
Subgroup-Specific Diagnostic, Prognostic, and Predictive Markers Influencing Pediatric Medulloblastoma Treatment

 ,
, 

Abstract
:1. Introduction
2. Wingless (WNT) Medulloblastoma
2.1. Targeting WNT/β-Catenin Signaling
2.2. Therapeutically Activating WNT/β-Catenin Signaling
3. Sonic Hedgehog (SHH) Medulloblastoma
3.1. Molecular Characteristics and Subtypes of SHH MB
3.2. Challenges in Treating SHH MB
3.3. Meeting the Challenges Posed by SOX2+ SHH MB Cancer Stem Cells
4. Group 3 Medulloblastoma
4.1. Molecular Characteristics and Subtypes of Group 3 MB
4.2. Risk Stratification, Prognostic Biomarkers, and Treatment Approaches
4.3. Pre-Clinical Group 3 Models Identify Druggable Pathways
5. Group 4 Medulloblastoma
5.1. Molecular Characteristics and Subtypes of Group 4 MB
5.2. Pre-Clinical Group 4 Models Identify Druggable Pathways
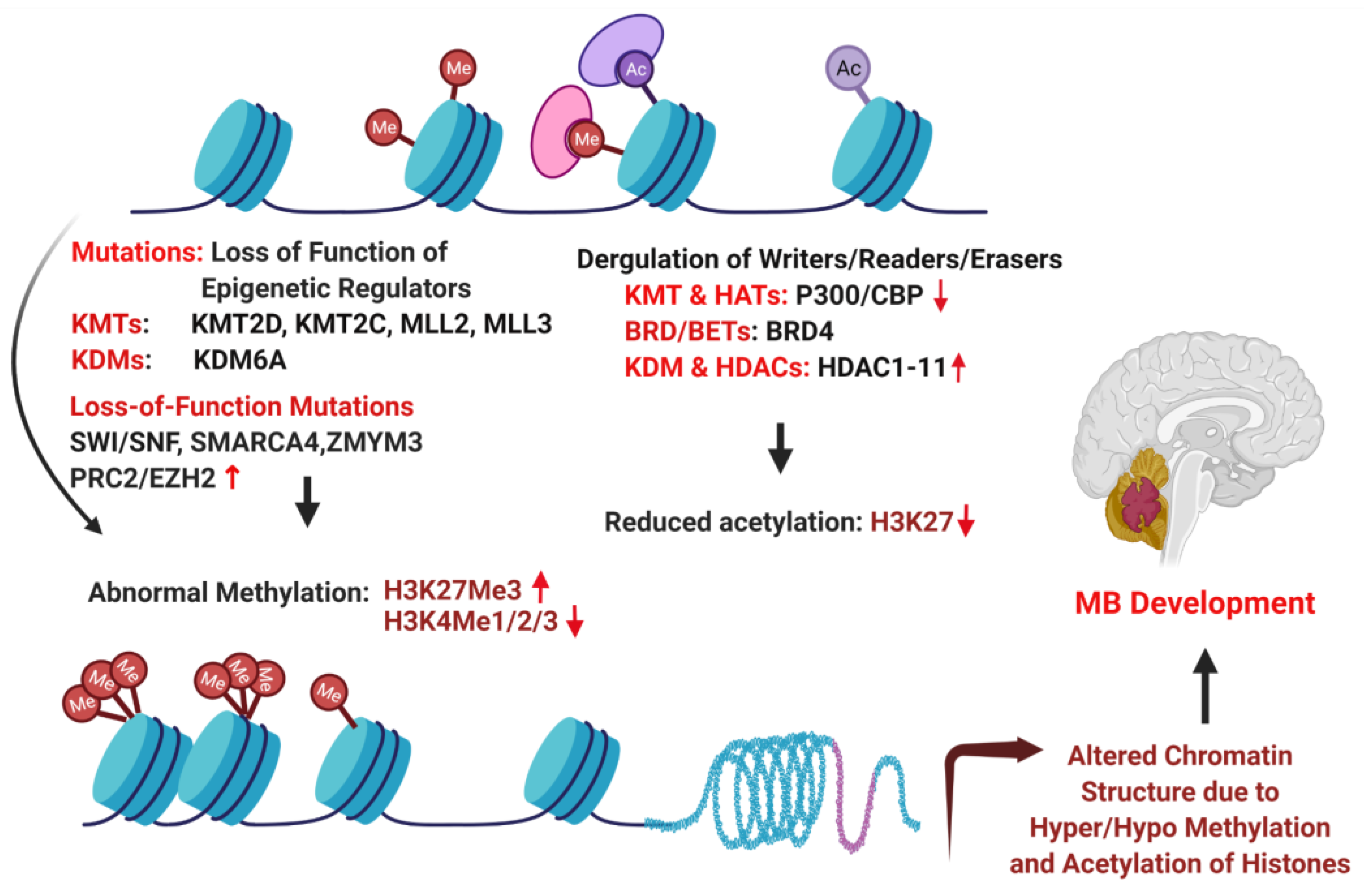
6. Epigenetic Drivers of MB
7. Drug Resistance and Recurrence in MBs
8. Non-Coding RNAs in MB
8.1. MiRNA
8.2. LncRNA
8.3. CircRNA
9. Future Directions and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-oncology 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [Green Version]
- Massimino, M.; Biassoni, V.; Gandola, L.; Garrè, M.L.; Gatta, G.; Giangaspero, F.; Poggi, G.; Rutkowski, S. Childhood medulloblastoma. Crit. Rev. Oncol./Hematol. 2016, 105, 35–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Jones, D.T.W.; Kool, M.; Robinson, G.; Gilbertson, R.J.; Cho, Y.-J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro. Oncol. 2013, 15 (Suppl. S2), ii1–ii56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.; Caracciolo, V.; Russo, G.; Reiss, K.; Giordano, A. Medulloblastoma: From molecular pathology to therapy. Clin. Cancer Res. 2008, 14, 971–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smoll, N.R.; Drummond, K.J. The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J. Clin. Neurosci. 2012, 19, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2011, 123, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Raffel, C. Medulloblastoma: Molecular Genetics and Animal Models. Neoplasia 2004, 6, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Ryzhova, M.V.; Zheludkova, O.G.; Kumirova, É.V.; Shishkina, L.V.; Panina, T.N.; Gorelyshev, S.K.; Khukhlaeva, E.A.; Mazerkina, N.A.; Matuev, K.B.; Medvedeva, O.A.; et al. Characteristics of medulloblastoma in children under age of three years. Zhurnal Vopr. Neirokhirurgii Im. N. N. Burd. 2013, 77, 3–10, discussion 11. [Google Scholar]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Fattet, S.; Haberler, C.; Legoix, P.; Varlet, P.; Lellouch-Tubiana, A.; Lair, S.; Manie, E.; Raquin, M.-A.; Bours, D.; Carpentier, S.; et al. Beta-catenin status in paediatric medulloblastomas: Correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J. Pathol. 2009, 218, 86–94. [Google Scholar] [CrossRef]
- Hovestadt, V.; Jones, D.T.W.; Picelli, S.; Wang, W.; Kool, M.; Northcott, P.A.; Sultan, M.; Stachurski, K.; Ryzhova, M.; Warnatz, H.-J.; et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 2014, 510, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.-J.; Koster, J.; Schouten-van Meeteren, A.; Van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Koster, J.; Bunt, J.; Hasselt, N.E.; Lakeman, A.; Van Sluis, P.; Troost, D.; Meeteren, N.S.-V.; Caron, H.N.; Cloos, J.; et al. Integrated Genomics Identifies Five Medulloblastoma Subtypes with Distinct Genetic Profiles, Pathway Signatures and Clinicopathological Features. PLoS ONE 2008, 3, e3088. [Google Scholar] [CrossRef]
- Korshunov, A.; Remke, M.; Kool, M.; Hielscher, T.; Northcott, P.A.; Williamson, D.; Pfaff, E.; Witt, H.; Jones, D.T.W.; Ryzhova, M.; et al. Biological and clinical heterogeneity of MYCN-amplified medulloblastoma. Acta Neuropathol. 2011, 123, 515–527. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.H.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stütz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef]
- Park, A.K.; Lee, S.-J.; Phi, J.H.; Wang, K.-C.; Kim, D.G.; Cho, B.-K.; Haberler, C.; Fattet, S.; Dufour, C.; Puget, S.; et al. Prognostic classification of pediatric medulloblastoma based on chromosome 17p loss, expression of MYCC and MYCN, and Wnt pathway activation. Neuro-oncology 2011, 14, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remke, M.; Hielscher, T.; Korshunov, A.; Northcott, P.A.; Bender, S.; Kool, M.; Westermann, F.; Benner, A.; Cin, H.; Ryzhova, M.; et al. FSTL5 is a marker of poor prognosis in non-WNT/non-SHH medulloblastoma. J. Clin. Oncol. 2011, 29, 3852–3861. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.; Parker, M.; Kranenburg, T.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Dubuc, A.M.; Ramaswamy, V.; Mack, S.C.; Gendoo, D.; Remke, M.; Wu, X.; Garzia, L.; Luu, B.; Cavalli, F.; et al. Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol. 2015, 129, 449–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottardo, N.G.; Hansford, J.R.; McGlade, J.P.; Alvaro, F.; Ashley, D.M.; Bailey, S.; Baker, D.L.; Bourdeaut, F.; Cho, Y.J.; Clay, M.; et al. Medulloblastoma Down Under 2013: A report from the third annual meeting of the International Medulloblastoma Working Group. Acta Neuropathol. 2014, 127, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, F.M.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, V.; Taylor, M.D. Medulloblastoma: From Myth to Molecular. J. Clin. Oncol. 2017, 35, 2355–2363. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.; Chintagumpala, M.; Ashley, D.; Kellie, S.; Kun, L.E.; Merchant, T.E.; Woo, S.; Wheeler, G.; Ahern, V.; Krasin, M.J.; et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): Long-term results from a prospective, multicentre trial. Lancet Oncol. 2006, 7, 813–820. [Google Scholar] [CrossRef]
- Packer, R.J.; Gajjar, A.; Vezina, G.; Rorke-Adams, L.; Burger, P.C.; Robertson, P.L.; Bayer, L.; LaFond, D.; Donahue, B.R.; Marymont, M.H.; et al. Phase III Study of Craniospinal Radiation Therapy Followed by Adjuvant Chemotherapy for Newly Diagnosed Average-Risk Medulloblastoma. J. Clin. Oncol. 2006, 24, 4202–4208. [Google Scholar] [CrossRef] [PubMed]
- Strother, D.; Lafay-Cousin, L. Adjuvant therapy for high-risk medulloblastoma: More is better? Neuro-oncology 2021, 23, 1048–1049. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.; Robinson, G.W.; Smith, K.S.; Lin, T.; Merchant, T.E.; Chintagumpala, M.; Mahajan, A.; Su, J.; Bouffet, E.; Bartels, U.; et al. Outcomes by Clinical and Molecular Features in Children With Medulloblastoma Treated With Risk-Adapted Therapy: Results of an International Phase III Trial (SJMB03). J. Clin. Oncol. 2021, 39, 822–835. [Google Scholar] [CrossRef]
- Dufour, C.; Foulon, S.; Geoffray, A.; Masliah-Planchon, J.; Figarella-Branger, D.; Bernier-Chastagner, V.; Padovani, L.; Guerrini-Rousseau, L.; Faure-Conter, C.; Icher, C.; et al. Prognostic relevance of clinical and molecular risk factors in children with high-risk medulloblastoma treated in the phase II trial PNET HR+5. Neuro-oncology 2020, 23, 1163–1172. [Google Scholar] [CrossRef]
- Leary, S.E.S.; Packer, R.J.; Li, Y.; Billups, C.A.; Smith, K.S.; Jaju, A.; Heier, L.; Burger, P.; Walsh, K.; Han, Y.; et al. Efficacy of Carboplatin and Isotretinoin in Children With High-risk Medulloblastoma: A Randomized Clinical Trial From the Children’s Oncology Group. JAMA Oncol. 2021, 7, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Tarbell, N.J.; Friedman, H.; Polkinghorn, W.R.; Yock, T.; Zhou, T.; Chen, Z.; Burger, P.; Barnes, P.; Kun, L. High-Risk Medulloblastoma: A Pediatric Oncology Group Randomized Trial of Chemotherapy Before or After Radiation Therapy (POG 9031). J. Clin. Oncol. 2013, 31, 2936–2941. [Google Scholar] [CrossRef] [Green Version]
- Lafay-Cousin, L.; Smith, A.; Chi, S.N.; Wells, E.; Madden, J.; Margol, A.; Ramaswamy, V.; Finlay, J.; Taylor, M.D.; Dhall, G.; et al. Clinical, Pathological, and Molecular Characterization of Infant Medulloblastomas Treated with Sequential High-Dose Chemotherapy. Pediatr. Blood Cancer 2016, 63, 1527–1534. [Google Scholar] [CrossRef] [Green Version]
- Rutkowski, S.; Bode, U.; Deinlein, F.; Ottensmeier, H.; Warmuth-Metz, M.; Soerensen, N.; Graf, N.; Emser, A.; Pietsch, T.; Wolff, J.E.; et al. Treatment of Early Childhood Medulloblastoma by Postoperative Chemotherapy Alone. N. Engl. J. Med. 2005, 352, 978–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, B.H.; Geyer, J.R.; Miller, D.C.; Curran, J.G.; Zhou, T.; Holmes, E.; Ingles, S.A.; Dunkel, I.J.; Hilden, J.; Packer, R.J.; et al. Pilot Study of Intensive Chemotherapy With Peripheral Hematopoietic Cell Support for Children Less Than 3 Years of Age With Malignant Brain Tumors, the CCG-99703 Phase I/II Study. A Report from the Children’s Oncology Group. Pediatr. Neurol. 2015, 53, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Lafay–Cousin, L.; Bouffet, E.; Hawkins, C.; Amid, A.; Huang, A.; Mabbott, D.J. Impact of Radiation Avoidance on Survival and Neurocognitive Outcome in Infant Medulloblastoma. Curr. Oncol. 2009, 16, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, J.R.; Sposto, R.; Jennings, M.; Boyett, J.M.; Axtell, R.A.; Breiger, D.; Broxson, E.; Donahue, B.; Finlay, J.L.; Goldwein, J.W.; et al. Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: A report from the Children’s Cancer Group. J. Clin. Oncol. 2005, 23, 7621–7631. [Google Scholar] [CrossRef] [PubMed]
- Grill, J.; Sainte-Rose, C.; Jouvet, A.; Gentet, J.-C.; Lejars, O.; Frappaz, D.; Doz, F.; Rialland, X.; Pichon, F.; Bertozzi, A.-I.; et al. Treatment of medulloblastoma with postoperative chemotherapy alone: An SFOP prospective trial in young children. Lancet Oncol. 2005, 6, 573–580. [Google Scholar] [CrossRef]
- Min, H.S.; Lee, J.Y.; Kim, S.-K.; Park, S.-H. Genetic Grouping of Medulloblastomas by Representative Markers in Pathologic Diagnosis. Transl. Oncol. 2013, 6, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Juraschka, K.; Taylor, M.D. Medulloblastoma in the age of molecular subgroups: A review. J. Neurosurg. Pediatr. 2019, 24, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Gibson, P.; Tong, Y.; Robinson, G.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.; Hogg, T.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010, 468, 1095–1099. [Google Scholar] [CrossRef]
- Ellison, D.W.; Dalton, J.; Kocak, M.; Nicholson, S.L.; Fraga, C.; Neale, G.; Kenney, A.M.; Brat, D.J.; Perry, A.; Yong, W.H.; et al. Medulloblastoma: Clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011, 121, 381–396. [Google Scholar] [CrossRef] [Green Version]
- Gilbertson, R.J. Medulloblastoma: Signalling a change in treatment. Lancet Oncol. 2004, 5, 209–218. [Google Scholar] [CrossRef]
- Wechsler-Reya, R.; Scott, M.P. The Developmental Biology of Brain Tumors. Annu. Rev. Neurosci. 2001, 24, 385–428. [Google Scholar] [CrossRef]
- Geron, L.; Salomao, K.B.; Borges, K.S.; Andrade, A.F.; Corrêa, C.A.; Scrideli, C.A.; Tone, L.G. Molecular characterization of Wnt pathway and function of beta-catenin overexpression in medulloblastoma cell lines. Cytotechnology 2018, 70, 1713–1722. [Google Scholar] [CrossRef]
- Dale, T.C. Signal transduction by the Wnt family of ligands. Biochem. J. 1998, 329 Pt 2, 209–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.E.; Soriano, S.; Xia, X.; Eberhart, C.G.; De Strooper, B.; Zheng, H.; Koo, E.H. Presenilin couples the paired phosphorylation of beta-catenin independent of axin: Implications for beta-catenin activation in tumorigenesis. Cell 2002, 110, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Rogers, H.A.; Miller, S.; Lowe, J.; Brundler, M.-A.; Coyle, B.; Grundy, R.G. An investigation of WNT pathway activation and association with survival in central nervous system primitive neuroectodermal tumours (CNS PNET). Br. J. Cancer 2009, 100, 1292–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, A.; Waha, A.; Berthold, F.; Wolter, M.; Reifenberger, J.; Hartmann, W.; Friedl, W.; Reifenberger, G.; Wiestler, O.D.; Pietsch, T. Somatic mutations ofWNT/wingless signaling pathway components in primitive neuroectodermal tumors. Int. J. Cancer 2001, 93, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Zurawel, R.H.; Chiappa, S.A.; Allen, C.; Raffel, C. Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer Res. 1998, 58, 896–899. [Google Scholar]
- Gilbertson, R.J.; Ellison, D.W. The Origins of Medulloblastoma Subtypes. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 341–365. [Google Scholar] [CrossRef]
- Baryawno, N.; Sveinbjornsson, B.; Eksborg, S.; Chen, C.S.; Kogner, P.; Johnsen, J.I. Small-molecule inhibitors of phosphatidylinositol 3-kinase/Akt signaling inhibit Wnt/beta-catenin pathway cross-talk and suppress medulloblastoma growth. Cancer Res. 2010, 70, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.-N.; Yu, M.-Y.; Kong, L.-M.; Wang, W.-H.; Yang, Y.-F.; Liu, J.-Q.; Qiu, M.-H.; Li, Y. Biflavone Ginkgetin, a Novel Wnt Inhibitor, Suppresses the Growth of Medulloblastoma. Nat. Prod. Bioprospecting 2015, 5, 91–97. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Li, Y.; Zhang, L.; Li, L.; Shen, Y.; Lin, L.; Zheng, W.; Chen, L.; Bian, X.; Ng, H.K.; et al. Curcumin suppresses cell proliferation through inhibition of the Wnt/beta-catenin signaling pathway in medulloblastoma. Oncol. Rep. 2014, 32, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wu, J.; Tan, C.; Klein, P.S. PP2A:B56epsilon is required for Wnt/beta-catenin signaling during embryonic development. Development 2003, 130, 5569–5578. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, F.; Scoppettuolo, M.N.; Carotenuto, M.; De Antonellis, P.; Di Dato, V.; De Vita, G.; Zollo, M. Norcantharidin impairs medulloblastoma growth by inhibition of Wnt/beta-catenin signaling. J. Neuro-oncol. 2012, 106, 59–70. [Google Scholar] [CrossRef]
- Ris, M.D.; Packer, R.; Goldwein, J.; Jones-Wallace, D.; Boyett, J.M. Intellectual Outcome After Reduced-Dose Radiation Therapy Plus Adjuvant Chemotherapy for Medulloblastoma: A Children’s Cancer Group Study. J. Clin. Oncol. 2001, 19, 3470–3476. [Google Scholar] [CrossRef]
- Leary, S.E.; Olson, J.M. The molecular classification of medulloblastoma: Driving the next generation clinical trials. Curr. Opin. Pediatr. 2012, 24, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Thompson, E.M.; Ashley, D.; Landi, D. Current medulloblastoma subgroup specific clinical trials. Transl. Pediatr. 2020, 9, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Olmeda, D.; Castel, S.; Vilaro, S.; Cano, A. Beta-catenin regulation during the cell cycle: Implications in G2/M and apoptosis. Mol. Biol. Cell 2003, 14, 2844–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjihannas, M.V.; Bernkopf, D.B.; Bruckner, M.; Behrens, J. Cell cycle control of Wnt/beta-catenin signalling by conductin/axin2 through CDC20. EMBO Rep. 2012, 13, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poschl, J.; Bartels, M.; Ohli, J.; Bianchi, E.; Kuteykin-Teplyakov, K.; Grammel, D.; Ahlfeld, J.; Schüller, U. Wnt/beta-catenin signaling inhibits the Shh pathway and impairs tumor growth in Shh-dependent medulloblastoma. Acta Neuropathol. 2014, 127, 605–607. [Google Scholar] [CrossRef]
- Manoranjan, B.; Venugopal, C.; Bakhshinyan, D.; Adile, A.A.; Richards, L.; Kameda-Smith, M.M.; Whitley, O.; Dvorkin-Gheva, A.; Subapanditha, M.; Savage, N.; et al. Wnt activation as a therapeutic strategy in medulloblastoma. Nat. Commun. 2020, 11, 4323. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Jones, D.T.W.; Jaeger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, W.; Digon-Söntgerath, B.; Koch, A.; Waha, A.; Endl, E.; Dani, I.; Denkhaus, D.; Goodyer, C.G.; Sörensen, N.; Wiestler, O.D.; et al. Phosphatidylinositol 3′-Kinase/AKT Signaling Is Activated in Medulloblastoma Cell Proliferation and Is Associated with Reduced Expression ofPTEN. Clin. Cancer Res. 2006, 12, 3019–3027. [Google Scholar] [CrossRef] [Green Version]
- Eckerdt, F.; Clymer, J.; Bell, J.B.; Beauchamp, E.M.; Blyth, G.T.; Goldman, S.; Platanias, L.C. Pharmacological mTOR targeting enhances the antineoplastic effects of selective PI3Kalpha inhibition in medulloblastoma. Sci. Rep. 2019, 9, 12822. [Google Scholar] [CrossRef] [Green Version]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, G.W.; Rudneva, V.; Buchhalter, I.; Billups, C.A.; Waszak, S.; Smith, K.S.; Bowers, D.; Bendel, A.; Fisher, P.; Partap, S.; et al. Risk-adapted therapy for young children with medulloblastoma (SJYC07): Therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol. 2018, 19, 768–784. [Google Scholar] [CrossRef]
- Menyhárt, O.; Győrffy, B. Molecular stratifications, biomarker candidates and new therapeutic options in current medulloblastoma treatment approaches. Cancer Metastasis Rev. 2020, 39, 211–233. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Song, Q.; Day, B.W. Phase I and phase II sonidegib and vismodegib clinical trials for the treatment of paediatric and adult MB patients: A systemic review and meta-analysis. Acta Neuropathol. Commun. 2019, 7, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog–Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef]
- Petrirena, G.J.; Masliah-Planchon, J.; Sala, Q.; Pourroy, B.; Frappaz, D.; Tabouret, E.; Graillon, T.; Gentet, J.-C.; Delattre, O.; Chinot, O.; et al. Recurrent extraneural sonic hedgehog medulloblastoma exhibiting sustained response to vismodegib and temozolomide monotherapies and inter-metastatic molecular heterogeneity at progression. Oncotarget 2018, 9, 10175–10183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic Analysis of Smoothened Inhibitor Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Danial, C.; Sarin, K.Y.; Oro, A.E.; Chang, A.L.S. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin. Cancer Res. 2015, 22, 1325–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.-R.; Zhao, H.; Zhang, X.-S.; Lang, H.; Yu, K. Novel-smoothened inhibitors for therapeutic targeting of naïve and drug-resistant hedgehog pathway-driven cancers. Acta Pharmacol. Sin. 2018, 40, 257–267. [Google Scholar] [CrossRef]
- Kumar, V.; Chaudhary, A.K.; Dong, Y.; Zhong, H.A.; Mondal, G.; Lin, F.; Kumar, V.; Mahato, R.I. Design, Synthesis and Biological Evaluation of novel Hedgehog Inhibitors for treating Pancreatic Cancer. Sci. Rep. 2017, 7, 1665. [Google Scholar] [CrossRef] [Green Version]
- Robinson, G.W.; Kaste, S.C.; Chemaitilly, W.; Bowers, D.; Laughton, S.; Smith, A.; Gottardo, N.; Partap, S.; Bendel, A.; Wright, K.D.; et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 2017, 8, 69295–69302. [Google Scholar] [CrossRef] [Green Version]
- Ocasio, J.; Babcock, B.; Malawsky, D.; Weir, S.J.; Loo, L.; Simon, J.M.; Zylka, M.J.; Hwang, D.; Dismuke, T.; Sokolsky, M.; et al. scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat. Commun. 2019, 10, 5829. [Google Scholar] [CrossRef] [Green Version]
- Vanner, R.J.; Remke, M.; Gallo, M.; Selvadurai, H.J.; Coutinho, F.; Lee, L.; Kushida, M.; Head, R.; Morrissy, S.; Zhu, X.; et al. Quiescent Sox2+ Cells Drive Hierarchical Growth and Relapse in Sonic Hedgehog Subgroup Medulloblastoma. Cancer Cell 2014, 26, 33–47. [Google Scholar] [CrossRef] [Green Version]
- Ahlfeld, J.; Favaro, R.; Pagella, P.; Kretzschmar, H.A.; Nicolis, S.; Schüller, U. Sox2 Requirement in Sonic Hedgehog-Associated Medulloblastoma. Cancer Res. 2013, 73, 3796–3807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvadurai, H.J.; Luis, E.; Desai, K.; Lan, X.; Vladoiu, M.C.; Whitley, O.; Galvin, C.; Vanner, R.J.; Lee, L.; Whetstone, H.; et al. Medulloblastoma Arises from the Persistence of a Rare and Transient Sox2+ Granule Neuron Precursor. Cell Rep. 2020, 31, 107511. [Google Scholar] [CrossRef] [PubMed]
- Hagey, D.; Klum, S.; Kurtsdotter, I.; Zaouter, C.; Topcic, D.; Andersson, O.; Bergsland, M.; Muhr, J. SOX2 regulates common and specific stem cell features in the CNS and endoderm derived organs. PLoS Genet. 2018, 14, e1007224. [Google Scholar] [CrossRef] [Green Version]
- Hagey, D.; Muhr, J. Sox2 Acts in a Dose-Dependent Fashion to Regulate Proliferation of Cortical Progenitors. Cell Rep. 2014, 9, 1908–1920. [Google Scholar] [CrossRef] [Green Version]
- Metz, E.P.; Rizzino, A. Sox2 dosage: A critical determinant in the functions of Sox2 in both normal and tumor cells. J. Cell. Physiol. 2019, 234, 19298–19306. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.L.; Wilder, P.J.; Desler, M.; Rizzino, A. Elevating SOX2 Levels Deleteriously Affects the Growth of Medulloblastoma and Glioblastoma Cells. PLoS ONE 2012, 7, e44087. [Google Scholar] [CrossRef] [Green Version]
- Wuebben, E.L.; Wilder, P.J.; Cox, J.L.; Grunkemeyer, J.A.; Caffrey, T.; Hollingsworth, M.A.; Rizzino, A. SOX2 functions as a molecular rheostat to control the growth, tumorigenicity and drug responses of pancreatic ductal adenocarcinoma cells. Oncotarget 2016, 7, 34890–34906. [Google Scholar] [CrossRef] [Green Version]
- Metz, E.P.; Wilder, P.J.; Dong, J.; Datta, K.; Rizzino, A. Elevating SOX2 in prostate tumor cells upregulates expression of neuroendocrine genes, but does not reduce the inhibitory effects of enzalutamide. J. Cell. Physiol. 2019, 235, 3731–3740. [Google Scholar] [CrossRef] [PubMed]
- Metz, E.P.; Wuebben, E.L.; Wilder, P.J.; Cox, J.L.; Datta, K.; Coulter, D.; Rizzino, A. Tumor quiescence: Elevating SOX2 in diverse tumor cell types downregulates a broad spectrum of the cell cycle machinery and inhibits tumor growth. BMC Cancer 2020, 20, 941. [Google Scholar] [CrossRef]
- Shih, D.J.; Northcott, P.A.; Remke, M.; Korshunov, A.; Ramaswamy, V.; Kool, M.; Luu, B.; Yao, Y.; Wang, X.; Dubuc, A.M.; et al. Cytogenetic prognostication within medulloblastoma subgroups. J. Clin. Oncol. 2014, 32, 886–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lospinoso Severini, L.; Ghirga, F.; Bufalieri, F.; Quaglio, D.; Infante, P.; Di Marcotullio, L. The SHH/GLI signaling pathway: A therapeutic target for medulloblastoma. Expert Opin. Ther. Targets 2020, 24, 1159–1181. [Google Scholar] [CrossRef]
- Vladoiu, M.C.; El-Hamamy, I.; Donovan, L.K.; Farooq, H.; Holgado, B.L.; Sundaravadanam, Y.; Ramaswamy, V.; Hendrikse, L.; Kumar, S.; Mack, S.C.; et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 2019, 572, 67–73. [Google Scholar] [CrossRef]
- Perreault, S.; Ramaswamy, V.; Achrol, A.; Chao, K.; Liu, T.; Shih, D.; Remke, M.; Schubert, S.; Bouffet, E.; Fisher, P.; et al. MRI Surrogates for Molecular Subgroups of Medulloblastoma. Am. J. Neuroradiol. 2014, 35, 1263–1269. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.-J.; Tsherniak, A.; Tamayo, P.; Santagata, S.; Ligon, A.; Greulich, H.; Berhoukim, R.; Amani, V.; Goumnerova, L.; Eberhart, C.G.; et al. Integrative Genomic Analysis of Medulloblastoma Identifies a Molecular Subgroup That Drives Poor Clinical Outcome. J. Clin. Oncol. 2011, 29, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.M.; Hielscher, T.; Bouffet, E.; Remke, M.; Luu, B.; Gururangan, S.; McLendon, R.E.; Bigner, D.D.; Lipp, E.S.; Perreault, S.; et al. Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: A retrospective integrated clinical and molecular analysis. Lancet Oncol. 2016, 17, 484–495. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Xu, Q.; Cui, Y.; Li, N.; Bian, X.; Lv, S. Medulloblastoma stem cells: Promising targets in medulloblastoma therapy. Cancer Sci. 2016, 107, 583–589. [Google Scholar] [CrossRef]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stütz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.H.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014, 511, 428–434. [Google Scholar] [CrossRef]
- Menyhárt, O.; Giangaspero, F.; Győrffy, B. Molecular markers and potential therapeutic targets in non-WNT/non-SHH (group 3 and group 4) medulloblastomas. J. Hematol. Oncol. 2019, 12, 29. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.W.; Jaeger, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.-J.; Pugh, T.; Hovestadt, V.; Stütz, A.M.; et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe, E.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bautista, F.; Fioravantti, V.; de Rojas, T.; Carceller, F.; Madero, L.; Lassaletta, A.; Moreno, L. Medulloblastoma in children and adolescents: A systematic review of contemporary phase I and II clinical trials and biology update. Cancer Med. 2017, 6, 2606–2624. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Chiba, Y.; Murakami, R.; Matsumoto, K.; Kawauchi, M.; Fujihara, R. Blood–brain barrier and blood–cerebrospinal fluid barrier in normal and pathological conditions. Brain Tumor Pathol. 2016, 33, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Voskamp, M.J.; Li, S.; van Daalen, K.R.; Crnko, S.; Broeke, T.T.; Bovenschen, N. Immunotherapy in Medulloblastoma: Current State of Research, Challenges, and Future Perspectives. Cancers 2021, 13, 5387. [Google Scholar] [CrossRef]
- Pei, Y.; Moore, C.E.; Wang, J.; Tewari, A.K.; Eroshkin, A.; Cho, Y.-J.; Witt, H.; Korshunov, A.; Read, T.-A.; Sun, J.L.; et al. An Animal Model of MYC-Driven Medulloblastoma. Cancer Cell 2012, 21, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawauchi, D.; Robinson, G.; Uziel, T.; Gibson, P.; Rehg, J.; Gao, C.; Finkelstein, D.; Qu, C.; Pounds, S.; Ellison, D.W.; et al. A Mouse Model of the Most Aggressive Subgroup of Human Medulloblastoma. Cancer Cell 2012, 21, 168–180. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.M.; Kuijper, S.; Lindsey, J.; Petrie, K.; Schwalbe, E.; Barker, K.; Boult, J.K.; Williamson, D.; Ahmad, Z.; Hallsworth, A.; et al. Combined MYC and P53 Defects Emerge at Medulloblastoma Relapse and Define Rapidly Progressive, Therapeutically Targetable Disease. Cancer Cell 2014, 27, 72–84. [Google Scholar] [CrossRef]
- Pei, Y.; Liu, K.-W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC- Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Hanaford, A.R.; Archer, T.C.; Price, A.; Kahlert, U.; Maciaczyk, J.; Nikkhah, G.; Kim, J.W.; Ehrenberger, T.; Clemons, P.A.; Dančík, V.; et al. DiSCoVERing innovative therapies for rare tumors: Combining genetically accurate disease models with in silico analysis to identify novel therapeutic targets. Clin. Cancer Res. 2016, 22, 3903–3914. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Murad, N.; Xu, Z.; Zhang, P.; Okonechnikov, K.; Kool, M.; Hinojosa, S.R.; Lazarski, C.; Zheng, P.; Liu, Y.; et al. MYC Drives Group 3 Medulloblastoma through Transformation of Sox2+ Astrocyte Progenitor Cells. Cancer Res. 2019, 79, 1967–1980. [Google Scholar] [CrossRef] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandopadhayay, P.; Bergthold, G.; Nguyen, B.; Schubert, S.; Gholamin, S.; Tang, Y.; Bolin, S.; Schumacher, S.E.; Zeid, R.; Masoud, S.; et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin. Cancer Res. 2014, 20, 912–925. [Google Scholar] [CrossRef] [Green Version]
- Bolin, S.; Borgenvik, A.; Persson, C.U.; Sundström, A.; Qi, J.; Bradner, J.E.; Weiss, W.A.; Cho, Y.-J.; Weishaupt, H.; Swartling, F.J. Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene 2018, 37, 2850–2862. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, N.K.; Kling, M.J.; Griggs, C.N.; Kesherwani, V.; Shukla, M.; McIntyre, E.M.; Ray, S.; Liu, Y.; McGuire, T.R.; Sharp, J.G.; et al. A Novel Combination Approach Targeting an Enhanced Protein Synthesis Pathway in MYC-driven (Group 3) Medulloblastoma. Mol. Cancer Ther. 2020, 19, 1351–1362. [Google Scholar] [CrossRef]
- Morfouace, M.; Shelat, A.; Jacus, M.; Freeman, B.B.; Turner, D.; Robinson, S.; Zindy, F.; Wang, Y.-D.; Finkelstein, D.; Ayrault, O.; et al. Pemetrexed and Gemcitabine as Combination Therapy for the Treatment of Group3 Medulloblastoma. Cancer Cell 2014, 25, 516–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, E.M.; Keir, S.T.; Venkatraman, T.; Lascola, C.; Yeom, K.W.; Nixon, A.B.; Liu, Y.; Picard, D.; Remke, M.; Bigner, D.D.; et al. The role of angiogenesis in Group 3 medulloblastoma pathogenesis and survival. Neuro-oncology 2017, 19, 1217–1227. [Google Scholar] [CrossRef]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPalpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Korshunov, A.; Pfister, S.; Taylor, M. The clinical implications of medulloblastoma subgroups. Nat. Rev. Neurol. 2012, 8, 340–351. [Google Scholar] [CrossRef]
- Northcott, P.A.; Hielscher, T.; Dubuc, A.; Mack, S.C.; Shih, D.J.H.; Remke, M.; Al-Halabi, H.; Albrecht, S.; Jabado, N.; Eberhart, C.G.; et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011, 122, 231–240. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.H.; Remke, M.; Cho, Y.-J.; Kool, M.; Hawkins, C.; Eberhart, C.G.; Dubuc, A.; Guettouche, T.; Cardentey, Y.; et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol. 2011, 123, 615–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roussel, M.F.; Robinson, G.W. Role of MYC in Medulloblastoma. Cold Spring Harb. Perspect. Med. 2013, 3, a014308. [Google Scholar] [CrossRef] [Green Version]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef]
- Pugh, T.J.; Weeraratne, S.D.; Archer, T.C.; Krummel, D.A.P.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L.; et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012, 488, 106–110. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Adamski, J.; Bartels, U.; Tabori, U.; Wang, X.; Huang, A.; Hawkins, C.; Mabbott, D.; Laperriere, N.; et al. Medulloblastoma subgroup-specific outcomes in irradiated children: Who are the true high-risk patients? Neuro-oncology 2016, 18, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Forget, A.; Martignetti, L.; Puget, S.; Calzone, L.; Brabetz, S.; Picard, D.; Montagud, A.; Liva, S.; Sta, A.; Dingli, F.; et al. Aberrant ERBB4-SRC Signaling as a Hallmark of Group 4 Medulloblastoma Revealed by Integrative Phosphoproteomic Profiling. Cancer Cell 2018, 34, 379–395.e7. [Google Scholar] [CrossRef] [Green Version]
- Archer, T.C.; Ehrenberger, T.; Mundt, F.; Gold, M.P.; Krug, K.; Mah, C.K.; Mahoney, E.L.; Daniel, C.J.; LeNail, A.; Ramamoorthy, D.; et al. Proteomics, Post-translational Modifications, and Integrative Analyses Reveal Molecular Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2018, 34, 396–410.e8. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Dubuc, A.M.; Remke, M.; Korshunov, A.; Northcott, P.A.; Zhan, S.H.; Mendez-Lago, M.; Kool, M.; Jones, D.T.W.; Unterberger, A.; Morrissy, A.S.; et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 2012, 125, 373–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, B.T.; Li, C.; Morgan, M.A.; Theurillat, I.; Finkelstein, D.; Wright, S.; Hyle, J.; Smith, S.M.; Fan, Y.; Wang, Y.-D.; et al. Inactivation of Ezh2 Upregulates Gfi1 and Drives Aggressive Myc-Driven Group 3 Medulloblastoma. Cell Rep. 2017, 18, 2907–2917. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Furlan, A.; Agbazahou, F.; Henry, M.; Gonzalez-Pisfil, M.; Le Nézet, C.; Champelovier, D.; Fournier, M.; Vandenbunder, B.; Bidaux, G.; Héliot, L. P-TEFb and Brd4: Actors of the transcription pause release as therapeutical targets. Med. Sci. 2018, 34, 685–692. [Google Scholar]
- Song, H.; Bhakat, R.; Kling, M.J.; Coulter, D.W.; Chaturvedi, N.K.; Ray, S.; Joshi, S.S. Targeting cyclin-dependent kinase 9 sensitizes medulloblastoma cells to chemotherapy. Biochem. Biophys. Res. Commun. 2019, 520, 250–256. [Google Scholar] [CrossRef]
- Shi, J.; Vakoc, C.R. The Mechanisms behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henssen, A.; Thor, T.; Odersky, A.; Heukamp, L.; El-Hindy, N.; Beckers, A.; Slpeleman, F.; Althoff, K.; Schäfers, S.; Schramm, A.; et al. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget 2013, 4, 2080–2095. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Li, M.; Zhang, X.; Jones, S.; Leary, R.J.; Lin, J.C.-H.; Boca, S.M.; Carter, H.; Samayoa, J.; Bettegowda, C.; et al. The Genetic Landscape of the Childhood Cancer Medulloblastoma. Science 2011, 331, 435–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, B.G.; Wang, X.; Shen, X.; McKenna, E.S.; Lemieux, M.; Cho, Y.-J.; Koellhoffer, E.; Pomeroy, S.L.; Orkin, S.H.; Roberts, C.W. Epigenetic Antagonism between Polycomb and SWI/SNF Complexes during Oncogenic Transformation. Cancer Cell 2010, 18, 316–328. [Google Scholar] [CrossRef] [Green Version]
- Leung, W.C.; Makharashvili, N.; Agarwal, P.; Chiu, L.-Y.; Pourpre, R.; Cammarata, M.B.; Cannon, J.R.; Sherker, A.; Durocher, D.; Brodbelt, J.S.; et al. ZMYM3 regulates BRCA1 localization at damaged chromatin to promote DNA repair. Genes Dev. 2017, 31, 260–274. [Google Scholar] [CrossRef] [Green Version]
- Kodan, A.; Futamata, R.; Kimura, Y.; Kioka, N.; Nakatsu, T.; Kato, H.; Ueda, K. ABCB1/MDR1/P-gp employs an ATP-dependent twist-and-squeeze mechanism to export hydrophobic drugs. FEBS Lett. 2021, 595, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Kara, A.; Özgür, A.; Nalbantoğlu, S.; Karadağ, A. DNA repair pathways and their roles in drug resistance for lung adenocarcinoma. Mol. Biol. Rep. 2021, 48, 3813–3825. [Google Scholar] [CrossRef]
- Bobola, M.S.; Finn, L.S.; Ellenbogen, R.G.; Geyer, J.R.; Berger, M.S.; Braga, J.M.; Meade, E.H.; Gross, M.E.; Silber, J.R. Apurinic/Apyrimidinic Endonuclease Activity Is Associated with Response to Radiation and Chemotherapy in Medulloblastoma and Primitive Neuroectodermal Tumors. Clin. Cancer Res. 2005, 11, 7405–7414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kievit, F.M.; Stephen, Z.R.; Wang, K.; Dayringer, C.J.; Sham, J.G.; Ellenbogen, R.G.; Silber, J.R.; Zhang, M. Nanoparticle mediated silencing of DNA repair sensitizes pediatric brain tumor cells to gamma-irradiation. Mol. Oncol. 2015, 9, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Xi, S.; Chen, Y.; Pramanik, S.; Zeng, J.; Roychoudhury, S.; Harris, H.; Akbar, A.; Elhag, S.S.; Coulter, D.W.; et al. Histone chaperone FACT complex inhibitor CBL0137 interferes with DNA damage repair and enhances sensitivity of medulloblastoma to chemotherapy and radiation. Cancer Lett. 2021, 520, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sui, Y.; Li, Q.; Zhao, Y.; Dong, X.; Yang, J.; Liang, Z.; Han, Y.; Tang, Y.; Ma, J. Effective inhibition of MYC-amplified group 3 medulloblastoma by FACT-targeted curaxin drug CBL0137. Cell Death Dis. 2020, 11, 1029. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Lindner, D.J.; Coleman, C.J.; Wildey, G.; Dowlati, A.; Stark, G.R. The FACT inhibitor CBL0137 Synergizes with Cisplatin in Small-Cell Lung Cancer by Increasing NOTCH1 Expression and Targeting Tumor-Initiating Cells. Cancer Res. 2018, 78, 2396–2406. [Google Scholar] [CrossRef] [Green Version]
- Dermawan, J.K.T.; Hitomi, M.; Silver, D.J.; Wu, Q.; Sandlesh, P.; Sloan, A.E.; Purmal, A.A.; Gurova, K.V.; Rich, J.N.; Lathia, J.D.; et al. Pharmacological Targeting of the Histone Chaperone Complex FACT Preferentially Eliminates Glioblastoma Stem Cells and Prolongs Survival in Preclinical Models. Cancer Res. 2016, 76, 2432–2442. [Google Scholar] [CrossRef] [Green Version]
- Frock, R.L.; Sadeghi, C.; Meng, J.; Wang, J.L. DNA End Joining: G0-ing to the Core. Biomolecules 2021, 11, 1487. [Google Scholar] [CrossRef] [PubMed]
- Elbakry, A.; Lobrich, M. Homologous Recombination Subpathways: A Tangle to Resolve. Front. Genet. 2021, 12, 723847. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Y.; Xiong, J.; Lan, B.; Wang, X.; Liu, J.; Lin, J.; Fei, Z.; Zheng, X.; Chen, C. Role of PRKDC in cancer initiation, progression, and treatment. Cancer Cell Int. 2021, 21, 563. [Google Scholar] [CrossRef]
- Hwang, J.W.; Kim, S.-N.; Myung, N.; Song, D.; Han, G.; Bae, G.-U.; Bedford, M.T.; Kim, Y.K. PRMT5 promotes DNA repair through methylation of 53BP1 and is regulated by Src-mediated phosphorylation. Commun. Biol. 2020, 3, 428. [Google Scholar] [CrossRef]
- Clarke, T.L.; Sanchez-Bailon, M.P.; Chiang, K.; Reynolds, J.J.; Ruiz, J.H.; Bandeiras, T.; Matias, P.; Maslen, S.L.; Skehel, J.M.; Stewart, G.; et al. PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol. Cell 2017, 65, 900–916.e7. [Google Scholar] [CrossRef]
- Chaturvedi, N.K.; Mahapatra, S.; Kesherwani, V.; Kling, M.J.; Shukla, M.; Ray, S.; Kanchan, R.; Perumal, N.; McGuire, T.R.; Sharp, J.G.; et al. Role of protein arginine methyltransferase 5 in group 3 (MYC-driven) Medulloblastoma. BMC Cancer 2019, 19, 1056. [Google Scholar] [CrossRef]
- Owens, J.L.; Beketova, E.; Liu, S.; Tinsley, S.L.; Asberry, A.M.; Deng, X.; Huang, J.; Li, C.; Wan, J.; Hu, C.-D. PRMT5 Cooperates with pICln to Function as a Master Epigenetic Activator of DNA Double-Strand Break Repair Genes. iScience 2019, 23, 100750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Wu, H.-J.; Hsu, J.-M.; Chang, S.-S.; Labaff, A.M.; Li, C.-W.; Wang, Y.; Hsu, J.L.; Hung, M.-C. Long non-coding RNAs: Versatile master regulators of gene expression and crucial players in cancer. Am. J. Transl. Res. 2012, 4, 127–150. [Google Scholar] [PubMed]
- Joshi, P.; Katsushima, K.; Zhou, R.; Meoded, A.; Stapleton, S.; Jallo, G.; Raabe, E.; Eberhart, C.G.; Perera, R.J. The therapeutic and diagnostic potential of regulatory noncoding RNAs in medulloblastoma. Neuro-oncol. Adv. 2019, 1. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Ameres, S.L.; Zamore, P. Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, E.; De Smaele, E.; Miele, E.; Laneve, P.; Po, A.; Pelloni, M.; Paganelli, A.; Di Marcotullio, L.; Caffarelli, E.; Screpanti, I.; et al. Concerted microRNA control of Hedgehog signalling in cerebellar neuronal progenitor and tumour cells. EMBO J. 2008, 27, 2616–2627. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, E.; De Smaele, E.; Po, A.; Di Marcotullio, L.; Tosi, E.; Espinola, M.S.B.; Di Rocco, C.; Riccardi, R.; Giangaspero, F.; Farcomeni, A.; et al. MicroRNA profiling in human medulloblastoma. Int. J. Cancer 2008, 124, 568–577. [Google Scholar] [CrossRef]
- Grunder, E.; D’Ambrosio, R.; Fiaschetti, G.; Abela, L.; Arcaro, A.; Zuzak, T.; Ohgaki, H.; Lv, S.-Q.; Shalaby, T.; Grotzer, M. MicroRNA-21 suppression impedes medulloblastoma cell migration. Eur. J. Cancer 2011, 47, 2479–2490. [Google Scholar] [CrossRef]
- Ray, S.; Coulter, D.W.; Gray, S.D.; Sughroue, J.A.; Roychoudhury, S.; McIntyre, E.M.; Chaturvedi, N.K.; Bhakat, K.K.; Joshi, S.S.; McGuire, T.R.; et al. Suppression of STAT3 NH2-terminal domain chemosensitizes medulloblastoma cells by activation of protein inhibitor of activated STAT3 via de-repression by microRNA-21. Mol. Carcinog. 2018, 57, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Shirsat, N.V.; Gokhale, A.; Kunder, R.; Goel, A.; Sarin, R.; Moiyadi, A.; Shenoy, A.; Mamidipally, C.; Noronha, S.; Kannan, S. Distinctive microRNA signature of medulloblastomas associated with the WNT signaling pathway. J. Cancer Res. Ther. 2010, 6, 521–529. [Google Scholar] [CrossRef]
- Kunder, R.; Jalali, R.; Sridhar, E.; Moiyadi, A.; Goel, N.; Goel, A.; Gupta, T.; Krishnatry, R.; Kannan, S.; Kurkure, P.; et al. Real-time PCR assay based on the differential expression of microRNAs and protein-coding genes for molecular classification of formalin-fixed paraffin embedded medulloblastomas. Neuro-oncology 2013, 15, 1644–1651. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Fernandez-L, A.; Hagan, J.P.; Ellison, D.W.; Grajkowska, W.; Gillespie, Y.; Grundy, R.; Van Meter, T.; Rutka, J.T.; Croce, C.M.; et al. The miR-17/92 polycistron is up-regulated in sonic hedgehog-driven medulloblastomas and induced by N-myc in sonic hedgehog-treated cerebellar neural precursors. Cancer Res. 2009, 69, 3249–3255. [Google Scholar] [CrossRef] [Green Version]
- Gershanov, S.; Toledano, H.; Michowiz, S.; Barinfeld, O.; Pinhasov, A.; Goldenberg-Cohen, N.; Salmon-Divon, M. MicroRNA-mRNA expression profiles associated with medulloblastoma subgroup 4. Cancer Manag. Res. 2018, 10, 339–352. [Google Scholar] [CrossRef] [Green Version]
- McDonald, J.D.; Daneshvar, L.; Willert, J.R.; Matsumura, K.; Waldman, F.; Cogen, P.H. Physical mapping of chromosome 17p13.3 in the region of a putative tumor suppressor gene important in medulloblastoma. Genomics 1994, 23, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, T.; Koch, A.; Wiestler, O.D. Molecular genetic studies in medulloblastomas: Evidence for tumor suppressor genes at the chromosomal regions 1q31-32 and 17p13. Klin. Padiatr. 1997, 209, 150–155. [Google Scholar] [CrossRef]
- Cogen, P.H.; McDonald, J.D. Tumor suppressor genes and medulloblastoma. J. Neuro-oncol. 1996, 29, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Cvekl, A.; Zavadil, J.; Birshtein, B.K.; Grotzer, M. Analysis of transcripts from 17p13.3 in medulloblastoma suggests ROX/MNT as a potential tumour suppressor gene. Eur. J. Cancer 2004, 40, 2525–2532. [Google Scholar] [CrossRef] [PubMed]
- Hoff, C.; Seranski, P.; Mollenhauer, J.; Korn, B.; Detzel, T.; Reinhardt, R.; Ramser, J.; Poustka, A. Physical and Transcriptional Mapping of the 17p13.3 Region That Is Frequently Deleted in Human Cancer. Genomics 2000, 70, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Briggs, K.J.; Corcoran-Schwartz, I.M.; Zhang, W.; Harcke, T.; Devereux, W.L.; Baylin, S.B.; Eberhart, C.G.; Watkins, D.N. Cooperation between the Hic1 and Ptch1 tumor suppressors in medulloblastoma. Genes Dev. 2008, 22, 770–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanchan, R.K.; Perumal, N.; Atri, P.; Chirravuri Venkata, R.; Thapa, I.; Klinkebiel, D.L.; Donson, A.M.; Perry, D.; Punsoni, M.; Talmon, G.A.; et al. MiR-1253 exerts tumor-suppressive effects in medulloblastoma via inhibition of CDK6 and CD276 (B7-H3). Brain Pathol. 2020, 30, 732–745. [Google Scholar] [CrossRef]
- Xu, Q.F.; Pan, Y.W.; Li, L.C.; Zhou, Z.; Huang, Q.L.; Pang, J.C.; Zhu, X.P.; Ren, Y.; Yang, H.; Ohgaki, H.; et al. MiR-22 is frequently downregulated in medulloblastomas and inhibits cell proliferation via the novel target PAPST1. Brain Pathol. 2014, 24, 568–583. [Google Scholar] [CrossRef] [PubMed]
- Perumal, N.; Kanchan, R.K.; Doss, D.; Bastola, N.; Atri, P.; Chirravuri-Venkata, R.; Thapa, I.; Vengoji, R.; Maurya, S.K.; Klinkebiel, D.; et al. MiR-212-3p functions as a tumor suppressor gene in group 3 medulloblastoma via targeting nuclear factor I/B (NFIB). Acta Neuropathol. Commun. 2021, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liang, J.; Liu, Z.; Zhang, C.; Wang, Y.; Watson, A.H.; Zhou, C.; Zhang, F.; Wu, K.; Zhang, F.; et al. The Role of CD276 in Cancers. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Shen, M.; Yao, H.; Chen, X.; Xiao, Z. Long Noncoding RNA TP73-AS1 Modulates Medulloblastoma Progression In Vitro And In Vivo By Sponging miR-494-3p And Targeting EIF5A2. OncoTargets Ther. 2019, 12, 9873–9885. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, T.; Wang, S.; Xiong, Y.; Zhang, R.; Zhang, X.; Zhao, J.; Yang, A.-G.; Wang, L.; Jia, L. Nkx2-2as Suppression Contributes to the Pathogenesis of Sonic Hedgehog Medulloblastoma. Cancer Res. 2017, 78, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Katsushima, K.; Lee, B.; Kunhiraman, H.; Zhong, C.; Murad, R.; Yin, J.; Liu, B.; Garancher, A.; Gonzalez-Gomez, I.; Monforte, H.L.; et al. The long noncoding RNA lnc-HLX-2-7 is oncogenic in Group 3 medulloblastomas. Neuro-oncology 2020, 23, 572–585. [Google Scholar] [CrossRef]
- Laneve, P.; Po, A.; Favia, A.; Legnini, I.; Alfano, V.; Rea, J.; Di Carlo, V.; Bevilacqua, V.; Miele, E.; Mastronuzzi, A.; et al. The long noncoding RNA linc-NeD125 controls the expression of medulloblastoma driver genes by microRNA sponge activity. Oncotarget 2017, 8, 31003–31015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutlu, M.; Tekin, C.; Aksoy, S.A.; Taskapilioglu, M.O.; Kaya, S.; Balcin, R.N.; Ocak, P.E.; Kocaeli, H.; Bekar, A.; Tolunay, S.; et al. Long non-coding RNAs as a predictive markers of group 3 medulloblastomas. Neurol. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kesherwani, V.; Shukla, M.; Coulter, D.W.; Sharp, J.G.; Joshi, S.S.; Chaturvedi, N.K. Long non-coding RNA profiling of pediatric Medulloblastoma. BMC Med. Genom. 2020, 13, 87. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Kuo, H.-C. The emerging roles and functions of circular RNAs and their generation. J. Biomed. Sci. 2019, 26, 29. [Google Scholar] [CrossRef] [PubMed]
- Lv, T.; Miao, Y.F.; Jin, K.; Han, S.; Xu, T.Q.; Qiu, Z.L.; Zhang, X.H. Dysregulated circular RNAs in medulloblastoma regulate proliferation and growth of tumor cells via host genes. Cancer Med. 2018, 7, 6147–6157. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
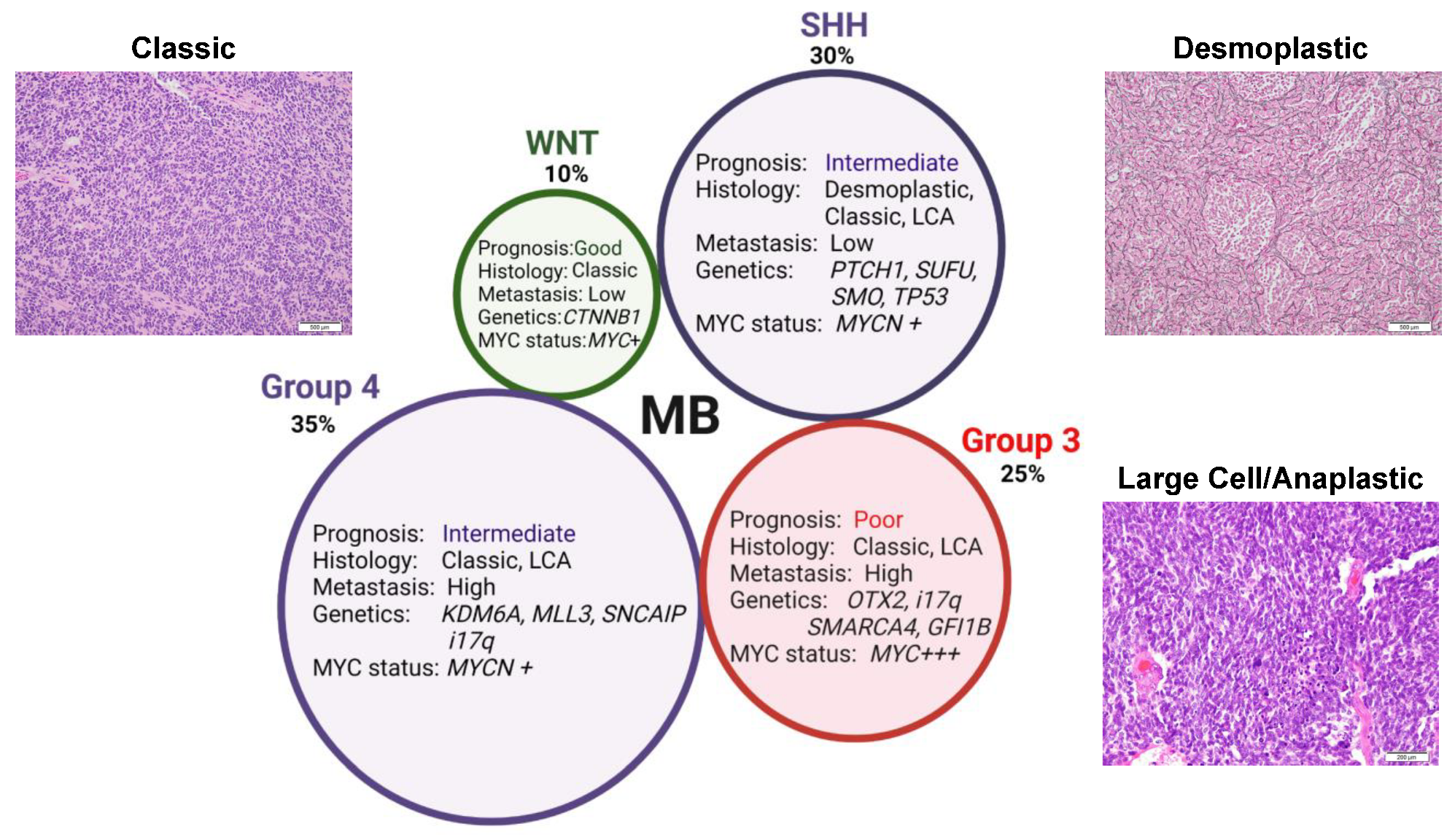
| Subgroup | Subtype | Frequency |
Median Age (yrs) | Distinguishing Genetic Events | Metastasis Incidence |
5-Year Overall Survival |
|---|---|---|---|---|---|---|
| WNT | WNTα | 70% | 10 | >95% monosomy chromosome 6 | 9% | 97% |
| WNTβ | 30% | 20 | ~30% monosomy chromosome 6 | 21% | 100% | |
| SHH | SHHα | 29% | 8 | Loss of 9q, 10q, 17p; gain of 9p; enriched in MYCN, GLI2 amp and TP53 mutations (35%) | 20% | 70% ** |
| SHHβ | 16% | 1.9 | Significant gain in chromosome 2; focal PTEN deletions (25%) | 33% | 67% | |
| SHHγ | 21% | 1.3 | Low copy number alterations | 9% | 88% | |
| SHHδ | 34% | 26 | Enriched in TERT mutations (90%) | 9% | 89% | |
| Group 3 | Grp 3α | 46% | 4.8 | i17q; loss of 8q and 17p | 43% | 66% |
| Grp 3β | 26% | 7.6 | OTX2 gain and DDX31 loss; activation of GFI1 and GFI1B oncogenes | 20% | 56% | |
| Grp 3γ | 28% | 5 | i17q; 8q gain and MYC amp | 39% | 42% | |
| Group 4 | Grp 4α | 30% | 8.2 | i17q; loss of 8p; 7q gain; MYCN and CDK6 amp | 40% | 67% |
| Grp 4β | 33% | 10 | i17q; 17p loss; SNCAIP dup | 41% | 75% | |
| Grp 4γ | 37% | 7 | i17q; loss of 8p; 7q gain; CDK6 amp | 39% | 83% |
| Average Risk | High Risk | |
|---|---|---|
| Age at diagnosis | 3 years old | <3 years old |
| Extend of post-operative disease (by MRI) | 1.5 cm | >1.5 cm |
| Presence of metastasis | No | Yes |
| 5-year event-free survival | 85% | 60–70% |
| Intensity of craniospinal irradiation | 23.4 Gy | 36–39 Gy * |
| Adjuvant chemotherapy | cisplatin, vincristine, cyclophosphamide or lomustine | cisplatin, vincristine, cyclophosphamide, and lomustine |
| Clinical Trial | Subgroups | Interventions | Phase |
|---|---|---|---|
| NCT01878617 | All | WNT: low dose CSI and lower dose cyclophosphamide SHH: vismodegib after standard chemotherapy G3/G4: adding permetrexed + gemcitabine to standard chemotherapy G3/G4: reduced dose cyclophosphamide | II |
| NCT02066220 | WNT | Low-risk: radiotherapy without carboplatin and reduced-intensity maintenance chemotherapy Standard-risk: radiotherapy with carboplatin and maintenance chemotherapy | II/III |
| NCT02359565 | Recurrent or Refractory MB | Pembrolizumab (MK-3475) every 21 days for 34 cycles | I |
| NCT02644460 | Recurrent or Refractory MB | Abemaciclib twice daily for 28 days with dose escalation for up to 2 years | I |
| NCT02724579 | WNT | Reduced CSI and no vincristine during radiotherapy followed by reduced-dose maintenance chemotherapy | II |
| NCT03130959 | Recurrent or Refractory MB | A: Nivolumab B: Nivolumab + Ipilimumab | II |
| NCT03155620 | Recurrent or Refractory MB | Targeted therapy based on genetic testing | II |
| NCT03173950 | Recurrent | Nivolumab weekly for 16 doses | II |
| NCT03213678 | Recurrent MB | Samotolisib twice daily for 28 days for up to 2 years | II |
| NCT03434262 | Recurrent or Refractory MB | SHH: Ribociclib + Sonidegib WNT/SHH: Ribociclib + Trametinib G3/G4: Ribociclib + gemcitabine | I |
| NCT04023669 | Recurrent or Refractory MB | All: Prexasertib + cyclophosphamide monthly up to 24 months G3/G4: Prexasertib + gemcitabine monthly up to 24 months | I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ray, S.; Chaturvedi, N.K.; Bhakat, K.K.; Rizzino, A.; Mahapatra, S. Subgroup-Specific Diagnostic, Prognostic, and Predictive Markers Influencing Pediatric Medulloblastoma Treatment. Diagnostics 2022, 12, 61. https://doi.org/10.3390/diagnostics12010061
Ray S, Chaturvedi NK, Bhakat KK, Rizzino A, Mahapatra S. Subgroup-Specific Diagnostic, Prognostic, and Predictive Markers Influencing Pediatric Medulloblastoma Treatment. Diagnostics. 2022; 12(1):61. https://doi.org/10.3390/diagnostics12010061
Chicago/Turabian StyleRay, Sutapa, Nagendra K. Chaturvedi, Kishor K. Bhakat, Angie Rizzino, and Sidharth Mahapatra. 2022. "Subgroup-Specific Diagnostic, Prognostic, and Predictive Markers Influencing Pediatric Medulloblastoma Treatment" Diagnostics 12, no. 1: 61. https://doi.org/10.3390/diagnostics12010061