
Hypersensitivity Pneumonitis: A Pictorial Review Based on the New ATS/JRS/ALAT Clinical Practice Guideline for Radiologists and Pulmonologists


Abstract
:1. Introduction
2. Epidemiology
3. Definition, Classification and Clinical Features of HP
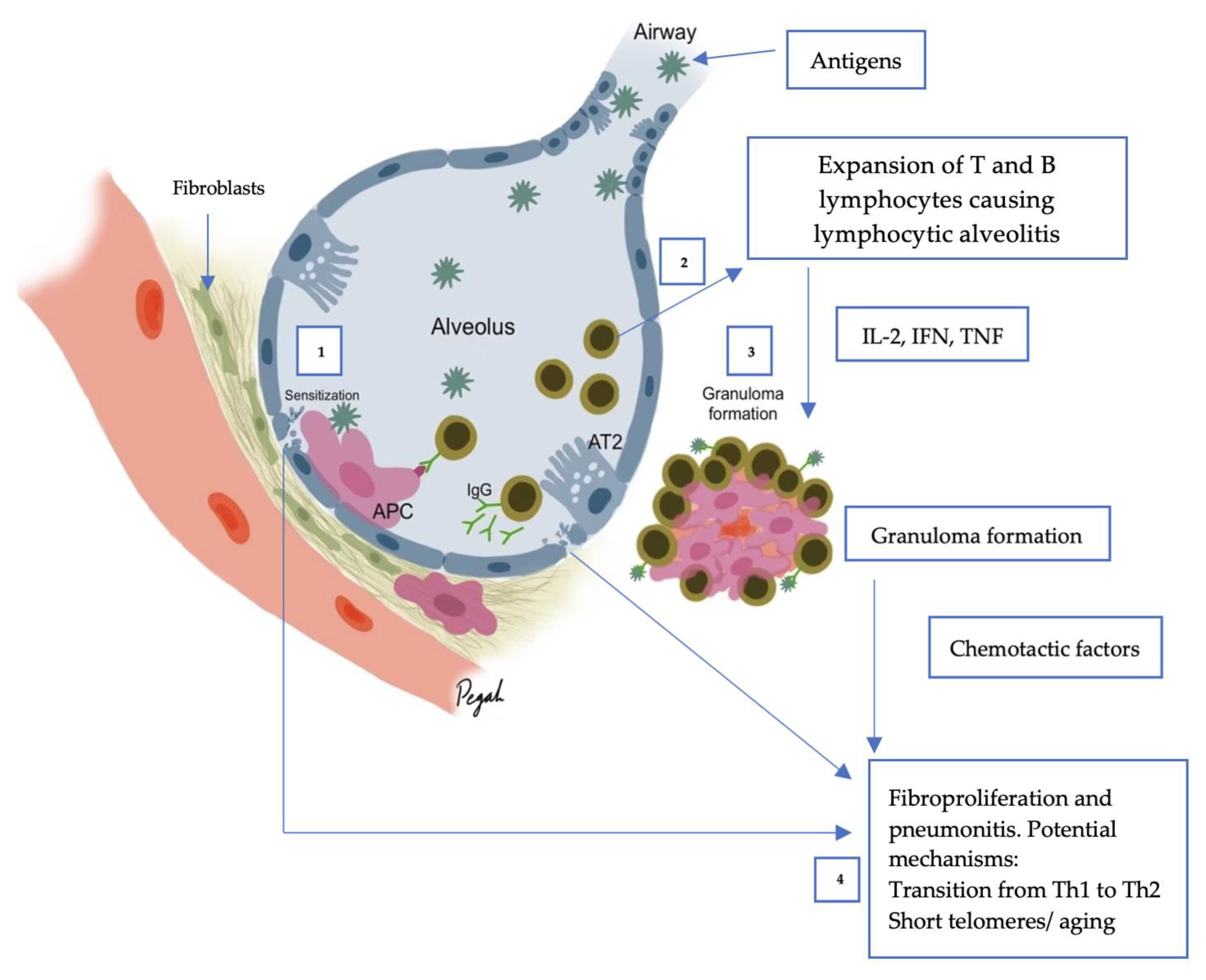
4. Pathogenesis of HP

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Source |
|---|---|
| Trichosporon spp. | Damo wooden hoses (Japan) |
| Argan | Cosmetic factory, hair salons |
| Chinchilla | Pet chinchilla |
| Aureobasidium pullulans | Domestic fungal contamination |
| Rhizopus spp. | Sawmill worker |
| Mucor spp. | Moldy wood |
| Beryllium, Cobalt, Zinc | Batteries, hard metal alloys, zinc fumes |
| Thermoactinomyces spp. | Farm environment, domestic bacterial contamination, garbage exposure |
| Saccharopolyspora rectivirgula | Farm environment, esparto grass |
| Nontuberculous mycobacterium | Hot tub |
| Wallemia sebi | Farm environment |
| Pseudomonas spp. | Cork factory, home humidifier |
| Protein of bloom, droppings, feather | Chickens, Budgerigars, Pigeons, Cockatiels |
| Aspergillus spp., Penicillium spp., Cladosporium spp. | Mold dust |
| Fungi and molds | Contaminated water |
| Acinetobacter spp. | Contaminated machine fluid |
| Achromobacter | Contaminated humidifiers |
| Bacillus spp. | Contaminated water, sawduct, moist |
| Streptomyces albus | Contaminated compost, mushroom, hay dusty soil |
| Methyl acrylates | Dental technicians |
| Isocyanate acid anhydrides | Plastic, paint, glue, varnish, resins |
| Aspergillus fumigatus, Thermophilic actinomycetes | Organic waste, soil |
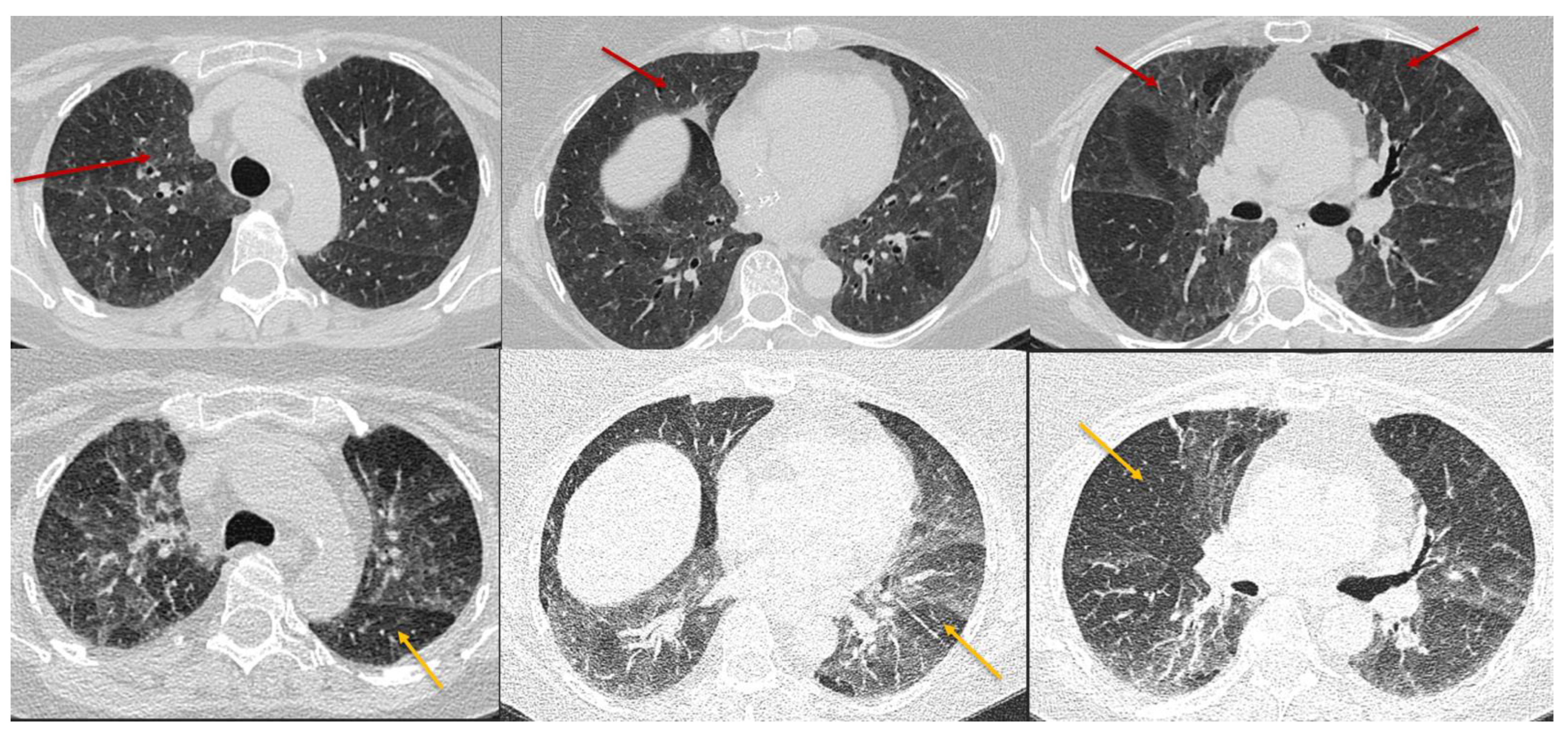
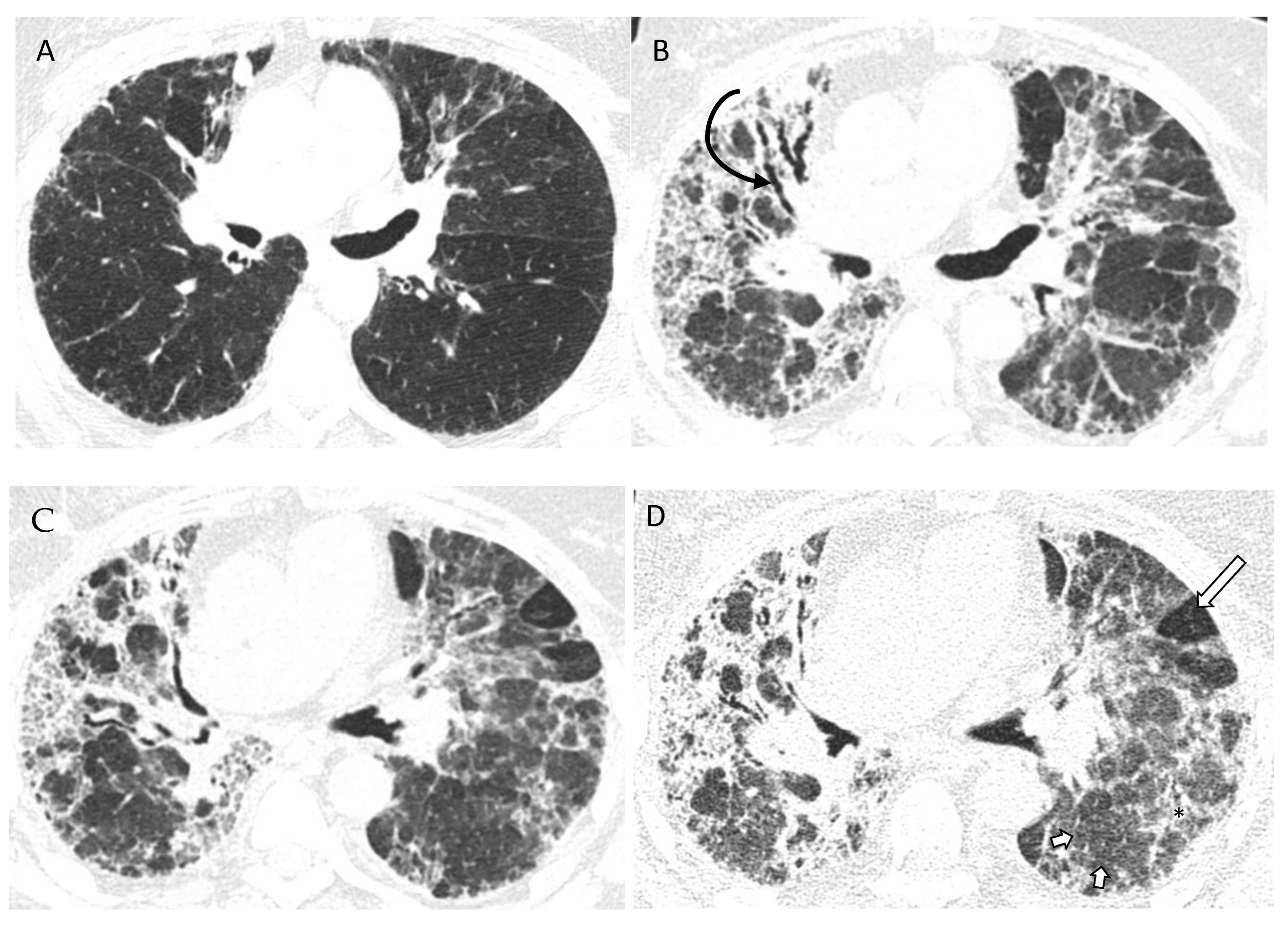
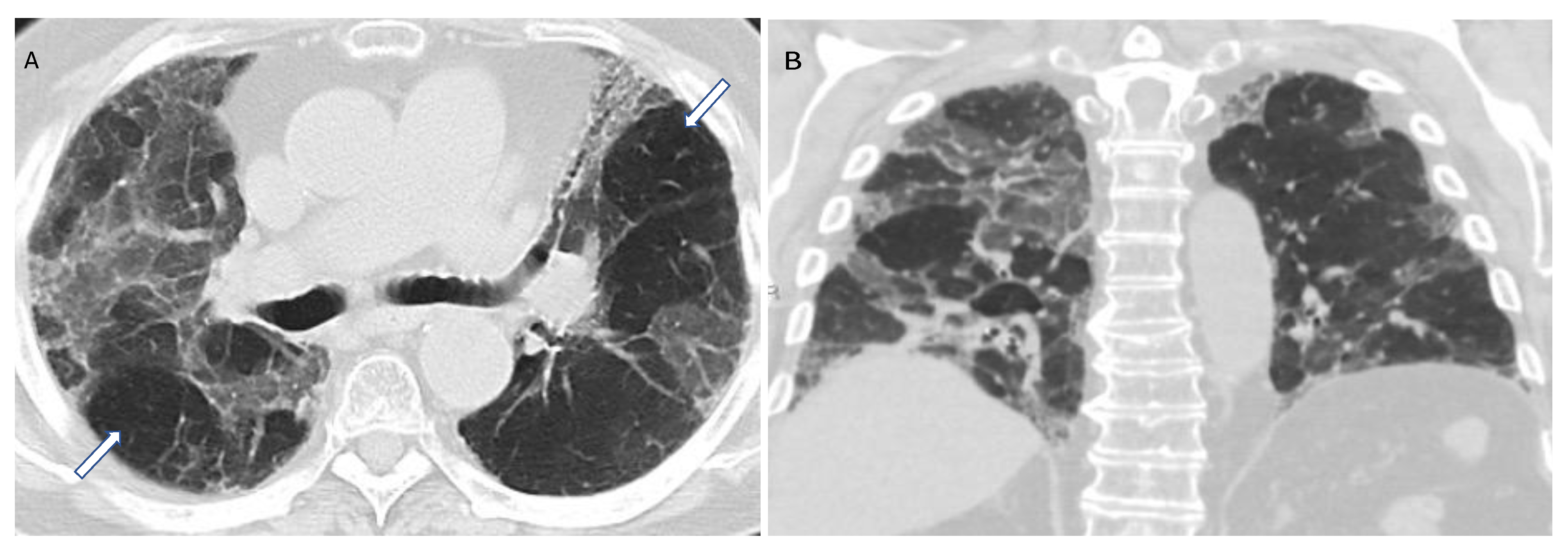
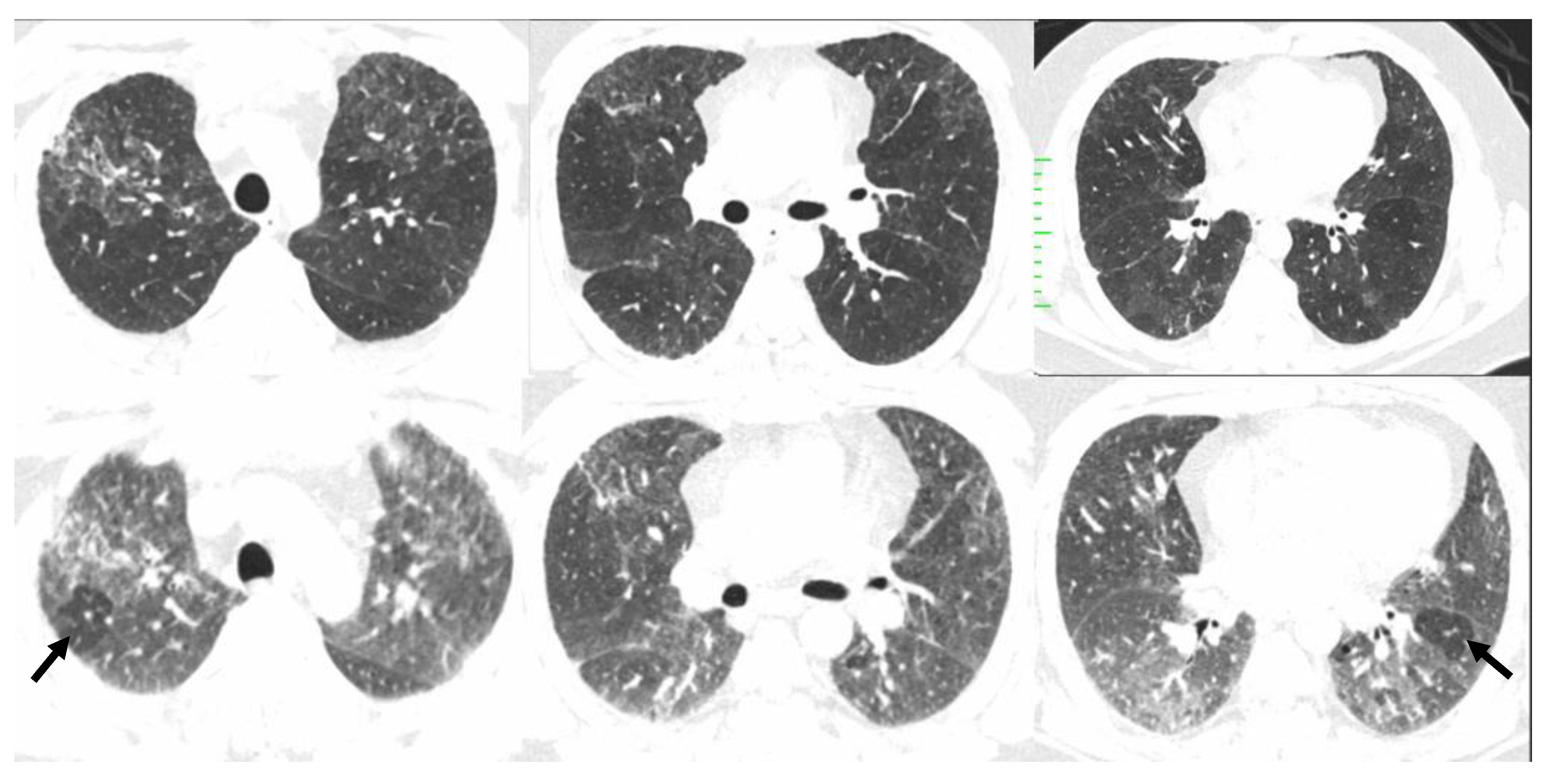
5. Imaging of HP
5.1. Chest HRCT Scanning Protocol
5.2. HRCT for Diagnosis of HP
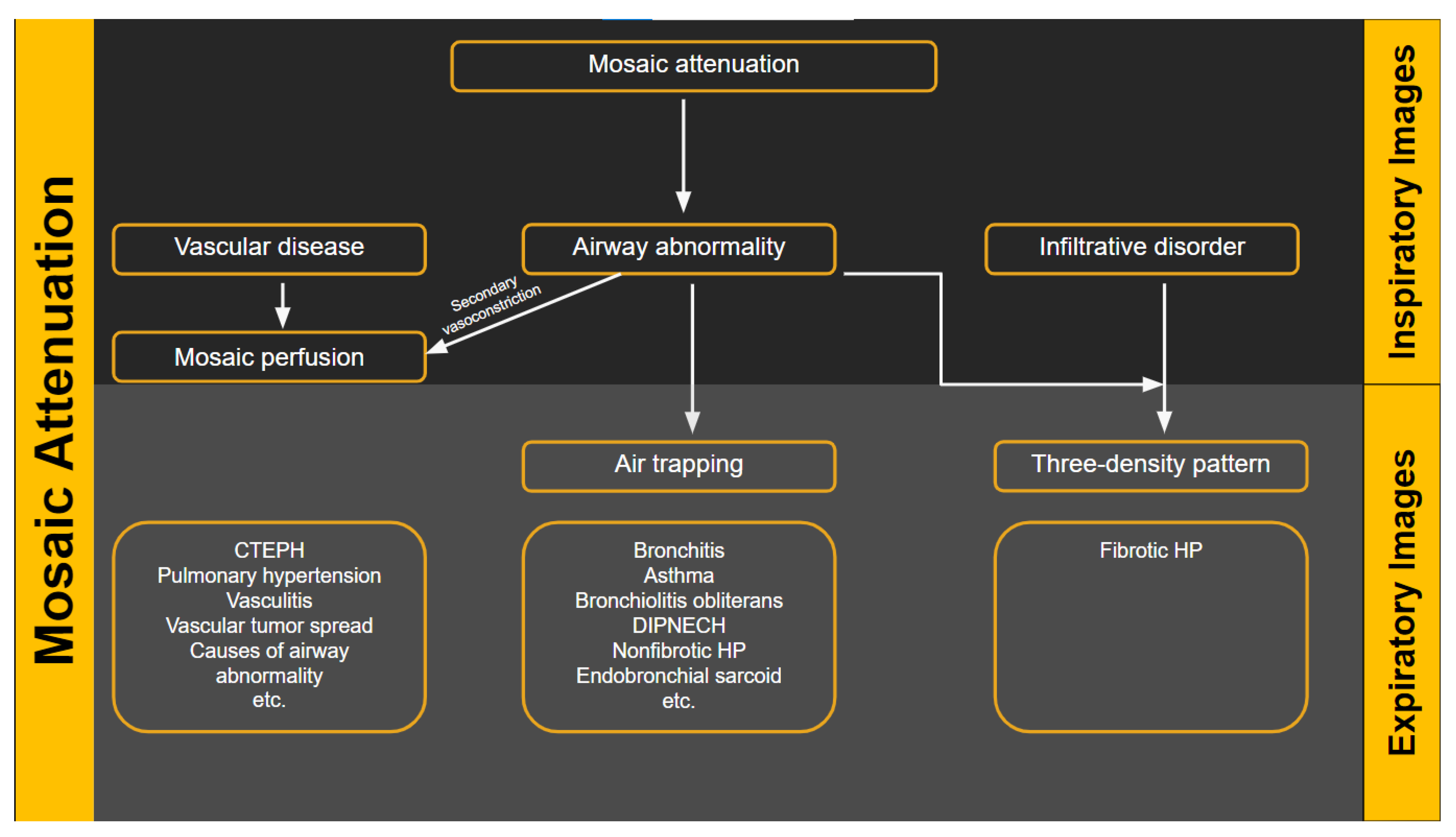
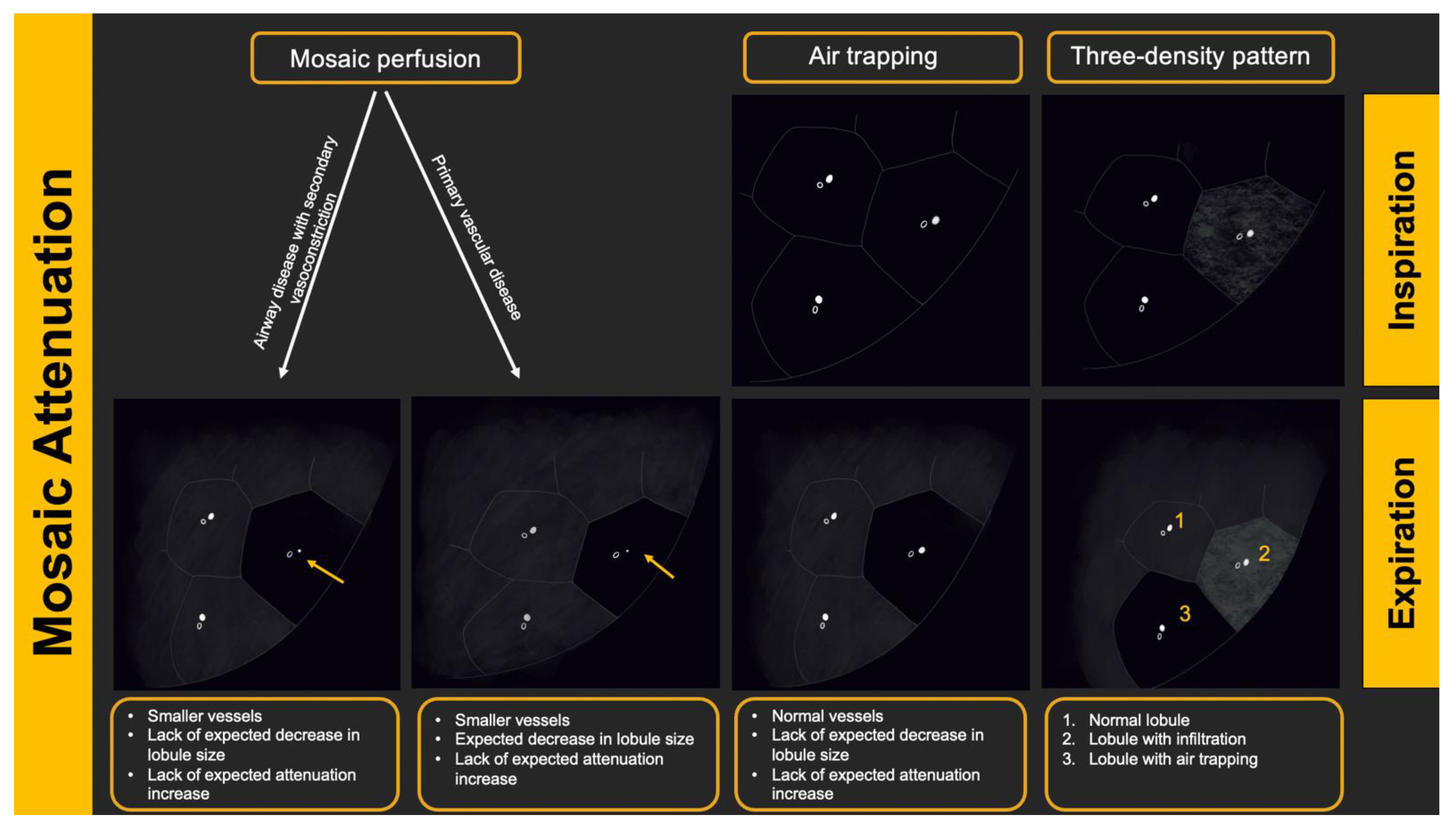
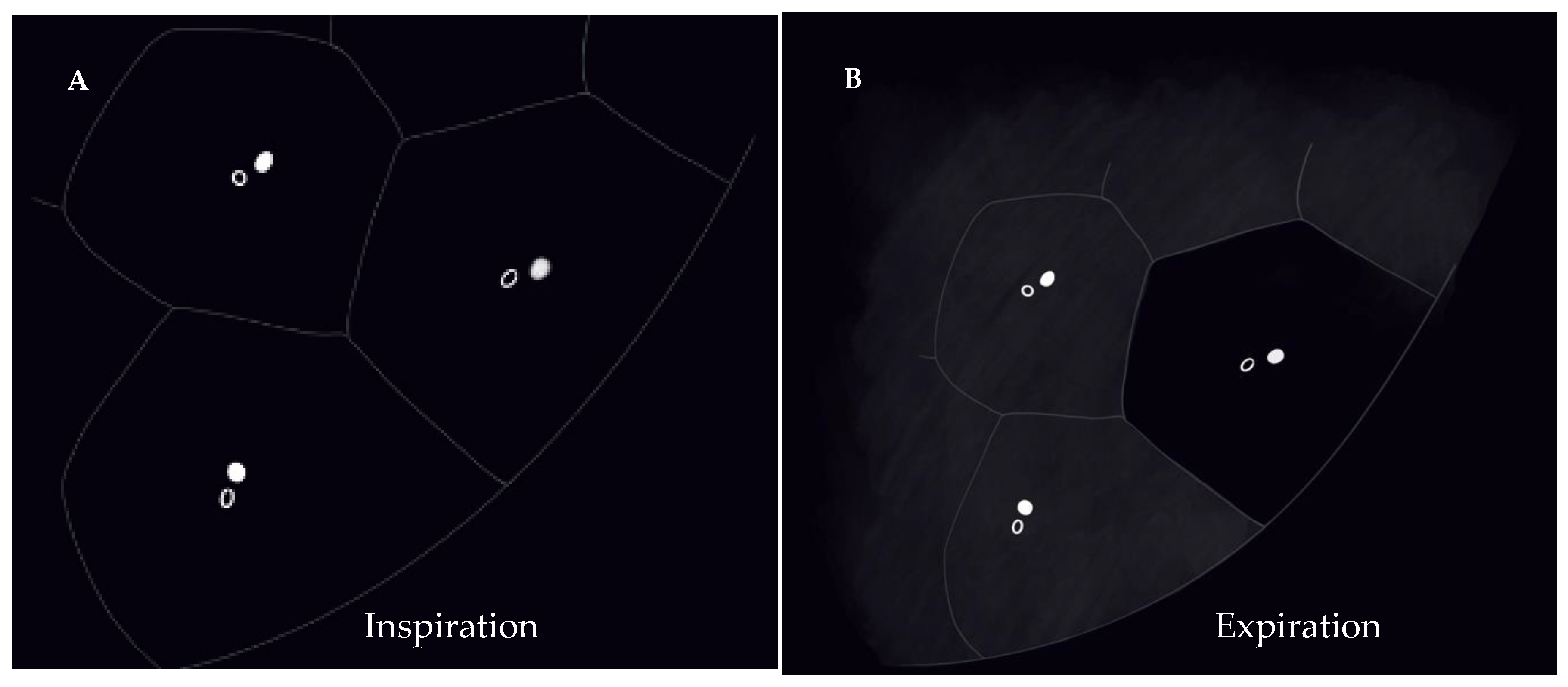
5.3. Radiologic Terms for Heterogeneous Lung Attenuation
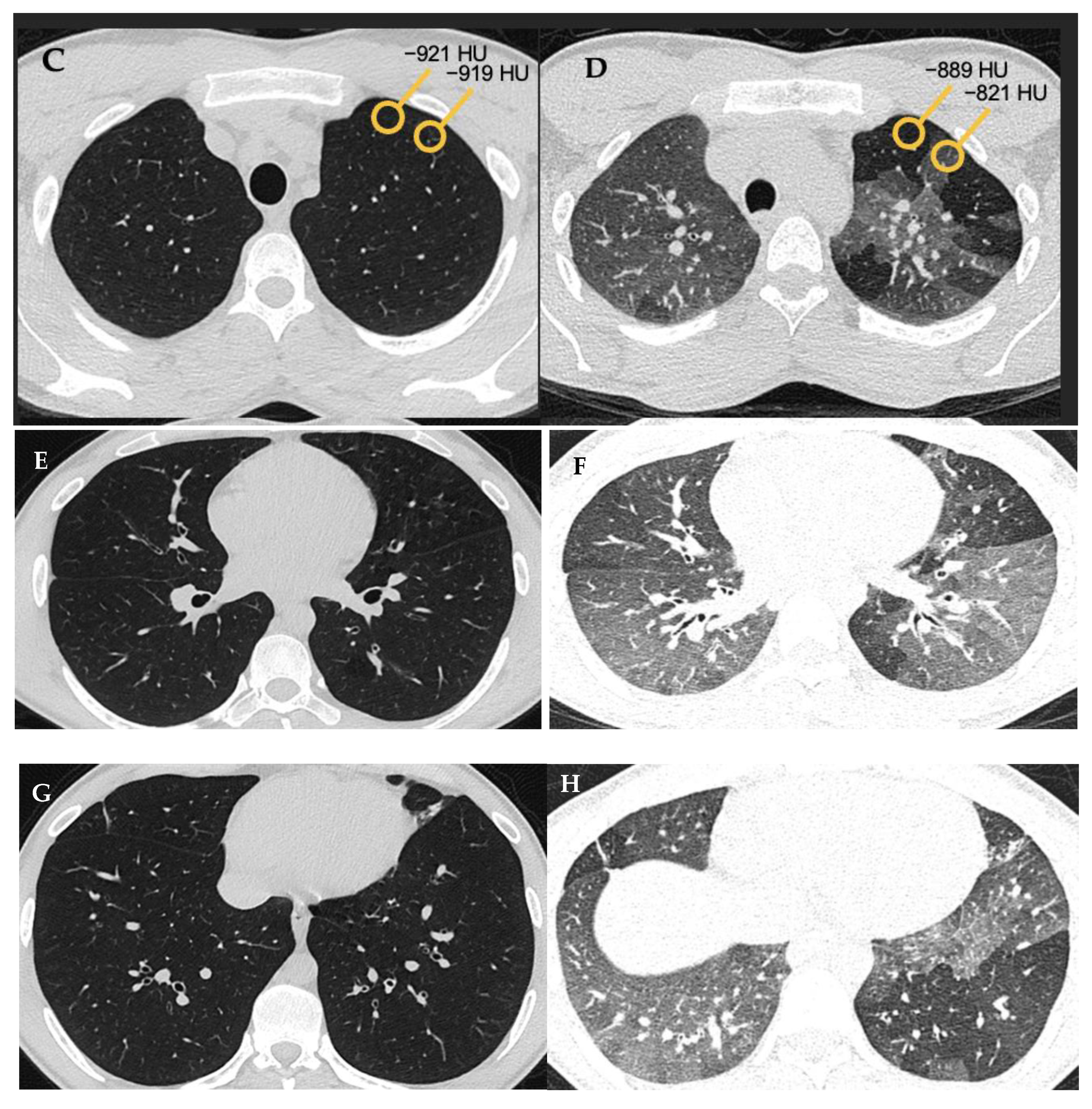
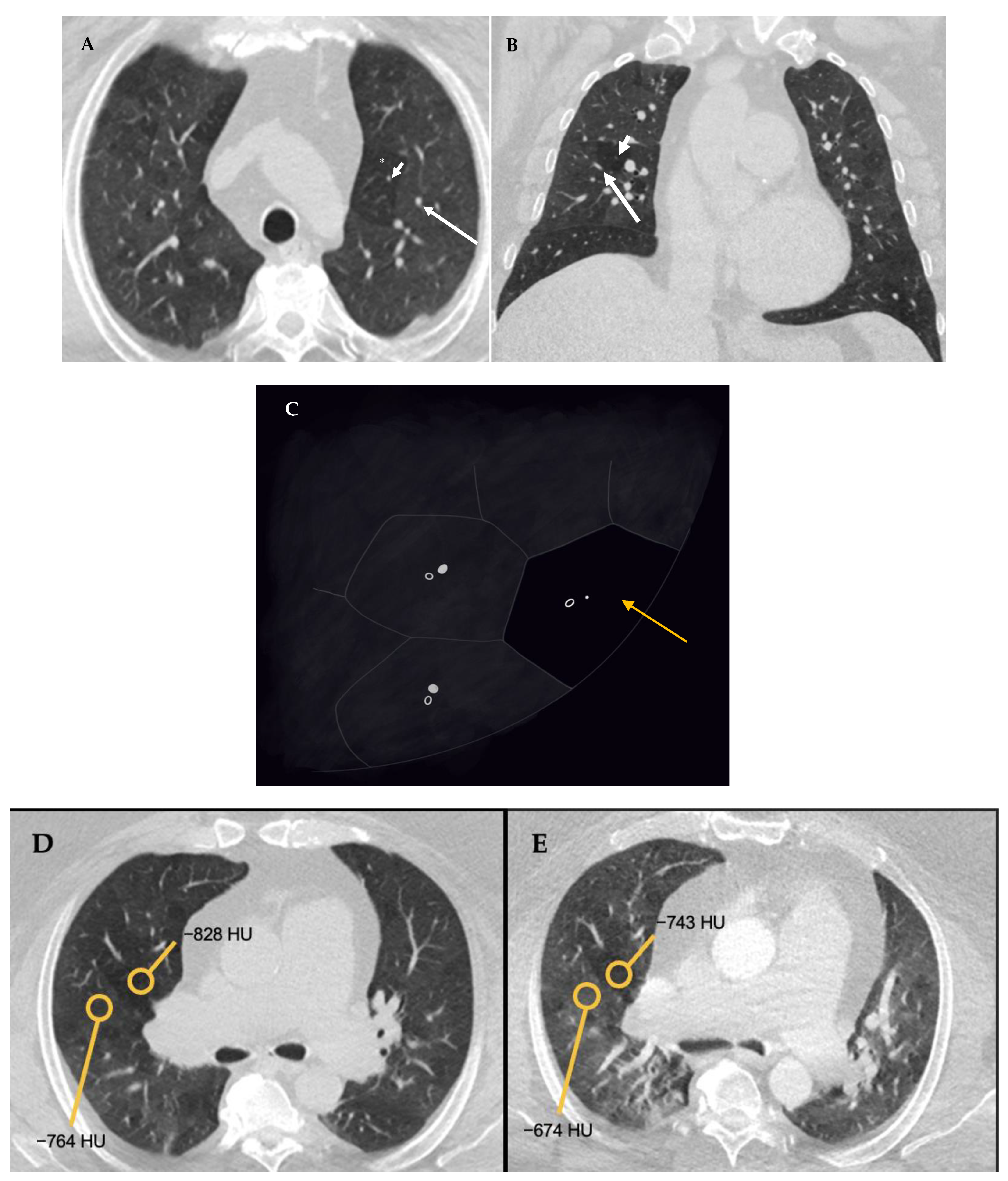
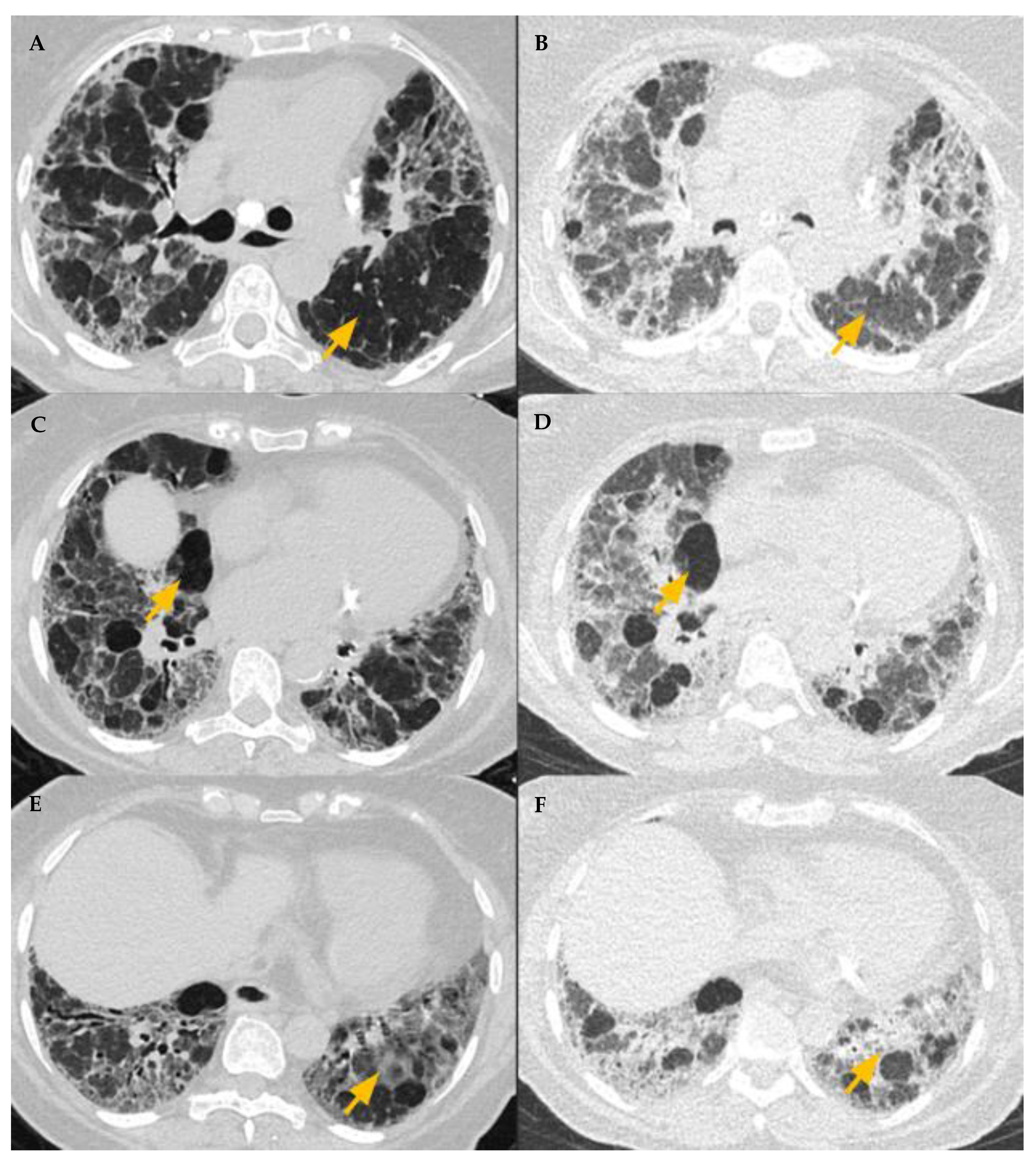
5.4. HRCT Patterns of HP
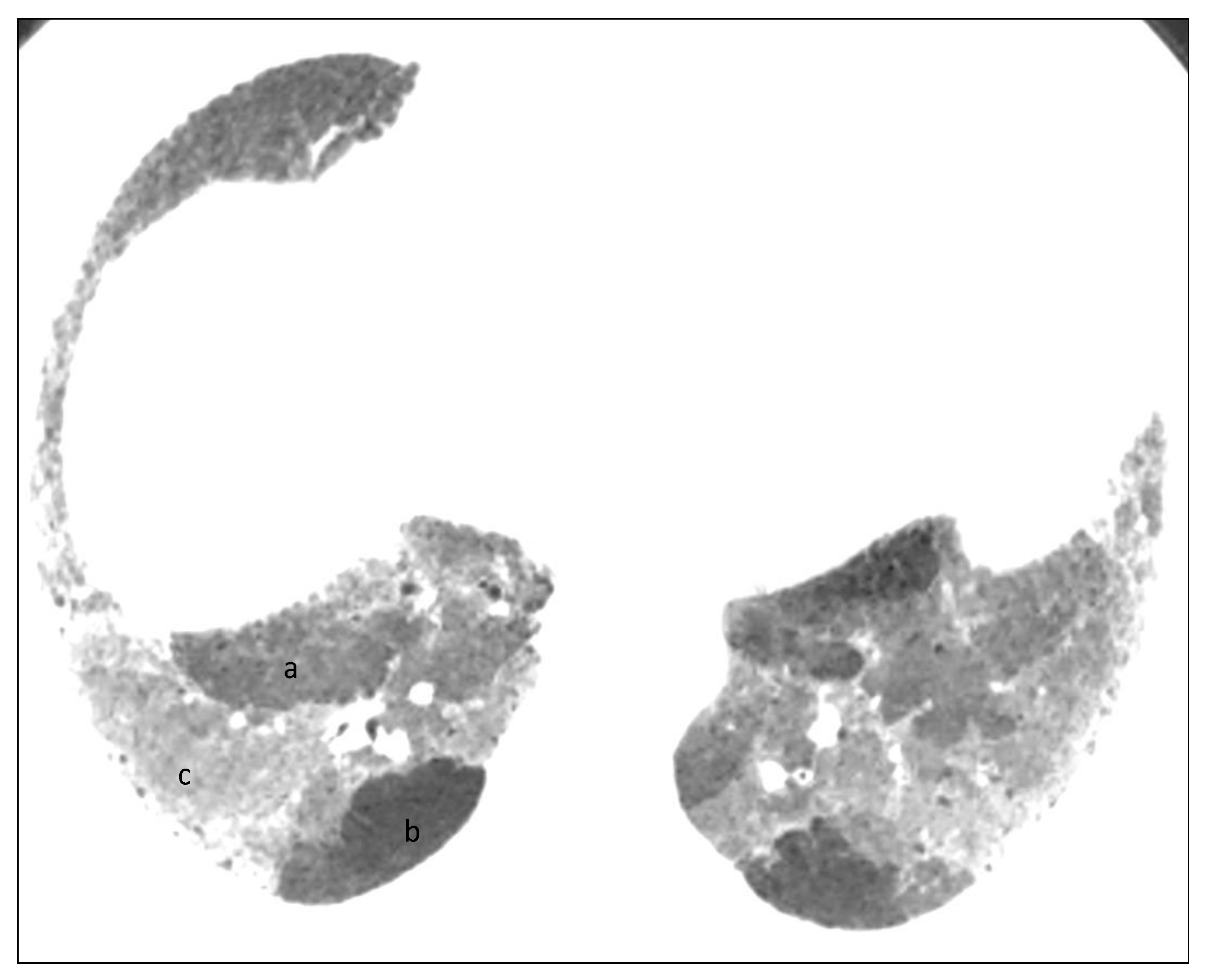
6. Histopathologic Features of HP
7. Diagnostic Algorithm and Challenging Scenarios
8. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buendia-Roldan, I.; Aguilar-Duran, H.; Johannson, K.; Selman, M. Comparing the performance of two recommended criteria for establishing a diagnosis for hypersensitivity pneumonitis. Am. J. Respir. Crit. Care Med. 2021, 204, 865–868. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Ryerson, C.; Myers, J.; Kreuter, J.; Vasakova, M.; Bargagli, E.; Chung, J.; Collins, B.; Bendstrup, E.; et al. Diagnosis of hypersensitivity pneumonitis in adults. An official ATS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit. Care Med. 2020, 202, e36–e69. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.; Egan, J.; Martinez, F.; Behr, J.; Brown, K.; Colby, T.; Cordier, J.; Flaherty, K.; Lynch, D. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [Green Version]
- Lynch, D.A.; Newell, J.; Logan, P.; King, T., Jr.; Muller, N. Can CT distinguish hypersensitivity pneumonitis from idiopathic pulmonary fibrosis? AJR. Am. J. Roentgenol. 1995, 165, 807–811. [Google Scholar] [CrossRef]
- Silva, C.I.S.; Muller, N.; Lynch, D.A.; Curran-Everett, D.; Brown, K.; Lee, K.; Chung, M.; Churg, A. Chronic hypersensitivity pneumonitis: Differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology 2008, 246, 288–297. [Google Scholar] [CrossRef]
- Fernández Pérez, E.R.; Kong, A.; Raimundo, K.; Koelsch, T.; Kulkarni, R.; Cole, A. Epidemiology of hypersensitivity pneumonitis among an insured population in the United States: A claims-based cohort analysis. Ann. Am. Thorac. Soc. 2018, 15, 460–469. [Google Scholar] [CrossRef]
- Pereira, C.A.; Gimenez, A.; Kuranishi, L.; Storrer, K. Chronic hypersensitivity pneumonitis. J. Asthma Allergy 2016, 9, 171. [Google Scholar] [CrossRef]
- Hochhegger, B.; Marchiori, E.; Zanon, M.; Rubin, A.S.; Fragomeni, R.; Altmayer, S.; Carvalho, C.R.R.; Baldi, B.G. Imaging in idiopathic pulmonary fibrosis: Diagnosis and mimics. Clinics 2019, 74, e225. [Google Scholar] [CrossRef]
- Pérez, E.R.F.; Travis, W.D.; Lynch, D.A.; Brown, K.K.; Selman, M.; Ryu, J.H.; Wells, A.U.; Tony Huang, Y.C.; Pereira, C.A.C. Diagnosis and evaluation of hypersensitivity pneumonitis: CHEST guideline and expert panel report. Chest 2021, 160, e97–e156. [Google Scholar] [CrossRef]
- Nogueira, R.; Melo, N.; Novais E Bastos, H.; Martins, N.; Delgado, L.; Morais, A.; Mota, P.C. Hypersensitivity pneumonitis: Antigen diversity and disease implications. Pulmonology 2019, 25, 97–108. [Google Scholar] [CrossRef]
- Richerson, H.B.; Bernstein, I.L.; Fink, J.N.; Hunninghake, G.W.; Novey, H.S.; Reed, C.E.; Salvaggio, J.E.; Schuyler, M.R.; Schwartz, H.J.; Stechschulte, D. Guidelines for the clinical evaluation of hypersensitivity pneumonitis: Report of the Subcommittee on Hypersensitivity Pneumonitis. J. Allergy Clin. Immunol. 1989, 84, 839–844. [Google Scholar] [CrossRef]
- Ojanguren, I.; Morell, F.; Ramon, M.A.; Villar, A.; Romero, C.; Cruz, M.J.; Munoz, X. Long-term outcomes in chronic hypersensitivity pneumonitis. Allergy 2019, 74, 944–952. [Google Scholar] [CrossRef]
- Leone, P.M.; Richeldi, L. Current Diagnosis and Management of Hypersensitivity Pneumonitis. Tuberc. Respir. Dis. 2020, 83, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Nishida, T.; Kawate, E.; Ishiguro, T.; Kanauchi, T.; Shimizu, Y.; Takayangi, N. Antigen avoidance and outcome of nonfibrotic and fibrotic hypersensitivity pneumonitis. ERJ Open Res. 2022, 8, 00474. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A.; King, T.E., Jr. Hypersensitivity pneumonitis: Insights in diagnosis and pathobiology. Am. J. Respir. Crit. Care Med. 2012, 186, 314–324. [Google Scholar] [CrossRef] [Green Version]
- Vasakova, M.; Selman, M.; Morell, F.; Sterclova, M.; Molina-Molina, M.; Raghu, G. Hypersensitivity pneumonitis: Current concepts of pathogenesis and potential targets for treatment. Am. J. Respir. Crit. Care Med. 2019, 200, 301–308. [Google Scholar] [CrossRef]
- Camarena, A.; Juárez, A.; Mejía, M.; Estrada, A.; Carrillo, G.; Falfán, R.; Zuñiga, J.; Navarro, C.; Granados, J.; Selman, M. Major histocompatibility complex and tumor necrosis factor-α polymorphisms in pigeon breeder’s disease. Am. J. Respir. Crit. Care Med. 2001, 163, 1528–1533. [Google Scholar] [CrossRef]
- Newton, C.A.; BaTra, K.; Torrealba, J.; Kozlitina, J.; Glazer, C.S.; Aravena, C.; Meyer, K.; Raghu, G.; Collard, H.R.; Garcia, C.K. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur. Respir. J. 2016, 48, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Adegunsoye, A.; Chung, J.H.; Ventura, I.B.; Jablonski, R.; Montner, S.; Vij, R.; Hines, S.E.; Strek, M.E. Characteristics and prevalence of domestic and occupational inhalational exposures across interstitial lung diseases. Chest 2021, 160, 209–218. [Google Scholar] [CrossRef]
- Cormier, Y.; Israël-Assayag, E. The role of viruses in the pathogenesis of hypersensitivity pneumonitis. Curr. Opin. Pulm. Med. 2000, 6, 420–423. [Google Scholar] [CrossRef]
- McSharry, C.; Banham, S.; Boyd, G. Effect of cigarette smoking on the antibody response to inhaled antigens and the prevalence of extrinsic allergic alveolitis among pigeon breeders. Clin. Exp. Allergy 1985, 15, 487–494. [Google Scholar] [CrossRef]
- Ohtsuka, Y.; Munakata, M.; Tanimura, K.; Ukita, H.; Kusaka, H.; Masaki, Y.; Doi, I.; Ohe, M.; Amishima, M.; Homma, Y. Smoking promotes insidious and chronic farmer’s lung disease, and deteriorates the clinical outcome. Intern. Med. 1995, 34, 966–971. [Google Scholar] [CrossRef] [Green Version]
- Pérez, E.R.F.; Koelsch, T.L.; Groshong, S.T.; Lynch, D.A.; Brown, K.K. Clinical decision-making in hypersensitivity pneumonitis: Diagnosis and management. In Seminars in Respiratory and Critical Care Medicine; Thieme Medical Publishers: Leipzig, Germany, 2020. [Google Scholar]
- Selman, M.; Pardo, A. When things go wrong: Exploring possible mechanisms driving the progressive fibrosis phenotype in interstitial lung diseases. Eur. Respir. J. 2021, 58, 2004507. [Google Scholar] [CrossRef]
- Petnak, T.; Moua, T. Exposure assessment in hypersensitivity pneumonitis: A comprehensive review and proposed screening questionnaire. ERJ Open Res. 2020, 6, 00230. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Radiology, A.C.O. ACR-STR Practice Parameter for the Performance of High-Resolution Computed Tomography (HRCT) of the Lungs in Adults; The American College of Radiology: Reston, VA, USA, 2019. [Google Scholar]
- Pontana, F.; Billard, A.S.; Duhamel, A.; Schmidt, B.; Faivre, J.B.; Hachulla, E.; Matran, R.; Remy, J.; Remy-Jardin, M. Effect of Iterative Reconstruction on the Detection of Systemic Sclerosis-related Interstitial Lung Disease: Clinical Experience in 55 Patients. Radiology 2016, 279, 297–305. [Google Scholar] [CrossRef]
- Winslow, J.; Zhang, Y.; Samei, E. A method for characterizing and matching CT image quality across CT scanners from different manufacturers. Med. Phys. 2017, 44, 5705–5717. [Google Scholar] [CrossRef]
- King, T.; Nicholson, A. Hypersensitivity pneumonitis (extrinsic allergic alveolitis): Clinical manifestations and diagnosis. Up-to-Date. Retrieved Febr. 2019, 26, 2021. [Google Scholar]
- Moua, T.; Pentak, T.; Charokopos, A.; Baqir, M.; Ryu, J.H. Challenges in the Diagnosis and Management of Fibrotic Hypersensitivity Pneumonitis: A Practical Review of Current Approaches. J. Clin. Med. 2022, 11, 1473. [Google Scholar] [CrossRef]
- Chavez-Galan, L.; Buendia-Roldan, I.; Castillo-Castillo, K.; Preciado-Garcia, M.; Ocana-Guzman, R.; Salgado, A.; Gaxiola, M.; Selman, M. Decreased expression of transmembrane TNFR2 in lung leukocytes subpopulations of patients with non-fibrotic hypersensitivity pneumonitis compared with the fibrotic disease. Clin. Immunol. 2020, 215, 108424. [Google Scholar] [CrossRef]
- Baqir, M.; White, D.; Ryu, J.H. Emphysematous changes in hypersensitivity pneumonitis: A retrospective analysis of 12 patients. Respir. Med. Case Rep. 2018, 24, 25–29. [Google Scholar] [CrossRef]
- Kouranos, V.; Jacob, J.; Nicholson, A.; Renzoni, E. Fibrotic hypersensitivity pneumonitis: Key issues in diagnosis and management. J. Clin. Med. 2017, 6, 62. [Google Scholar] [CrossRef] [Green Version]
- Hansell, D.M.; Bankier, A.A.; MacMahon, H.; McLoud, T.C.; Muller, N.L.; Remy, J. Fleischner Society: Glossary of terms for thoracic imaging. Radiology 2008, 246, 697–722. [Google Scholar] [CrossRef] [Green Version]
- Webb, W.R.; Müller, N.L.; Naidich, D.P. Standardized terms for high-resolution computed tomography of the lung: A proposed glossary. J. Thorac. Imaging 1993, 8, 167–175. [Google Scholar] [CrossRef]
- Chung, M.H.; Edinburg, K.J.; Webb, E.M.; McCowin, M.; Webb, W.R. Mixed infiltrative and obstructive disease on high-resolution CT: Differential diagnosis and functional correlates in a consecutive series. J. Thorac. Imaging 2001, 16, 69–75. [Google Scholar] [CrossRef]
- Barnett, J.; Molyneaux, P.L.; Rawal, B.; Abdullah, R.; Hare, S.S.; Vancheeswaran, R.; Desai, S.R.; Maher, T.M.; Wells, A.U.; Devaraj, A. Variable utility of mosaic attenuation to distinguish fibrotic hypersensitivity pneumonitis from idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 54, 1900531. [Google Scholar] [CrossRef]
- Koster, M.A.; Thomson, C.C.; Collins, B.F.; Jenkins, A.R.; Ruminjo, J.K.; Raghu, G. Diagnosis of hypersensitivity pneumonitis in adults 2020 clinical practice guideline: Summary for clinicians. Ann. Am. Thorac. Soc. 2021, 18, 559–566. [Google Scholar] [CrossRef]
- Fernández Pérez, E.R.; Swigris, J.J.; Forssen, A.V.; Tourin, O.; Solomon, J.J.; Huie, T.J.; Olson, A.L.; Brown, K.K. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest 2013, 144, 1644–1651. [Google Scholar] [CrossRef] [Green Version]
- Johannson, K.A.; Elicker, B.M.; Vittinghoff, E.; Assayag, D.; Boer, K.; Golden, J.A.; Jones, K.D.; King, T.E., Jr.; Koth, L.L.; Lee, J.S. A diagnostic model for chronic hypersensitivity pneumonitis. Thorax 2016, 71, 951–954. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.T.; Chatzkel, J.; Hewitt, M.G. Expiratory air trapping on thoracic computed tomography. A diagnostic subclassification. Ann. Am. Thorac. Soc. 2014, 11, 874–881. [Google Scholar] [CrossRef]
- Groner, L.K.; Green, D.B.; Weisman, S.V.; Legasto, A.C.; Toy, D.; Gruden, J.F.; Escalon, J.G. Thoracic Manifestations of Rheumatoid Arthritis. RadioGraphics 2021, 41, 32–55. [Google Scholar] [CrossRef]
- Hochhegger, B.; Sanches, F.D.; Altmayer, S.P.L.; Pacini, G.S.; Zanon, M.; Guedes, A.C.B.; Watte, G.; Meirelles, G.; Barros, M.C.; Marchiori, E. Air trapping in usual interstitial pneumonia pattern at CT: Prevalence and prognosis. Sci. Rep. 2018, 8, 17267. [Google Scholar] [CrossRef] [Green Version]
- Silva, C.I.S.; Muller, N.L.; Hansell, D.M.; Lee, K.S.; Nicholson, A.G.; Wells, A.U. Nonspecific interstitial pneumonia and idiopathic pulmonary fibrosis: Changes in pattern and distribution of disease over time. Radiology 2008, 247, 251–259. [Google Scholar] [CrossRef]
- Morisset, J.; Johannson, K.A.; Jones, K.D.; Wolters, P.J.; Collard, H.R.; Walsh, S.L.F.; Ley, B.; HP Delphi Collaborators. Identification of diagnostic criteria for chronic hypersensitivity pneumonitis. An International Modified Delphi Survey. Am. J. Respir. Crit. Care Med. 2018, 197, 1036–1044. [Google Scholar] [CrossRef]













| Age | Unknown Antigen | VC, DLCO * | Lymphocytes (%) in BAL | |
|---|---|---|---|---|
| Non-fibrotic HP | Younger | Less common | Low | Higher |
| Fibrotic HP | Older | More common | Lower | Lower |
| Typical HP Pattern (Suggests a Diagnosis of HP) | Compatible with HP | Indeterminate for HP |
|---|---|---|
| At least one finding indicative of small airway disease | Not applicable | |
| Air trapping | ||
| Ill-defined <5 mm centrilobular nodules | ||
| At least one finding indicative of parenchymal infiltration | ||
| Mosaic attenuation | Diffuse and subtle GGO | |
| GGOs | Airspace consolidation | |
| Lung cysts | ||
| Distribution of findings | ||
| Craniocaudal: diffuse +/− basal sparing | Craniocaudal: diffuse (variant: lower lobe predominance) | |
| Axial: diffuse | Axial: diffuse (variant: peribronchovascular) | |
| Typical HP Pattern (Suggests a Diagnosis of HP) | Compatible with HP | Indeterminate for HP |
|---|---|---|
| At least one finding indicative of small airway disease | At least one finding indicative of small airway disease | Neither Typical nor Compatible with HP |
| Three-density pattern | Three-density pattern | HRCT Patterns: UIP pattern Probably UIP pattern Indeterminate for UIP Fibrotic NSIP pattern OP like pattern Truly indeterminate pattern |
| Air trapping | Air trapping | |
| Ill-defined <5 mm centrilobular nodules | Ill-defined <5 mm centrilobular nodules | |
| At least one finding indicative of pulmonary fibrosis | Variant pattern of fibrosis | |
| Coarse reticulations with distortion | UIP pattern of fibrosis | |
| Traction bronchiectasis | Extensive GGO and superimposed subtle fibrosis | |
| Honeycombing (not dominant) | ||
| Distribution of findings | Variant distribution of fibrosis | |
| Random axially and craniocaudally | Craniocaudal: Upper lung zone predominant | |
| Mid zone predominant | Axial: peribronchovascular, subpleural | |
| Relative sparing of the bases |
| HP | Probable HP | Indeterminate for HP |
|---|---|---|
| Nonfibrotic HP | ||
| All three following features in at least one biopsy site | Both of the following features in at least one biopsy site | Presence of one of the following in at least one biopsy site |
| Cellular interstitial pneumonia | Cellular interstitial pneumonia | Cellular interstitial pneumonia |
| Cellular bronchiolitis | Cellular bronchiolitis | Cellular bronchiolitis |
| Poorly formed non-necrotizing granulomas | Selected IIP* patterns | |
| Absence of features suggesting any alternative diagnosis in any biopsy site | Absence of features suggesting any alternative diagnosis in any biopsy site | Absence of features suggesting any alternative diagnosis in any biopsy site |
| Fibrotic HP | ||
| All three following features in at least one biopsy site | Both of the following features in at least one biopsy site | Presence of the following in at least one biopsy site |
| Chronic fibrosing interstitial pneumonia | Chronic fibrosing interstitial pneumonia | Chronic fibrosing interstitial pneumonia |
| Airway-centered fibrosis | Airway-centered fibrosis | |
| Poorly formed non-necrotizing granulomas | ||
| Absence of features suggesting any alternative diagnosis in any biopsy site † | Absence of features suggesting any alternative diagnosis in any biopsy site | Absence of features suggesting any alternative diagnosis in any biopsy site |
| HRCT | ||||||
|---|---|---|---|---|---|---|
| Typical for HP | Compatible with HP | Indeterminate for HP | ||||
| History of exposure and/or serum IgG testing | Exposure + | Exposure − | Exposure + | Exposure − | Exposure + | Exposure − |
| No BAL or BAL without lymphocytosis and either no or indeterminate histopathology | Moderate confidence | Low confidence | Low confidence | Not excluded | Not excluded | Not excluded |
| BAL lymphocytosis without histopathology sampling | High confidence | Moderate confidence | Moderate confidence | Low confidence | Low confidence | Not excluded |
| BAL lymphocytosis with indeterminate histopathology | Definite | High confidence | Moderate confidence | Moderate confidence | Low confidence | Not excluded |
| Probable HP histopathology | Definite | High confidence | High confidence | Moderate confidence | Moderate confidence | Low confidence |
| Typical HP histopathology | Definite | Definite | Definite | Definite | Definite | High confidence |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dabiri, M.; Jehangir, M.; Khoshpouri, P.; Chalian, H. Hypersensitivity Pneumonitis: A Pictorial Review Based on the New ATS/JRS/ALAT Clinical Practice Guideline for Radiologists and Pulmonologists. Diagnostics 2022, 12, 2874. https://doi.org/10.3390/diagnostics12112874
Dabiri M, Jehangir M, Khoshpouri P, Chalian H. Hypersensitivity Pneumonitis: A Pictorial Review Based on the New ATS/JRS/ALAT Clinical Practice Guideline for Radiologists and Pulmonologists. Diagnostics. 2022; 12(11):2874. https://doi.org/10.3390/diagnostics12112874
Chicago/Turabian StyleDabiri, Mona, Maham Jehangir, Pegah Khoshpouri, and Hamid Chalian. 2022. "Hypersensitivity Pneumonitis: A Pictorial Review Based on the New ATS/JRS/ALAT Clinical Practice Guideline for Radiologists and Pulmonologists" Diagnostics 12, no. 11: 2874. https://doi.org/10.3390/diagnostics12112874