
Recessive Dystrophic Epidermolysis bullosa due to Hemizygous 40 kb Deletion of COL7A1 and the Proximate PFKFB4 Gene Focusing on the Mutation c.425A>G Mimicking Homozygous Status

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. NGS-Based Mutation Analysis
2.2. Immunofluorescence (IF) Antigen Mapping
2.3. PCR-Based STR Typing
2.4. Copy Number Analysis via Multiplex Ligation-Dependent Probe Amplification (MLPA)
2.5. Linear Amplification Mediated PCR (LAM-PCR)
2.6. Confirmation of the 40 kb Deletion with PCR
3. Results
3.1. Immunofluorescence (IF) Antigen Mapping
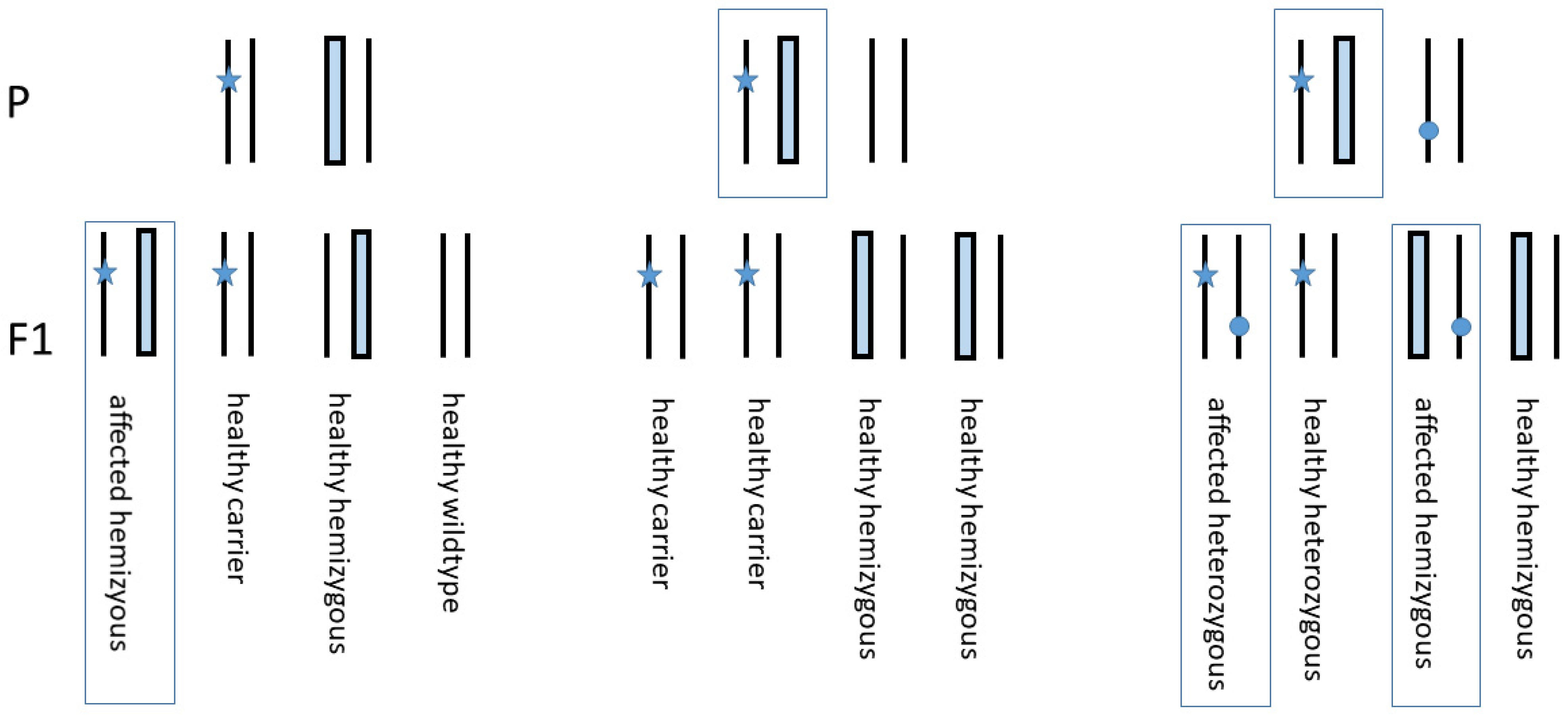
3.2. Genotyping Discrepancy of c.425A>G and c.6654C>G within the Family
3.3. Paternity Testing
3.4. STR Marker Test on Chromosome 3 Adjacent COL7A1
3.5. Copy Number Analysis Revealed COL7A1 Hemizygosity of the Patient
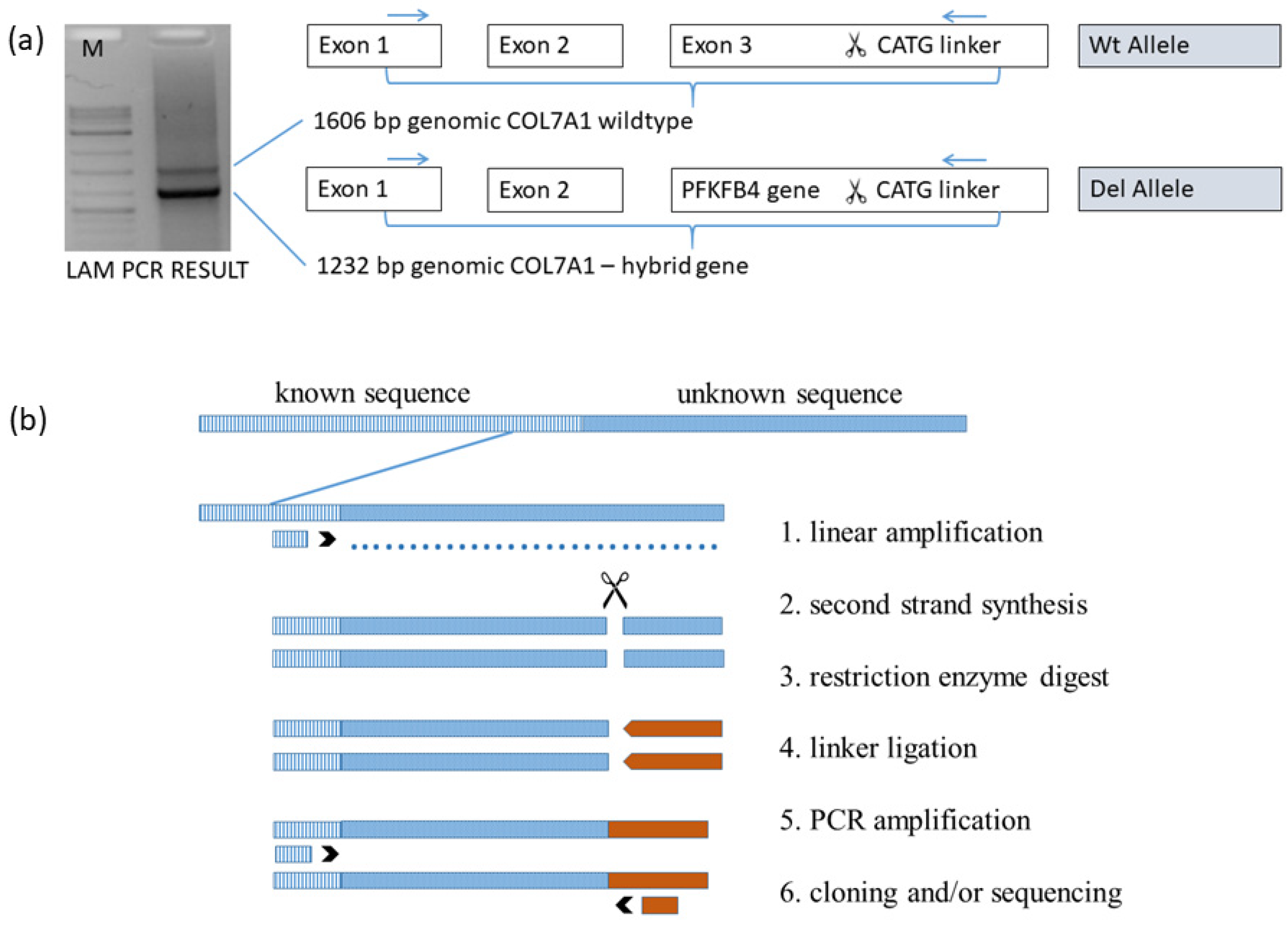
3.6. Linear Amplification Mediated PCR Exactly Defined the Breaking Points of Deletion
3.7. Confirmation of the 40 kb Deletion via PCR
4. Discussion
Genetic Counselling
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GRCh38.p13. | Genome Reference Consortium Human Build 38 patch release 13 |
| COL7A1 | Collagen type VII, alpha 1 |
| PFKFB4 | 6-Phosphofructo-2-kinase/fructose2,6-biphosphatase 4 |
| UCN2 | Urocortin 2 |
| UPD | Uniparental isodisomy |
| LAM-PCR | Linear amplification-mediated PCR |
| MLPA | Multiplex ligation-dependent probe amplification |
References
- Fine, J.-D.; Eady, R.A.; Bauer, E.A.; Bauer, J.W.; Bruckner-Tuderman, L.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; Leigh, I.; et al. The classification of inherited Epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J. Am. Acad. Dermatol. 2008, 58, 931–950. [Google Scholar] [CrossRef]
- Christiano, A.M.; Hoffman, G.G.; Chung-Honet, L.C.; Lee, S.; Cheng, W.; Uitto, J.; Greenspan, D.S. Structural Organization of the Human Type VII Collagen Gene (COL7A1), Composed of More Exons Than Any Previously Characterized Gene. Genomics 1994, 21, 169–179. [Google Scholar] [CrossRef]
- McGrath, J.A.; Ishida-Yamamoto, A.; O’Grady, A.; Leigh, I.M.; Eady, R.A. Structural Variations in Anchoring Fibrils in Dystrophic Epidermolysis bullosa: Correlation with Type VII Collagen Expression. J. Investig. Dermatol. 1993, 100, 366–372. [Google Scholar] [CrossRef] [Green Version]
- Hilal, L.; Rochat, A.; Duquesnoy, P.; Blanchet-Bardon, C.; Wechsler, J.; Martin, N.; Christiano, A.M.; Barrandon, Y.; Uitto, J.; Goossens, M.; et al. A homozygous insertion–deletion in the type VII collagen gene (COL7A1) in Hallopeau–Siemens dystrophic Epidermolysis bullosa. Nat. Genet. 1993, 5, 287–293. [Google Scholar] [CrossRef]
- Hovnanian, A.; Rochat, A.; Bodemer, C.; Petit, E.; Rivers, C.A.; Prost, C.; Fraitag, S.; Christiano, A.M.; Uitto, J.; Lathrop, M.; et al. Characterization of 18 New Mutations in COL7A1 in Recessive Dystrophic Epidermolysis bullosa Provides Evidence for Distinct Molecular Mechanisms Underlying Defective Anchoring Fibril Formation. Am. J. Hum. Genet. 1997, 61, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Uitto, J.; Pulkkinen , L.; Christiano, A.M. Molecular basis of the dystrophic and junctional forms of Epidermolysis bullosa: Mutations in the type VII collagen and kalinin (laminin 5) genes. J. Investig. Dermatol. 1994, 103, 39S–46S. [Google Scholar]
- Whittock, N.V.; Ashton, G.H.; Mohammedi, R.; Mellerio, J.E.; Mathew, C.; Abbs, S.J.; Eady, R.A.; McGrath, J. Comparative Mutation Detection Screening of the Type VII Collagen Gene (COL7A1) Using the Protein Truncation Test, Fluorescent Chemical Cleavage of Mismatch, and Conformation Sensitive Gel Electrophoresis. J. Investig. Dermatol. 1999, 113, 673–686. [Google Scholar] [CrossRef] [Green Version]
- Dang, N.; Murrell, D.F. Mutation analysis and characterization of COL7A1 mutations in dystrophic Epidermolysis bullosa. Exp. Dermatol. 2008, 17, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Jarvikallio, A.; Pulkkinen, L.; Uitto, J. Molecular basis of dystrophic Epidermolysis bullosa: Mutations in the type VII collagen gene (COL7A1). Hum. Mutat. 1997, 10, 338–347. [Google Scholar] [CrossRef]
- Kotzot, D.; Utermann, G. Uniparental disomy (UPD) other than 15: Phenotypes and bibliography updated. Am. J. Med Genet. Part A 2005, 136A, 287–305. [Google Scholar] [CrossRef]
- Lin, S.-P.; Huang, S.-Y.; Tu, M.-E.; Wu, Y.-H.; Lin, C.-Y.; Lin, H.-Y.; Lee-Chen, G.-J. Netherton syndrome: Mutation analysis of two Taiwanese families. Arch. Dermatol. Res. 2007, 299, 145–150. [Google Scholar] [CrossRef]
- Numata, S.; Hamada, T.; Teye, K.; Matsuda, M.; Ishii, N.; Karashima, T.; Kabashima, K.; Furumura, M.; Ohata, C.; Hashimoto, T. Complete Maternal Isodisomy of Chromosome 5 in a Japanese Patient with Netherton Syndrome. J. Investig. Dermatol. 2014, 134, 849–852. [Google Scholar] [CrossRef] [Green Version]
- Castori, M.; Floriddia, G.; Pisaneschi, E.; Covaciu, C.; Paradisi, M.; Torrente, I.; Castiglia, D. Complete maternal isodisomy causing reduction to homozygosity for a novel LAMB3 mutation in Herlitz junctional Epidermolysis bullosa. J. Dermatol. Sci. 2008, 51, 58–61. [Google Scholar] [CrossRef]
- Higgins, R.; Jensen, A.; Wachstein, J.; Bruckner-Tuderman, L.; Spiegel, R.; Traber, H.; Achermann, J.; Schaller, M.; Fehrenbacher, B.; Röcken, M.; et al. Uniparental Disomy of Chromosome 2 Unmasks New ITGA6 Recessive Mutation and Results in a Lethal Junctional Epidermolysis bullosa in a Newborn. Acta Derm. Venereol. 2020, 100, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Sawamura, D.; Goto, M.; Nakamura, H.; McMillan, J.R.; Park, S.; Kono, S.; Hasegawa, S.; Paku, S.; Nakamura, T.; et al. Epidermolysis bullosa Simplex Associated with Pyloric Atresia Is a Novel Clinical Subtype Caused by Mutations in the Plectin Gene (PLEC1). J. Mol. Diagn. 2005, 7, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Pulkkinen, L.; Bullrich, F.; Czarnecki, P.; Weiss, L.; Uitto, J. Maternal Uniparental Disomy of Chromosome 1 with Reduction to Homozygosity of the LAMB3 Locus in a Patient with Herlitz Junctional Epidermolysis bullosa. Am. J. Hum. Genet. 1997, 61, 611–619. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, Y.; Pulkkinen, L.; Chao, S.-C.; Nakajima, H.; Nakano, Y.; Shimizu, H.; Uitto, J. Complete Paternal Uniparental Isodisomy of Chromosome 1: A Novel Mechanism for Herlitz Junctional Epidermolysis bullosa. J. Investig. Dermatol. 2000, 115, 307–311. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, Y.; Pulkkinen, L.; Shimizu, H.; Lin, L.; Hagiwara, S.; Nishikawa, T.; Uitto, J. Maternal Uniparental Meroisodisomy in the LAMB3 Region of Chromosome 1 Results in Lethal Junctional Epidermolysis bullosa. J. Investig. Dermatol. 1998, 110, 828–831. [Google Scholar] [CrossRef] [PubMed]
- Fassihi, H.; Wessagowit, V.; Ashton, G.H.S.; Moss, C.; Ward, R.; Denyer, J.; Mellerio, J.E.; McGrath, J.A. Complete paternal uniparental isodisomy of chromosome 1 resulting in Herlitz junctional Epidermolysis bullosa. Clin. Exp. Dermatol. 2005, 30, 71–74. [Google Scholar] [CrossRef]
- Fassihi, H.; Lu, L.; Wessagowit, V.; Ozoemena, L.C.; Jones, C.A.; Dopping-Hepenstal, P.J.; Foster, L.; Atherton, D.J.; Mellerio, J.E.; McGrath, J.A. Complete Maternal Isodisomy of Chromosome 3 in a Child with Recessive Dystrophic Epidermolysis bullosa but No Other Phenotypic Abnormalities. J. Investig. Dermatol. 2006, 126, 2039–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hon, K.E.; Burd, A.; Choi, P.C.; Luk, N.T. Epidermolysis bullosa in three Chinese neonates. J. Dermatol. Treat. 2007, 18, 306–311. [Google Scholar] [CrossRef]
- Aradhya, S.; Lewis, R.; Bonaga, T.; Nwokekeh, N.; Stafford, A.; Boggs, B.; Hruska, K.; Smaoui, N.; Compton, J.G.; Richard, G.; et al. Exon-level array CGH in a large clinical cohort demonstrates increased sensitivity of diagnostic testing for Mendelian disorders. Genet. Med. 2012, 14, 594–603. [Google Scholar] [CrossRef] [Green Version]
- Kern, J.; Grüninger, G.; Imsak, R.; Müller, M.; Schumann, H.; Kiritsi, D.; Emmert, S.; Borozdin, W.; Kohlhase, J.; Bruckner-Tuderman, L.; et al. Forty-two novelCOL7A1mutations and the role of a frequent single nucleotide polymorphism in theMMP1promoter in modulation of disease severity in a large European dystrophic Epidermolysis bullosa cohort. Br. J. Dermatol. 2009, 161, 1089–1097. [Google Scholar] [CrossRef]
- Lee, M.; Xu, G.; Wang, K.; Wang, H.; Zhang, J.; Tang, Z.; Lin, Z.; Yang, Y. Recessive dystrophic Epidermolysis bullosa caused by ade novointerstitial deletion spanningCOL7A1and a hemizygous splicing mutation intrans. Clin. Exp. Dermatol. 2016, 41, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Titeux, M.; Mejía, J.E.; Mejlumian, L.; Bourthoumieu, S.; Mirval, S.; Tonasso, L.; Heller, M.; Prost-Squarcioni, C.; Hovnanian, A. Recessive dystrophic Epidermolysis bullosa caused by COL7A1 hemizygosity and a missense mutation with complex effects on splicing. Hum. Mutat. 2006, 27, 291–292. [Google Scholar] [CrossRef] [PubMed]
- Csikos, M.; Szocs, H.; Laszik, A.; Mecklenbeck, S.; Horvath, A.; Karpati, S.; Bruckner-Tuderman, L. High frequency of the 425AG splice-site mutation and novel mutations of the COL7A1 gene in central Europe: Significance for future mutation detection strategies in dystrophic Epidermolysis bullosa. Br. J. Dermatol. 2005, 152, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Gardella, R.; Belletti, L.; Zoppi, N.; Marini, D.; Barlati, S.; Colombi, M. Identification of two splicing mutations in the collagen type VII gene (COL7A1) of a patient affected by the localisata variant of recessive dystrophic Epidermolysis bullosa. Am. J. Hum. Genet. 1996, 59, 292–300. [Google Scholar] [PubMed]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.-D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus reclassification of inherited Epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quantifiler® HP and Trio DNA Quantification Kits User Guide; Revision E. Applied Biosystems by Life Technologies: Carlsbad, CA, USA, 2015.
- AmpFlSTR® NGM SElect™ PCR Amplification Kit User Guide; 03/2012. Applied Biosystems by Life Technologies: Waltham, MA, USA, 2012.
- GeneMapper ID-X, Version 1.4; Life Technologies: Carlsbad, CA, USA, 2012.
- ISO. General Requirements for the Competence of Testing and Calibration Laboratories (ISO/IEC 17025;2005); ISO: Berlin, Germany, 2005; p. 73. [Google Scholar]
- Baur, M.-P.; Fimmers, R.; Spitz, W. Program for Biostatstical Kinship Analysis, Version 2.1.0.54-DEU-04.02.18; Institute for Medical Biometrics, Informatics and Epidemiology: Essen, Germany, 2016.
- Schmidt, M.; Schwarzwaelder, K.; Bartholomae, C.; Zaoui, K.; Ball, C.; Pilz, I.; Braun, S.; Glimm, H.; von Kalle, C. High-resolution insertion-site analysis by linear amplification–mediated PCR (LAM-PCR). Nat. Methods 2007, 4, 1051–1057. [Google Scholar] [CrossRef]
- Huang, N.; Lee, I.; Marcotte, E.; Hurles, M.E. Characterising and Predicting Haploinsufficiency in the Human Genome. PLoS Genet. 2010, 6, e1001154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotowski, K.; Rosik, J.; Machaj, F.; Supplitt, S.; Wiczew, D.; Jabłońska, K.; Wiechec, E.; Ghavami, S.; Dzięgiel, P. Role of PFKFB3 and PFKFB4 in Cancer: Genetic Basis, Impact on Disease Development/Progression, and Potential as Therapeutic Targets. Cancers 2021, 13, 909. [Google Scholar] [CrossRef]
- Lu, H.; Chen, S.; You, Z.; Xie, C.; Huang, S.; Hu, X. PFKFB4 negatively regulated the expression of histone acetyltransferase GCN5 to mediate the tumorigenesis of thyroid cancer. Dev. Growth Differ. 2020, 62, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Bonamonte, D.; Filoni, A.; De Marco, A.; Lospalluti, L.; Nacchiero, E.; Ronghi, V.; Colagrande, A.; Giudice, G.; Cazzato, G. Squamous Cell Carcinoma in Patients with Inherited Epidermolysis bullosa: Review of Current Literature. Cells 2022, 11, 1365. [Google Scholar] [CrossRef]
- Filoni, A.; Cicco, G.; Cazzato, G.; Bosco, A.; Lospalluti, L.; Tucci, M.; Cimmino, A.; Foti, C.; Marzullo, A.; Bonamonte, D. Immune Disregulation in Cutaneous Squamous Cell Carcinoma of Patients with Recessive Dystrophic Epidermolysis bullosa: A Single Pilot Study. Life 2022, 12, 213. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MS Marker | Mother | Patient | Father | |||
|---|---|---|---|---|---|---|
| D10S1248 | 14 | 15 | 14 | 14 | 14 | 14 |
| vWA | 17 | 16 | 17 | 15 | 15 | 15 |
| D16S539 | 11 | 12 | 11 | 9 | 14 | 9 |
| D2S1338 | 25 | 18 | 25 | 26 | 17 | 26 |
| AMEL | X | X | X | Y | X | Y |
| D8S1179 | 14 | 12 | 14 | 13 | 11 | 13 |
| D21S11 | 30 | 29 | 30 | 29 | 29 | 29 |
| D18S51 | 16 | 13 | 16 | 18 | 21 | 18 |
| D22S1045 | 16 | 15 | 16 | 11 | 15 | 11 |
| D19S433 | 13 | 14 | 13 | 14 | 15 | 14 |
| TH01 | 8 | 7 | 8 | 9.3 | 9 | 9.3 |
| FGA | 22 | 20.2 | 22 | 22 | 24 | 22 |
| D2S441 | 14 | 14 | 14 | 14 | 14 | 14 |
| D3S1358 | 18 | 14 | 18 | 15 | 15 | 15 |
| D1S1656 | 15 | 16.3 | 15 | 18.3 | 16 | 18.3 |
| D12S391 | 18 | 18 | 18 | 16 | 17.3 | 16 |
| SE33 | 14 | 19 | 14 | 14 | 29.2 | 14 |
| D3S1581 | 86 | 111 | 86 | 86 | 84 | 86 |
| * COL7A1 c.6654C>G (ex84) | var | wt | var | del | wt | del |
| * COL7A1 c.425A>G (ex3) | mut | wt | mut | del | wt | del |
| D3S3629 | 246 | 244 | 246 | 250 | 250 | 250 |
| D3S1289 | 200 | 189 | 200 | 200 | 202 | 200 |
| Gene Symbol 1 | Gene Name 1 | Protein Function | OMIM 2 |
|---|---|---|---|
| COL7A1 | Collagen type VII, alpha 1 (Epidermolysis bullosa, dystrophic, dominant and recessive) | Assembles into anchoring fibrils, ensuring adherence of epidermis to the underlying dermis. | 120120 |
| UCN2 | Urocortin 2 | Specific ligand for the type 2 CRH receptor, which mediates stress coping responses during the recovery phase of stress. | 605902 |
| PFKFB4 | 6-phosphofructo-2-kinase/fructose- -2,6-biphosphatase 4 | Regulates the steady-state concentration of fructose-2,6-bisphosphate in the glycolysis pathway. | 605320 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klausegger, A.; Jeschko, N.; Grammer, M.; Cemper-Kiesslich, J.; Neuhuber, F.; Diem, A.; Breitenbach-Koller, H.; Sander, G.; Kotzot, D.; Bauer, J.W.; et al. Recessive Dystrophic Epidermolysis bullosa due to Hemizygous 40 kb Deletion of COL7A1 and the Proximate PFKFB4 Gene Focusing on the Mutation c.425A>G Mimicking Homozygous Status. Diagnostics 2022, 12, 2460. https://doi.org/10.3390/diagnostics12102460
Klausegger A, Jeschko N, Grammer M, Cemper-Kiesslich J, Neuhuber F, Diem A, Breitenbach-Koller H, Sander G, Kotzot D, Bauer JW, et al. Recessive Dystrophic Epidermolysis bullosa due to Hemizygous 40 kb Deletion of COL7A1 and the Proximate PFKFB4 Gene Focusing on the Mutation c.425A>G Mimicking Homozygous Status. Diagnostics. 2022; 12(10):2460. https://doi.org/10.3390/diagnostics12102460
Chicago/Turabian StyleKlausegger, Alfred, Niklas Jeschko, Markus Grammer, Jan Cemper-Kiesslich, Franz Neuhuber, Anja Diem, Hannelore Breitenbach-Koller, Gabriele Sander, Dieter Kotzot, Johann Wolfgang Bauer, and et al. 2022. "Recessive Dystrophic Epidermolysis bullosa due to Hemizygous 40 kb Deletion of COL7A1 and the Proximate PFKFB4 Gene Focusing on the Mutation c.425A>G Mimicking Homozygous Status" Diagnostics 12, no. 10: 2460. https://doi.org/10.3390/diagnostics12102460