
Clinical and Genetic Characterisation of Cystic Fibrosis Patients in Latvia: A Twenty-Five-Year Experience

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. CF Screening Results
3.2. CFTR Genotype
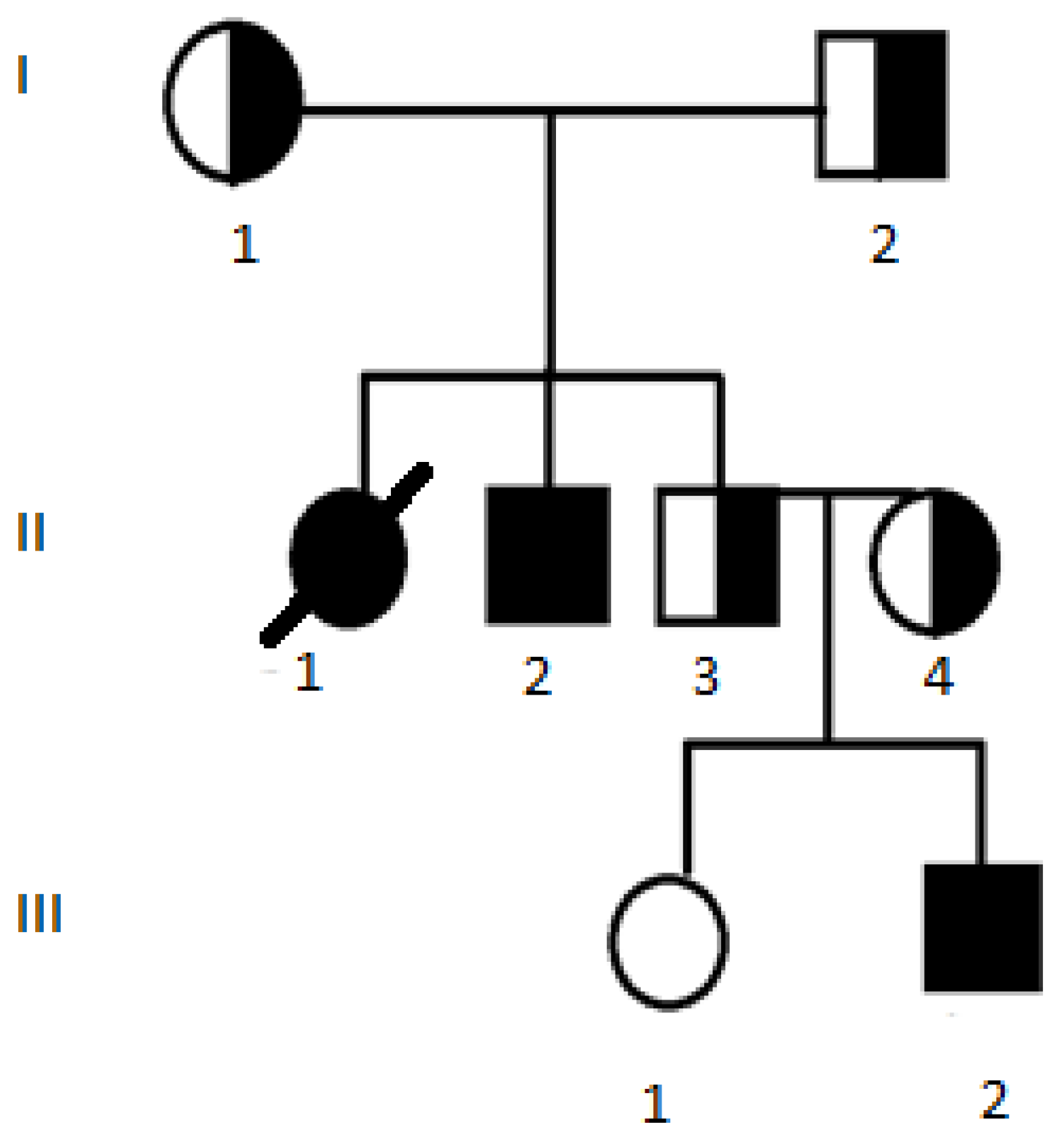
3.2.1. Case 1 with Genotype c.[1521_1523delCTT];[169T>C] p.[(Phe508del)];[(Trp57Arg)], Legacy Nomenclature dF508/W57R
3.2.2. Case 2 with Genotype c.[3844T>C];[2012delT] p.[(Trp1282Arg)];[(Leu671Ter)], Legacy Nomenclature W1282R/2143delT
3.2.3. Cases 3 and 4 with Genotype c.[1521_1523delCTT];[3844T>C] p.[(Phe508del)];[(Trp1282Arg)], Legacy Nomenclature dF508/W1282R
3.2.4. Cases 5 and 6 with Genotype c.[1521_1523delCTT];[(743+1_744-1)_(1584+1_1585-1)dup] p.[(Phe508del)];[(?)], Legacy Nomenclature dF508/dup6b-10
3.2.5. Case 7 with Genotype c.[503C>A];[4004T>C] p.[(Ser168Ter)];[(Leu1335Pro)], Legacy Nomenclature Ser168Ter/L1335P
3.3. Clinical and Laboratory Characteristics of CF Patients under Regular Follow-Up
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bareil, C.; Bergougnoux, A. CFTR Gene Variants, Epidemiology and Molecular Pathology. Arch. Pediatrie Organe Off. Soc. Fr. Pediatrie 2020, 27, eS8–eS12. [Google Scholar] [CrossRef]
- Scotet, V.; Gutierrez, H.; Farrell, P.M. Newborn Screening for CF across the Globe-Where Is It Worthwhile? Int. J. Neonatal Screen. 2020, 6, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banjar, H.H.; Tuleimat, L.; El Seoudi, A.A.A.; Mogarri, I.; Alhaider, S.; Nizami, I.Y.; Almaghamsi, T.; Alkaf, S.A.; Moghrabi, N. Genotype patterns for mutations of the cystic fibrosis transmembrane conductance regulator gene: A retrospective descriptive study from Saudi Arabia. Ann. Of. Saudi Med. 2020, 40, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR Biology toward Combinatorial Pharmacotherapy: Expanded Classification of Cystic Fibrosis Mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, M.; Matsvay, A.; Glazova, O.; Krasovskiy, S.; Usacheva, M.; Amelina, E.; Chernyak, A.; Ivanov, M.; Musienko, S.; Prodanov, T.; et al. Targeted Sequencing Reveals Complex, Phenotype-Correlated Genotypes in Cystic Fibrosis. BMC Medical Genomics 2018, 11 (Suppl. S1), 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobadilla, J.L.; Macek, M., Jr.; Fine, J.P.; Farrell, P.M. Cystic Fibrosis: A Worldwide Analysis of CFTR Mutations-Correlation with Incidence Data and Application to Screening. Hum. Mutat. 2002, 19, 575–606. [Google Scholar] [CrossRef] [PubMed]
- Ziętkiewicz, E.; Rutkiewicz, E.; Pogorzelski, A.; Klimek, B.; Voelkel, K.; Witt, M. CFTR Mutations Spectrum and the Efficiency of Molecular Diagnostics in Polish Cystic Fibrosis Patients. PLoS ONE 2014, 9, e89094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, M.D. How to determine the mechanism of action of CFTR modulator compounds: A gateway to theranostics. Eur. J. Med. Chem. 2020, 210, 112989. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Amaral, M.D. Progress in Therapies for Cystic Fibrosis. Lancet Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Classification of CFTR Mutation Classes. Lancet Respir. Med. 2016, 4, e37–e38. [Google Scholar] [CrossRef]
- De Boeck, K. Cystic Fibrosis in the Year 2020: A Disease with a New Face. Acta Paediatr. 2020, 109, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Lace, B.; Grīnblate, S.; Korņejeva, L.; Švābe, V.; Grauduma, I.; Vēvere, P.; Lugovska, R.; Krams, A.; Martinsons, A. Neonatal Cystic Fibrosis Screening in Latvia: A Pilot Project. Proc. Latv. Acad. Sci. Sect. 2009, 63, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15.e1. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, M.; Narzi, L.; Piergentili, R.; Ferraguti, G.; Grandoni, F.; Quattrucci, S.; Strom, R. A 96-Well Formatted Method for Exon and Exon/Intron Boundary Full Sequencing of the CFTR Gene. Anal. Biochem. 2006, 353, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.C.; De Boeck, K.; Amaral, M.D. New Pharmacological Approaches for Cystic Fibrosis: Promises, Progress, Pitfalls. Pharmacol. Ther. 2015, 145, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, S.; Bobba, A.; Jurgelevičius, V.; Vacca, R.A.; Lattanzio, P.; Merafina, R.; Utkus, A.; Kučinskas, V.; Marra, E. Molecular Basis of Cystic Fibrosis in Lithuania: Incomplete CFTR Mutation Detection by PCR-Based Screening Protocols. Genet. Test. 2006, 10, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Petrova, N.V.; Kashirskaya, N.Y.; Vasilyeva, T.A.; Kondratyeva, E.I.; Zhekaite, E.K.; Voronkova, A.Y.; Sherman, V.D.; Galkina, V.A.; Ginter, E.K.; Kutsev, S.I.; et al. Analysis of CFTR Mutation Spectrum in Ethnic Russian Cystic Fibrosis Patients. Genes 2020, 11, 554. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Rab, A.; Pellicore, M.; Davis, E.F.; McCague, A.F.; Evans, T.A.; Joynt, A.T.; Lu, Z.; Cai, Z.; Raraigh, K.S.; et al. Residual Function of Cystic Fibrosis Mutants Predicts Response to Small Molecule CFTR Modulators. JCI Insight 2018, 3, e121159. [Google Scholar] [CrossRef]
- Raraigh, K.S.; Han, S.T.; Davis, E.; Evans, T.A.; Pellicore, M.J.; McCague, A.F.; Joynt, A.T.; Lu, Z.; Atalar, M.; Sharma, N.; et al. Functional Assays Are Essential for Interpretation of Missense Variants Associated with Variable Expressivity. Am. J. Hum. Genet. 2018, 102, 1062–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teder, M.; Klaassen, T.; Oitmaa, E.; Kaasik, K.; Metspalu, A. Distribution of CFTR Gene Mutations in Cystic Fibrosis Patients from Estonia. J. Med. Genet. 2000, 37, E16. [Google Scholar] [CrossRef] [PubMed]
- Petrova, G.; Yaneva, N.; Hrbková, J.; Libik, M.; Savov, A.; Macek, M. Identification of 99% of CFTR Gene Mutations in Bulgarian-, Bulgarian Turk-, and Roma Cystic Fibrosis Patients. Mol. Genet. Genom. Med. 2019, 7, e696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnunen, S.; Bonache, S.; Casals, T.; Monto, S.; Savilahti, E.; Kere, J.; Järvelä, I. Spectrum of Mutations in CFTR in Finland: 18 Years Follow-up Study and Identification of Two Novel Mutations. J. Cyst. Fibros. 2005, 4, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, N.; Balinova, N.; Marakhonov, A.; Vasilyeva, T.; Kashirskaya, N.; Galkina, V.; Ginter, E.; Kutsev, S.; Zinchenko, R. Ethnic Differences in the Frequency of CFTR Gene Mutations in Populations of the European and North Caucasian Part of the Russian Federation. Front. Genet. 2021, 12, 678374. [Google Scholar] [CrossRef] [PubMed]
- Quint, A.; Lerer, I.; Sagi, M.; Abeliovich, D. Mutation Spectrum in Jewish Cystic Fibrosis Patients in Israel: Implication to Carrier Screening. Am. J. Med. Genet. Part A 2005, 136A, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Soltysova, A.; Tarova, E.T.; Ficek, A.; Baldovic, M.; Polakova, H.; Kayserova, H.; Kadasi, L. Comprehensive Genetic Study of Cystic Fibrosis in Slovak Patients in 25 Years of Genetic Diagnostics. Clin. Respir. J. 2017, 12, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Loeber, J.; Platis, D.; Zetterström, R.; Almashanu, S.; Boemer, F.; Bonham, J.; Borde, P.; Brincat, I.; Cheillan, D.; Dekkers, E.; et al. Neonatal Screening in Europe Revisited: An ISNS Perspective on the Current State and Developments Since 2010. Int. J. Neonatal Screen. 2021, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Zybert, K.; Wozniacki, L.; Tomaszewska-Sobczyńska, A.; Wertheim-Tysarowska, K.; Czerska, K.; Ołtarzewski, M.; Sands, D. Clinical Characteristics of Rare CFTR Mutations Causing Cystic Fibrosis in Polish Population. Pediatric Pulmonol. 2020, 55, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Torresani, T.; Fingerhut, R.; Rueegg, C.S.; Gallati, S.; Kuehni, C.E.; Baumgartner, M.R.; Barben, J. Newborn Screening for Cystic Fibrosis in Switzerland--Consequences after Analysis of a 4 Months Pilot Study. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2013, 12, 667–674. [Google Scholar] [CrossRef] [Green Version]
- De Boeck, K.; Zolin, A.; Cuppens, H.; Olesen, H.; Viviani, L. The Relative Frequency of CFTR Mutation Classes in European Patients with Cystic Fibrosis. J. Cyst. Fibros. 2014, 13, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Garratt, A.; Hill, A. Worldwide Rates of Diagnosis and Effective Treatment for Cystic Fibrosis. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2022, 21, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambraia, A.; Junior, M.C.; Zembrzuski, V.M.; Junqueira, R.M.; Cabello, P.H.; de Cabello, G.M.K. Next-Generation Sequencing for Molecular Diagnosis of Cystic Fibrosis in a Brazilian Cohort. Dis. Markers 2021, 2021, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Amini, S.; Jafarrangraz, S.S.; Modaresi, M.; Gholami, K. Evaluation of adherence to medication treatment in pediatric patients with cystic fibrosis; a cross-sectional study. Eur. Respir. J. 2019, 54, pa4520. [Google Scholar] [CrossRef]
- A Goodfellow, N.; Hawwa, A.F.; Reid, A.J.; Horne, R.; Shields, M.D.; McElnay, J.C. Adherence to Treatment in Children and Adolescents with Cystic Fibrosis: A Cross-Sectional, Multi-Method Study Investigating the Influence of Beliefs about Treatment and Parental Depressive Symptoms. BMC Pulm. Med. 2015, 15, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yiallouros, P.K.; Matthaiou, A.; Anagnostopoulou, P.; Kouis, P.; Libik, M.; Adamidi, T.; Eleftheriou, A.; Demetriou, A.; Ioannou, P.; Tanteles, G.A.; et al. Demographic Characteristics, Clinical and Laboratory Features, and the Distribution of Pathogenic Variants in the CFTR Gene in the Cypriot Cystic Fibrosis (CF) Population Demonstrate the Utility of a National CF Patient Registry. Orphanet J. Rare Dis. 2021, 16, 409. [Google Scholar] [CrossRef] [PubMed]
- Kutney, K.A.; Sandouk, Z.; Desimone, M.; Moheet, A. Obesity in Cystic Fibrosis. J. Clin. Transl. Endocrinol. 2021, 26, 100276. [Google Scholar] [CrossRef]


{kind=link}
| Allele According to Legacy Nomenclature | Exon/ Intron NM_000492.3 | HGVS Nomenclature | Class of CFTR Mutation | N | % of CF-Causing Alleles in Latvia |
|---|---|---|---|---|---|
| W57R | 3 | c.169T>C p.(Trp57Arg) | II or III a | 2 | 1.5% |
| 394delTT | 3 | c.262_263delTT p.(Leu88IlefsX22) | I [15] | 1 | 0.8% |
| P67L | 3 | c.200C>T p.(Pro67Leu) | II-III [4] | 1 | 0.8% |
| L138ins | 4 | c.413_415dupTAC p.(Leu138dup) | II b | 1 | 0.8% |
| 621+1G>T | 4 | c.489+1G>T p.(?) | I [15] | 1 | 0.8% |
| Ser168Ter | 5 | c.503C>A p.(Ser168Ter) | I c | 1 | 0.8% |
| R334W | 8 | c.1001G>A p.(Arg334Gln) | III [15], II-III [4] | 1 | 0.8% |
| A455E | 10 | c.1364C>A p.(Ala455Glu) | II [15], II-III [4] | 1 | 0.8% |
| F508del | 11 | c.1521_1523delCTT p.(Phe508del) | II-III [15], II-III-VI [4] | 93 | 70.5% |
| 1677delTA | 11 | c.1545_1546delTA p.(Tyr515X) | I c | 2 | 1.5% |
| R553X | 11 | c.1657C>T p.(Arg553X) | I c | 2 | 1.5% |
| 2143delT | 14 | c.2012delT p.(Leu671Ter) | I c | 2 | 1.5% |
| 2184insA | 14 | c.2052dupA p.(Gln685ThrfsX5) | I c | 1 | 0.8% |
| R1006H | 20 | c.3197G>A p.(Arg1066His) | II-III [4] | 3 | 2.3% |
| W1282X | 23 | c.3846G>A p.(Trp1282Ter) | I-II-III-VI [4] | 2 | 1.5% |
| W1282R | 23 | c.3844T>C p.(Trp1282Arg) | II-III [16,17] | 2 | 1.5% |
| L1335P | 25 | c.4004T>C p.(Leu1335Pro) | II d | 3 | 2.3% |
| dele2,3 | covering 2nd and 3rd exons | c.54-5940_273+10250del21kb p.(Ser18ArgfsX16) | I [15] | 5 | 3.8% |
| CFTRdup6b-10 | covering 7th–11th exons | c.(743+1_744-1)_(1584+1_1585-1)dup p.(?) | I c | 1 | 0.8% |
| CFTRdel6b-10 | covering 7th–11th exons | c.(743+1_744-1)_(1584+1_1585-1)del p.(?) | I c | 1 | 0.8% |
| 3849+10kb C>T | intron between 22nd and 23rd exons | c.3718-2477C>T p.(?) | V [15] | 2 | 1.5% |
| not identified | 4 | 3.0% |
| Variant Legacy Name. | Variant HGVS Nomenclature | Latvia % | Estonia a % | Lithuania b % | Poland c % | Bulgaria d % | Finland e % | Russia (Ethnic) f % | Jewish g % |
|---|---|---|---|---|---|---|---|---|---|
| F508del | c.1521_1523delCTT p.(Phe508del) | 70.5 | 51.7 | 52 | 54.54 | 55 | 36 | 54.99 | 35.6 |
| dele2,3 | c.54-5940_273+10250del21kb p.(Ser18ArgfsX16) | 3.8 | 0 | 2 | 4.47 | 0 | 5.9 | 7.59 | 0 |
| R1006H | c.3197G>A p.(Arg1066His) | 2.3 | 0.017 | 0 | 0 | 0 | 0 | 0 | 0 |
| L1335P | c.4004T>C p.(Leu1335Pro) | 2.3 | 0 | 0 | 0 | 0.36 | 0 | 0.22 | 0 |
| W57R | c.169T>C p.(Trp57Arg) | 1.5 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| 1677delTA | c.1545_1546delTA p.(Tyr515X) | 1.5 | 0 | 0 | 0 | 1.07 | 0 | 0.18 | 0 |
| R553X | c.1657C>T p.(Arg553X) | 1.5 | 0 | 4.2 | 0 | 0 | 0 | 0.18 | 0 |
| 2143delT | c.2012delT p.(Leu671Ter) | 1.5 | 0 | 0 | 0 | 0 | 0 | 2.71 | 0 |
| W1282X | c.3846G>A p.(Trp1282Ter) | 1.5 | 0 | 1 | 0.61 | 1.43 | 0 | 1.16 | 31.3 |
| W1282R | c.3844T>C p.(Trp1282Arg) | 1.5 | 0 | 0 | 0 | 0 | 0 | 0.76 | 0 |
| 3849+10kb C>T | c.3718-2477C>T p.(?) | 1.5 | 0 | 0 | 3.93 | 1.43 | 0 | 2.35 | 4.6 |
| 394delTT | c.262_263delTT p.(Leu88IlefsX22) | 0.8 | 13.3 | 0 | 0 | 0 | 35 | 0.54 | 0 |
| P67L | c.200C>T p.(Pro67Leu) | 0.8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| L138ins | c.413_415dupTAC p.(Leu138dup) | 0.8 | 0 | 0 | 0 | 0 | 0 | 1.12 | 0 |
| 621+1G>T | c.489+1G>T p.(?) | 0.8 | 0 | 0 | 0.34 | 1.43 | 0 | 0.25 | 0 |
| Ser168Ter | c.503C>A p.(Ser168Ter) | 0.8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| R334W | c.1001G>A p.(Arg334Gln) | 0.8 | 0 | 0 | 0.41 | 0.71 | 0 | 0.69 | 0 |
| A455E | c.1364C>A p.(Ala455Glu) | 0.8 | 0 | 0 | 0 | 0 | 0 | 0.04 | 0.4 |
| 2184insA | c.2052dupA p.(Gln685ThrfsX5) | 0.8 | 0 | 0 | 1.02 | 2.89 | 0 | 2.24 | 0 |
| CFTRdup6b-10 | c.(743+1_744-1)_(1584+1_1585-1)dup p.(?) | 0.8 | 0 | 0 | 0 | 0 | 0 | 0.18 | 0 |
| CFTRdel6b-10 | c.(743+1_744-1)_(1584+1_1585-1)del p.(?) | 0.8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| not identified | 3.0 | 13.3 | 35.8 | 17.5 | 1.07 | 2.9 | 17.22 | 2.5 | |
| No. of alleles analysed | 132 | 60 | 98 | 1476 | 280 | 102 | 2768 | 281 | |
| Characteristic | Value |
|---|---|
| Number of patients | 45 |
| Male/female ratio | 19/26 |
| Mean age at follow-up (years) ± SD (min–max) | 13.2 ± 12.2 (1.4–35) |
| Mean age at diagnosis (years) (min–max) | 2.58 (0–14) |
| Number of patients older than 18 years (%) | 13 (28.9%) |
| Mean Z-score for BMI aged 2–17 years (min–max) | −0.4 (−2.3–1.4) |
| Mean BMI aged 18 years or older ± SD (min–max) | 19.0 (15.6–23.9) |
| Mean FEV1 (% predicted) ± SD (min–max) | 79.7 (18.3–111.9) |
| Prevalence of Pseudomonas aeruginosa infection (%) | 10 (22.2%) |
| Prevalence of Burkholderia species infection (%) | 1 (2.2%) |
| Prevalence of Staphylococcus aureus infection (%) | 28 (62.2%) |
| Prevalence of methicillin-resistant Staphylococcus aureus infection (%) | 1 (2.2%) |
| Prevalence of non-tuberculous Mycobacterium species infection | 0 |
| Prevalence of Stenotrophomonas maltophilia infection (%) | 3 (6.7%) |
| Prevalence of Haemophilus influenzae infection (%) | 3 (6.7%) |
| Prevalence of Achromobacter species infection (%) | 4 (8.9%) |
| CF liver disease (%) | 14 (31.1%) |
| CF-related diabetes mellitus (%) | 3 (6.7%) |
| Pancreatic sufficient cases (%) | 3 (6.7%) |
| Female patients that have given birth | 0 |
| Patients born with meconium ileus (%) | 7 (15.5%) |
| Patients that have undergone lung transplantation | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Auzenbaha, M.; Aleksejeva, E.; Taurina, G.; Kornejeva, L.; Kempa, I.; Svabe, V.; Gailite, L. Clinical and Genetic Characterisation of Cystic Fibrosis Patients in Latvia: A Twenty-Five-Year Experience. Diagnostics 2022, 12, 2893. https://doi.org/10.3390/diagnostics12112893
Auzenbaha M, Aleksejeva E, Taurina G, Kornejeva L, Kempa I, Svabe V, Gailite L. Clinical and Genetic Characterisation of Cystic Fibrosis Patients in Latvia: A Twenty-Five-Year Experience. Diagnostics. 2022; 12(11):2893. https://doi.org/10.3390/diagnostics12112893
Chicago/Turabian StyleAuzenbaha, Madara, Elina Aleksejeva, Gita Taurina, Liene Kornejeva, Inga Kempa, Vija Svabe, and Linda Gailite. 2022. "Clinical and Genetic Characterisation of Cystic Fibrosis Patients in Latvia: A Twenty-Five-Year Experience" Diagnostics 12, no. 11: 2893. https://doi.org/10.3390/diagnostics12112893