno, K.; Kato, S.; Tomita, A.; Kinoshita, T.; Nakamura, S.; Asano, N. Nodular lymphocyte predominant Hodgkin lymphoma: Clincopathological study of 25 cases from Japan with a reappraisal of tissue associated macrophages. Pathol. Int. 2015, 65, 652–660. [Google Scholar] [CrossRef]
- Marafioti, T.; Hummel, M.; Anagnostopoulos, I.; Foss, H.D.; Falini, B.; Delsol, G.; Isaacson, P.G.; Pileri, S.; Stein, H. Origin of nodular lymphocyte-predominant Hodgkin’s disease from a clonal expansion of highly mutated germinal-center B cells. N. Engl. J. Med. 1997, 337, 453–458. [Google Scholar] [CrossRef]
- Brune, V.; Tiacci, E.; Pfeil, I.; Doring, C.; Eckerle, S.; van Noesel, C.J.; Klapper, W.; Falini, B.; von Heydebreck, A.; Metzler, D.; et al. Origin and pathogenesis of nodular lymphocyte-predominant Hodgkin lymphoma as revealed by global gene expression analysis. J. Exp. Med. 2008, 205, 2251–2268. [Google Scholar] [CrossRef]
- Schuhmacher, B.; Bein, J.; Rausch, T.; Benes, V.; Tousseyn, T.; Vornanen, M.; Ponzoni, M.; Thurner, L.; Gascoyne, R.; Steidl, C.; et al. JUNB, DUSP2, SGK1, SOCS1 and CREBBP are frequently mutated in T-cell/histiocyte-rich large B-cell lymphoma. Haematologica 2019, 104, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, S.; Doring, C.; Vucic, E.; Chan, F.C.; Ennishi, D.; Tousseyn, T.; de Wolf-Peeters, C.; Perner, S.; Wlodarska, I.; Steidl, C.; et al. Array comparative genomic hybridization reveals similarities between nodular lymphocyte predominant Hodgkin lymphoma and T cell/histiocyte rich large B cell lymphoma. Br. J. Haematol. 2015, 169, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Franke, S.; Wlodarska, I.; Maes, B.; Vandenberghe, P.; Achten, R.; Hagemeijer, A.; De Wolf-Peeters, C. Comparative genomic hybridization pattern distinguishes T-cell/histiocyte-rich B-cell lymphoma from nodular lymphocyte predominance Hodgkin’s lymphoma. Am. J. Pathol. 2002, 161, 1861–1867. [Google Scholar] [CrossRef]
- Prakash, S.; Fountaine, T.; Raffeld, M.; Jaffe, E.S.; Pittaluga, S. IgD positive L&H cells identify a unique subset of nodular lymphocyte predominant Hodgkin lymphoma. Am. J. Surg. Pathol. 2006, 30, 585–592. [Google Scholar] [CrossRef]
- Paschold, L.; Willscher, E.; Bein, J.; Vornanen, M.; Eichenauer, D.A.; Simnica, D.; Thiele, B.; Wickenhauser, C.; Rosenwald, A.; Bernd, H.W.; et al. Evolutionary clonal trajectories in nodular lymphocyte-predominant Hodgkin lymphoma with high risk of transformation. Haematologica 2021, 106, 2654–2666. [Google Scholar] [CrossRef]
- Thurner, L.; Hartmann, S.; Fadle, N.; Regitz, E.; Kemele, M.; Kim, Y.J.; Bohle, R.M.; Nimmesgern, A.; von Muller, L.; Kempf, V.A.J.; et al. Lymphocyte predominant cells detect Moraxella catarrhalis-derived antigens in nodular lymphocyte-predominant Hodgkin lymphoma. Nat. Commun. 2020, 11, 2465. [Google Scholar] [CrossRef]
- Traverse-Glehen, A.; Pittaluga, S.; Gaulard, P.; Sorbara, L.; Alonso, M.A.; Raffeld, M.; Jaffe, E.S. Mediastinal gray zone lymphoma: The missing link between classic Hodgkin’s lymphoma and mediastinal large B-cell lymphoma. Am. J. Surg. Pathol. 2005, 29, 1411–1421. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Leroy, K.; Yu, X.; Gaulard, P.; Gascoyne, R.D.; Chan, W.C.; Zhao, T.; Haioun, C.; Greiner, T.C.; et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 2003, 198, 851–862. [Google Scholar] [CrossRef]
- Evens, A.M.; Kanakry, J.A.; Sehn, L.H.; Kritharis, A.; Feldman, T.; Kroll, A.; Gascoyne, R.D.; Abramson, J.S.; Petrich, A.M.; Hernandez-Ilizaliturri, F.J.; et al. Gray zone lymphoma with features intermediate between classical Hodgkin lymphoma and diffuse large B-cell lymphoma: Characteristics, outcomes, and prognostication among a large multicenter cohort. Am. J. Hematol. 2015, 90, 778–783. [Google Scholar] [CrossRef] [Green Version]
- Kanavaros, P.; Gaulard, P.; Charlotte, F.; Martin, N.; Ducos, C.; Lebezu, M.; Mason, D.Y. Discordant expression of immunoglobulin and its associated molecule mb-1/CD79a is frequently found in mediastinal large B cell lymphomas. Am. J. Pathol. 1995, 146, 735–741. [Google Scholar] [PubMed]
- Pilichowska, M.; Pittaluga, S.; Ferry, J.A.; Hemminger, J.; Chang, H.; Kanakry, J.A.; Sehn, L.H.; Feldman, T.; Abramson, J.S.; Kritharis, A.; et al. Clinicopathologic consensus study of gray zone lymphoma with features intermediate between DLBCL and classical HL. Blood Adv. 2017, 1, 2600–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakakibara, A.; Kohno, K.; Iwakoshi, A.; Moritani, S.; Fujishiro, A.; Kito, K.; Suzuki, Y.; Shimada, S.; Nakaguro, M.; Shimoyama, Y.; et al. Diagnostic utility of programmed cell death ligand 1 (clone SP142) in mediastinal composite lymphoma: A report of two cases. Pathol. Int. 2020, 70, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, C.; Copie-Bergman, C.; Damotte, D.; Ben-Neriah, S.; Burroni, B.; Cornillon, J.; Lemal, R.; Golfier, C.; Fabiani, B.; Chassagne-Clement, C.; et al. Gray-zone Lymphoma between cHL and Large B-Cell Lymphoma: A Histopathologic Series From the LYSA. Am. J. Surg. Pathol. 2019, 43, 341–351. [Google Scholar] [CrossRef]
- Sarkozy, C.; Chong, L.; Takata, K.; Chavez, E.A.; Miyata-Takata, T.; Duns, G.; Telenius, A.; Boyle, M.; Slack, G.W.; Laurent, C.; et al. Gene expression profiling of gray zone lymphoma. Blood Adv. 2020, 4, 2523–2535. [Google Scholar] [CrossRef]
- Campo, E.; Jaffe, E.S. Taking gray zone lymphomas out of the shadows. Blood 2021, 137, 1703–1704. [Google Scholar] [CrossRef]
- Eberle, F.C.; Salaverria, I.; Steidl, C.; Summers, T.A., Jr.; Pittaluga, S.; Neriah, S.B.; Rodriguez-Canales, J.; Xi, L.; Ylaya, K.; Liewehr, D.; et al. Gray zone lymphoma: Chromosomal aberrations with immunophenotypic and clinical correlations. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2011, 24, 1586–1597. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Khiabanian, H.; Fangazio, M.; Vasishtha, M.; Messina, M.; Holmes, A.B.; Ouillette, P.; Trifonov, V.; Rossi, D.; Tabbo, F.; et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014, 6, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.J.; Rosenwald, A.; Wright, G.; Lee, A.; Last, K.W.; Weisenburger, D.D.; Chan, W.C.; Delabie, J.; Braziel, R.M.; Campo, E.; et al. Transformation of follicular lymphoma to diffuse large B-cell lymphoma proceeds by distinct oncogenic mechanisms. Br. J. Haematol. 2007, 136, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Nicolae, A.; Pittaluga, S.; Abdullah, S.; Steinberg, S.M.; Pham, T.A.; Davies-Hill, T.; Xi, L.; Raffeld, M.; Jaffe, E.S. EBV-positive large B-cell lymphomas in young patients: A nodal lymphoma with evidence for a tolerogenic immune environment. Blood 2015, 126, 863–872. [Google Scholar] [CrossRef] [Green Version]
- Asano, N.; Yamamoto, K.; Tamaru, J.; Oyama, T.; Ishida, F.; Ohshima, K.; Yoshino, T.; Nakamura, N.; Mori, S.; Yoshie, O.; et al. Age-related Epstein–Barr virus (EBV)-associated B-cell lymphoproliferative disorders: Comparison with EBV-positive classic Hodgkin lymphoma in elderly patients. Blood 2009, 113, 2629–2636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahara, T.; Satou, A.; Ishikawa, E.; Kohno, K.; Kato, S.; Suzuki, Y.; Takahashi, E.; Ohashi, A.; Asano, N.; Tsuzuki, T.; et al. Clinicopathological analysis of neoplastic PD-L1-positive EBV(+) diffuse large B cell lymphoma, not otherwise specified, in a Japanese cohort. Virchows Arch. Int. J. Pathol. 2021, 478, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, E.; Kato, S.; Shimada, K.; Tanaka, T.; Suzuki, Y.; Satou, A.; Kohno, K.; Sakakibara, A.; Yamamura, T.; Nakamura, M.; et al. Clinicopathological analysis of primary intestinal diffuse large B-cell lymphoma: Prognostic evaluation of CD5, PD-L1, and Epstein–Barr virus on tumor cells. Cancer Med. 2018, 7, 6051–6063. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Tsuyuki, Y.; Suzuki, Y.; Shimada, K.; Kato, S.; Takahara, T.; Mori, M.; Nakaguro, M.; Sakakibara, A.; Nakamura, S.; et al. Clinicopathologic Analysis of Primary Adrenal Diffuse Large B-Cell Lymphoma: A Reappraisal of 23 Japanese Patients Based on EBV Association and PD-L1 Expression in Tumor Cells. Am. J. Surg. Pathol. 2021, 45, 1606–1615. [Google Scholar] [CrossRef]
- Miyagi, S.; Ishikawa, E.; Nakamura, M.; Shimada, K.; Yamamura, T.; Furukawa, K.; Tanaka, T.; Mabuchi, S.; Tsuyuki, Y.; Kohno, K.; et al. Reappraisal of Primary Epstein–Barr Virus (EBV)-positive Diffuse Large B-Cell Lymphoma of the Gastrointestinal Tract: Comparative Analysis Among Immunosuppressed and Nonimmunosuppressed Stage I and II–IV Patients. Am. J. Surg. Pathol. 2020, 44, 1173–1183. [Google Scholar] [CrossRef]
- Oyama, T.; Ichimura, K.; Suzuki, R.; Suzumiya, J.; Ohshima, K.; Yatabe, Y.; Yokoi, T.; Kojima, M.; Kamiya, Y.; Taji, H.; et al. Senile EBV+ B-cell lymphoproliferative disorders: A clinicopathologic study of 22 patients. Am. J. Surg. Pathol. 2003, 27, 16–26. [Google Scholar] [CrossRef]
- Dojcinov, S.D.; Venkataraman, G.; Raffeld, M.; Pittaluga, S.; Jaffe, E.S. EBV positive mucocutaneous ulcer--a study of 26 cases associated with various sources of immunosuppression. Am. J. Surg. Pathol. 2010, 34, 405–417. [Google Scholar] [CrossRef]
- Daroontum, T.; Kohno, K.; Eladl, A.E.; Satou, A.; Sakakibara, A.; Matsukage, S.; Yakushiji, N.; Ya-In, C.; Nakamura, S.; Asano, N.; et al. Comparison of Epstein–Barr virus-positive mucocutaneous ulcer associated with treated lymphoma or methotrexate in Japan. Histopathology 2018, 72, 1115–1127. [Google Scholar] [CrossRef]
- Satou, A.; Banno, S.; Hanamura, I.; Takahashi, E.; Takahara, T.; Nobata, H.; Katsuno, T.; Takami, A.; Ito, Y.; Ueda, R.; et al. EBV-positive mucocutaneous ulcer arising in rheumatoid arthritis patients treated with methotrexate: Single center series of nine cases. Pathol. Int. 2019, 69, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Kohno, K.; Suzuki, Y.; Elsayed, A.A.; Sakakibara, A.; Takahara, T.; Satou, A.; Kato, S.; Nakamura, S.; Asano, N. Immunohistochemical Assessment of the Diagnostic Utility of PD-L1 (Clone SP142) for Methotrexate-Associated Lymphoproliferative Disorders with an Emphasis of Neoplastic PD-L1 (Clone SP142)-Positive Classic Hodgkin Lymphoma Type. Am. J. Clin. Pathol. 2020, 153, 571–582. [Google Scholar] [CrossRef]
- Megahed, N.A.; Kohno, K.; Sakakibara, A.; Eladl, A.E.; Elsayed, A.A.; Wu, C.C.; Suzuki, Y.; Takahara, T.; Kato, S.; Nakamura, S.; et al. Anaplastic variant of diffuse large B-cell lymphoma: Reappraisal as a nodal disease with sinusoidal involvement. Pathol. Int. 2019, 69, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, P.; Chai, J.; Yu, K.; Xu, T.; Zhao, D.; Liu, Y.; Wang, Y.; Wang, K.; Ma, J.; et al. The clinicopathological and molecular features of sinusoidal large B-cell lymphoma. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2021, 34, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, Y.; Wang, Y.; Chen, G.; Chen, Q.; Xiao, H.; Liu, F.; Qi, C.; Yu, Z.; Li, X.; et al. Anaplastic Variant of Diffuse Large B-cell Lymphoma Displays Intricate Genetic Alterations and Distinct Biological Features. Am. J. Surg. Pathol. 2017, 41, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Atsaves, V.; Tsesmetzis, N.; Chioureas, D.; Kis, L.; Leventaki, V.; Drakos, E.; Panaretakis, T.; Grander, D.; Medeiros, L.J.; Young, K.H.; et al. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia 2017, 31, 1633–1637. [Google Scholar] [CrossRef]
- Savage, K.J.; Harris, N.L.; Vose, J.M.; Ullrich, F.; Jaffe, E.S.; Connors, J.M.; Rimsza, L.; Pileri, S.A.; Chhanabhai, M.; Gascoyne, R.D.; et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: Report from the International Peripheral T-Cell Lymphoma Project. Blood 2008, 111, 5496–5504. [Google Scholar] [CrossRef]
- Hsi, E.D.; Said, J.; Macon, W.R.; Rodig, S.J.; Ondrejka, S.L.; Gascoyne, R.D.; Morgan, E.A.; Dorfman, D.M.; Maurer, M.J.; Dogan, A. Diagnostic accuracy of a defined immunophenotypic and molecular genetic approach for peripheral T/NK-cell lymphomas. A North American PTCL study group project. Am. J. Surg. Pathol. 2014, 38, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Krenacs, L.; Wellmann, A.; Sorbara, L.; Himmelmann, A.W.; Bagdi, E.; Jaffe, E.S.; Raffeld, M. Cytotoxic cell antigen expression in anaplastic large cell lymphomas of T- and null-cell type and Hodgkin’s disease: Evidence for distinct cellular origin. Blood 1997, 89, 980–989. [Google Scholar] [CrossRef]
- Foss, H.D.; Anagnostopoulos, I.; Araujo, I.; Assaf, C.; Demel, G.; Kummer, J.A.; Hummel, M.; Stein, H. Anaplastic large-cell lymphomas of T-cell and null-cell phenotype express cytotoxic molecules. Blood 1996, 88, 4005–4011. [Google Scholar] [CrossRef]
- Dunleavy, K.; Wilson, W.H.; Jaffe, E.S. Angioimmunoblastic T cell lymphoma: Pathobiological insights and clinical implications. Curr. Opin. Hematol. 2007, 14, 348–353. [Google Scholar] [CrossRef]
- Ohshima, K.; Suzumiya, J.; Kato, A.; Tashiro, K.; Kikuchi, M. Clonal HTLV-I-infected CD4+ T-lymphocytes and non-clonal non-HTLV-I-infected giant cells in incipient ATLL with Hodgkin-like histologic features. Int. J. Cancer 1997, 72, 592–598. [Google Scholar] [CrossRef]
- Okada, K.; Takahara, T.; Suzuki, Y.; Kohno, K.; Sakakibara, A.; Satou, A.; Takahashi, E.; Nakamura, S. Histiocytic and dendritic cell neoplasms: Reappraisal of a Japanese series based on t(14;18) and neoplastic PD-L1 expression. Pathol. Int. 2021, 71, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Griffin, G.K.; Sholl, L.M.; Lindeman, N.I.; Fletcher, C.D.; Hornick, J.L. Targeted genomic sequencing of follicular dendritic cell sarcoma reveals recurrent alterations in NF-kappaB regulatory genes. Mod. Pathol. Off. J. United States Can. Acad. Pathol. Inc. 2016, 29, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, S.; Nagahama, M.; Kagami, Y.; Yatabe, Y.; Takeuchi, T.; Kojima, M.; Motoori, T.; Suzuki, R.; Taji, H.; Ogura, M.; et al. Hodgkin’s disease expressing follicular dendritic cell marker CD21 without any other B-cell marker: A clinicopathologic study of nine cases. Am. J. Surg. Pathol. 1999, 23, 363–376. [Google Scholar] [CrossRef]
- Nakamura, S.; Koshikawa, T.; Kitoh, K.; Nakayama, A.; Yamakawa, M.; Imai, Y.; Ishii, K.; Fujita, M.; Suchi, T. Interdigitating cell sarcoma: A morphologic and immunologic study of lymph node lesions in four cases. Pathol. Int. 1994, 44, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, A.; Takahashi, E.; Ishikawa, E.; Kohno, K.; Asano, N.; Nakamura, S. Neoplastic PD-L1 expression on interdigitating dendritic cell sarcoma: A supplementary study of a case report. Pathol. Int. 2018, 68, 577–578. [Google Scholar] [CrossRef]
- Lei, Y.; Zhao, S.; Jiang, M. Unexpected Favorable Outcome to PD-1 Antibody Plus Lenvatinib in a Patient with Recurrent Intestinal Follicular Dendritic Cell Sarcoma: A Case Report and Literature Review. Front. Immunol. 2021, 12, 653319. [Google Scholar] [CrossRef]
- Rudiger, T.; Ott, G.; Ott, M.M.; Muller-Deubert, S.M.; Muller-Hermelink, H.K. Differential diagnosis between classic Hodgkin’s lymphoma, T-cell-rich B-cell lymphoma, and paragranuloma by paraffin immunohistochemistry. Am. J. Surg. Pathol. 1998, 22, 1184–1191. [Google Scholar] [CrossRef]
- Sohani, A.R.; Jaffe, E.S.; Harris, N.L.; Ferry, J.A.; Pittaluga, S.; Hasserjian, R.P. Nodular lymphocyte-predominant hodgkin lymphoma with atypical T cells: A morphologic variant mimicking peripheral T-cell lymphoma. Am. J. Surg. Pathol. 2011, 35, 1666–1678. [Google Scholar] [CrossRef]
- Nayak, L.; Iwamoto, F.M.; LaCasce, A.; Mukundan, S.; Roemer, M.G.M.; Chapuy, B.; Armand, P.; Rodig, S.J.; Shipp, M.A. PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood 2017, 129, 3071–3073. [Google Scholar] [CrossRef] [Green Version]
- Tsuyuki, Y.; Ishikawa, E.; Kohno, K.; Shimada, K.; Ohka, F.; Suzuki, Y.; Mabuchi, S.; Satou, A.; Takahara, T.; Kato, S.; et al. Expression of programmed cell death ligand-1 by immune cells in the microenvironment is a favorable prognostic factor for primary diffuse large B-cell lymphoma of the central nervous system. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2021, 41, 99–108. [Google Scholar] [CrossRef]
- Takahara, T.; Ishikawa, E.; Suzuki, Y.; Kogure, Y.; Sato, A.; Kataoka, K.; Nakamura, S. PD-L1-expressing extranodal diffuse large B-cell lymphoma, NOS with and without PD-L1 3’-UTR structural variations. J. Clin. Exp. Hematop. JCEH 2022, 21028. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, K.; Chen, L.; Berglund, M.; Ren, W.; de Miranda, N.F.; Lisboa, S.; Fangazio, M.; Zhu, S.; Hou, Y.; Wu, K.; et al. Genetic basis of PD-L1 overexpression in diffuse large B-cell lymphomas. Blood 2016, 127, 3026–3034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakakibara, A.; Inagaki, Y.; Imaoka, E.; Sakai, Y.; Ito, M.; Ishikawa, E.; Shimada, S.; Shimada, K.; Suzuki, Y.; Nakamura, S.; et al. Divergence and heterogeneity of neoplastic PD-L1 expression: Two autopsy case reports of intravascular large B-cell lymphoma. Pathol. Int. 2019, 69, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Kohno, K.; Sakakibara, A.; Iwakoshi, A.; Hasegawa, M.; Adachi, S.; Ishikawa, E.; Suzuki, Y.; Shimada, S.; Nakaguro, M.; Shimoyama, Y.; et al. Syncytial variant of classic Hodgkin lymphoma: Four cases diagnosed with the aid of CD274/programmed cell death ligand 1 immunohistochemistry. Pathol. Int. 2020, 70, 108–115. [Google Scholar] [CrossRef]
- Rengstl, B.; Rieger, M.A.; Newrzela, S. On the origin of giant cells in Hodgkin lymphoma. Commun. Integr. Biol. 2014, 7, e28602. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Type of Genetic Alteration | Frequency (%) | Reference | |
|---|---|---|---|---|
| CHLs | NLPHL | |||
| Immune evasion | ||||
| PD-L1/PD-L1 | Gain/amplification | 30–97 | [37,44,61] | |
| B2M | SNV, indel | 39 | [61] | |
| CIITA | Translocation, SNV | 8 | [61] | |
| JAK/STAT activation | ||||
| JAK2 | Gain/amplification | 33 | [75] | |
| SOCS1 | SNV | 40–70 | 50 | [68] |
| STAT6 | SNV, gain | 30 | [61,62] | |
| PTPN1 | SNV, indel | 22 | [61] | |
| XPO1 | SNV, gain | 18–26 | [62] | |
| Constitutive NF-κB activation | ||||
| TNFAIP3 | SNV, indel | 57–74 | [61,64] | |
| REL | Gain/amplification | 50 | 50 | [63] |
| NFKBIA | SNV, indel | 17 | [61] | |
| NFKBIE | SNV, indel | 26 | [61] | |
| NIK | Gain/amplification | 25 | [67,70] | |
| BCL3 | Gain/translocation | 15 | [71] | |
| PI3K/AKT pathway activation | ||||
| GNA13 | SNV | 24–26 | [61,62] | |
| ITPKB | SNV | 16 | [61,62] | |
| MAPK/ERK pathway activation | ||||
| DUSP2 | SNV | 54 | [72] | |
| AP-1 regulation | ||||
| JUNB | SNV | 39 | [72] | |
| BCL6 disregulation | ||||
| BCL6 | Translocation | 48 | [73,74] | |
| Chromatin remodeling | ||||
| ARID1A | SNV, indel | 26 | [61] | |
| Unknown function | ||||
| SGK1 | SNV | 70 | [72] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahara, T.; Satou, A.; Tsuzuki, T.; Nakamura, S. Hodgkin Lymphoma: Biology and Differential Diagnostic Problem. Diagnostics 2022, 12, 1507. https://doi.org/10.3390/diagnostics12061507
Takahara T, Satou A, Tsuzuki T, Nakamura S. Hodgkin Lymphoma: Biology and Differential Diagnostic Problem. Diagnostics. 2022; 12(6):1507. https://doi.org/10.3390/diagnostics12061507
Chicago/Turabian StyleTakahara, Taishi, Akira Satou, Toyonori Tsuzuki, and Shigeo Nakamura. 2022. "Hodgkin Lymphoma: Biology and Differential Diagnostic Problem" Diagnostics 12, no. 6: 1507. https://doi.org/10.3390/diagnostics12061507