
Cancer Immunotherapy with Immune Checkpoint Inhibitors-Biomarkers of Response and Toxicity; Current Limitations and Future Promise

 , , , and
, , , and
Abstract
:1. Introduction
2. Approved Predictive Biomarkers of Response
2.1. Immunohistochemistry for PD-L1
2.2. Tumour Mutational Burden
2.3. Microsatellite Instability
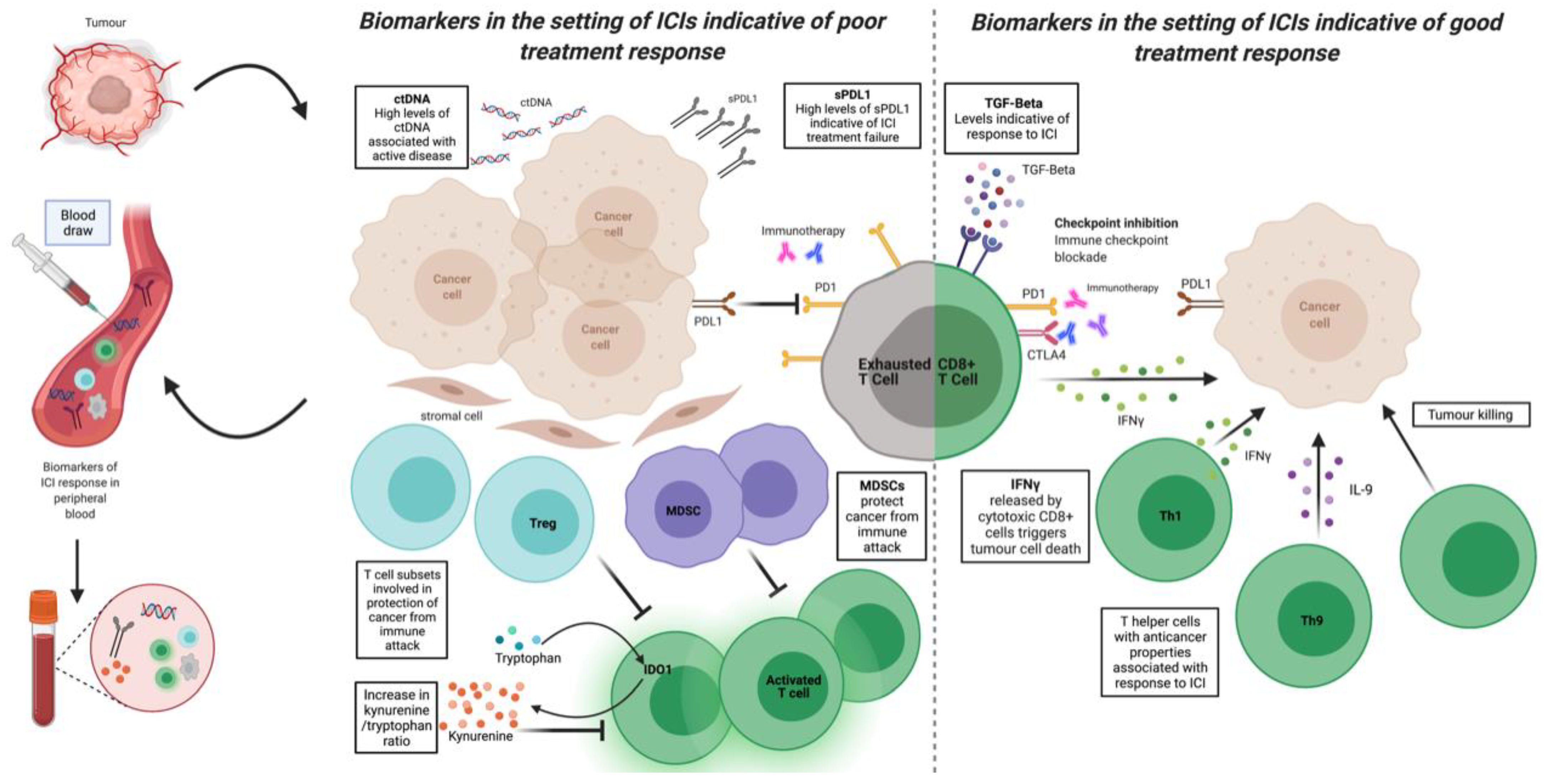
3. Novel Biomarkers of Response, Resistance, and Toxicity
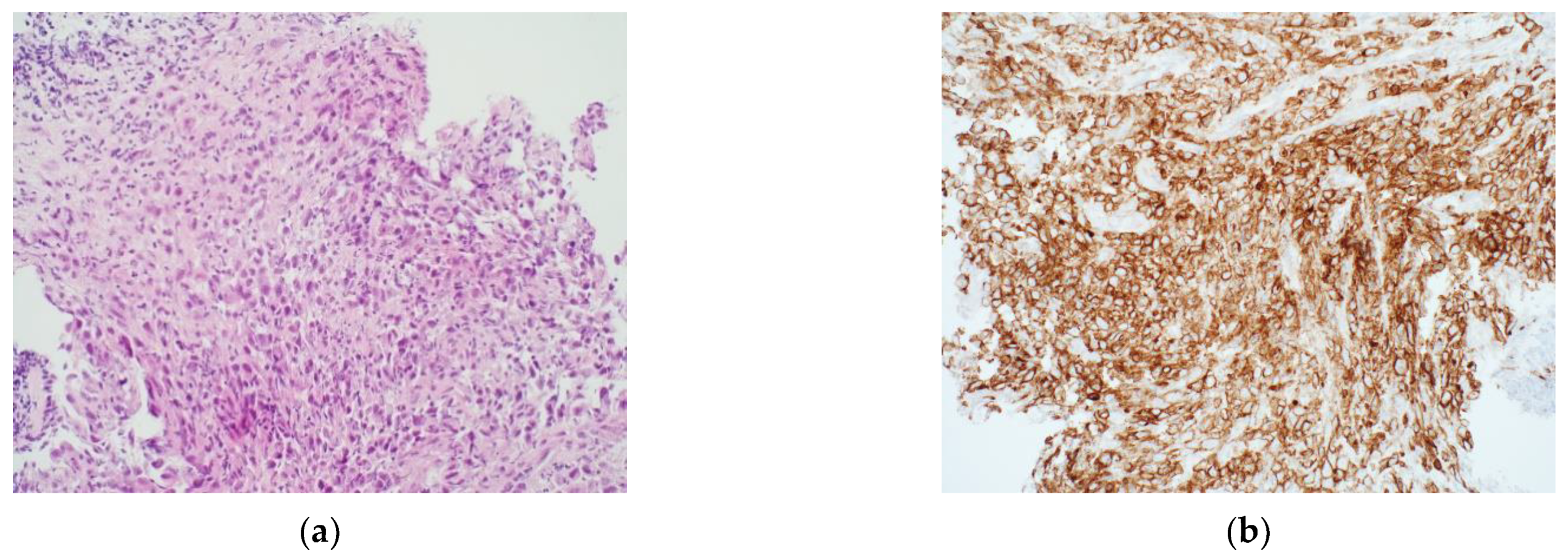
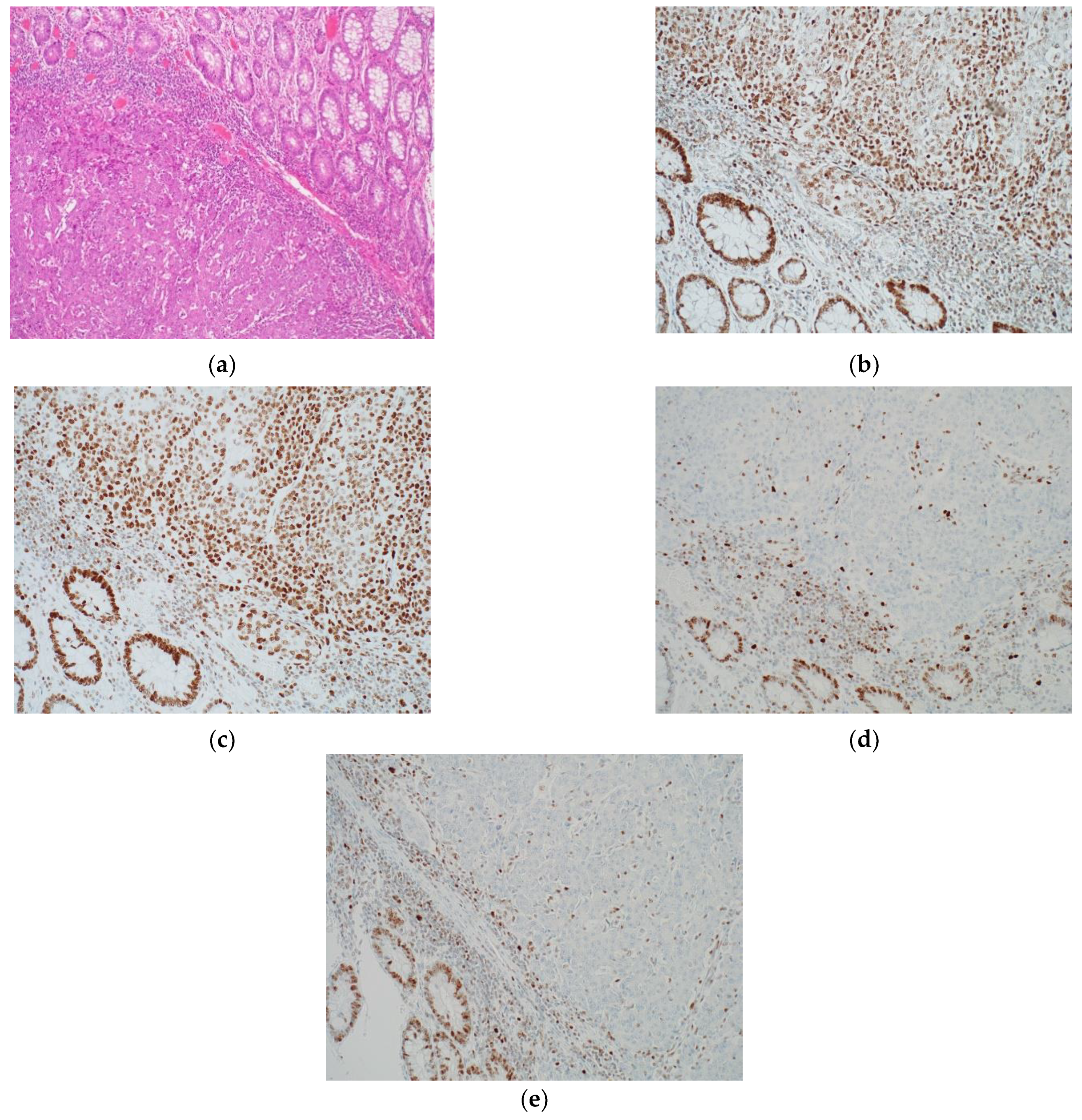
3.1. Immunohistochemistry
3.2. Systemic Markers of Inflammation
3.3. Cytokines, Chemokines, and Other Soluble Immune Markers
3.4. Immune Metabolism
3.5. Flow Cytometry of Circulating Immune Cells
3.6. Next Generation Sequencing
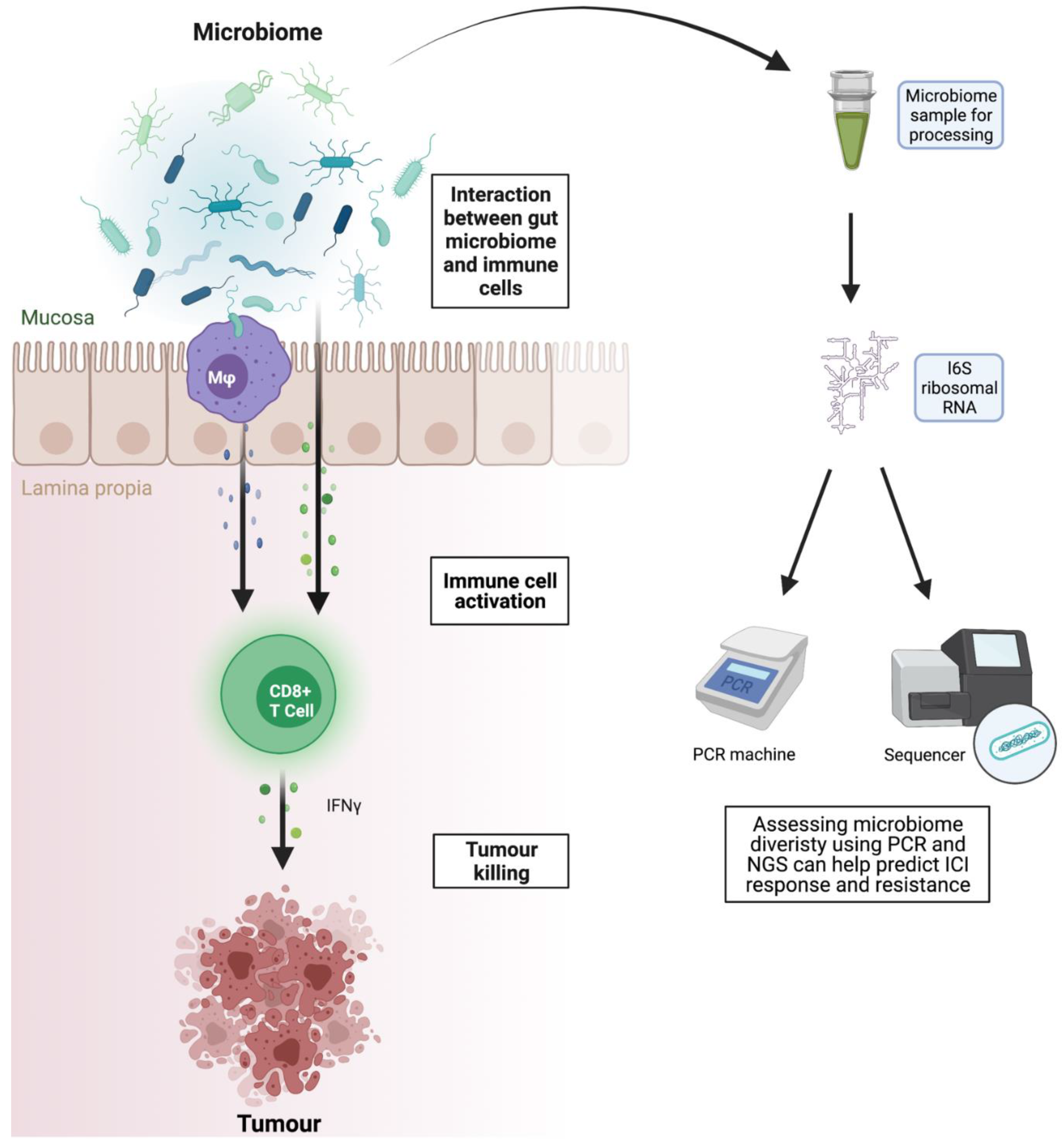
3.7. Microbiome as Biomarker and Therapeutic Target
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shankar, B.; Zhang, J.; Naqash, A.R.; Forde, P.M.; Feliciano, J.L.; Marrone, K.A.; Ettinger, D.S.; Hann, C.L.; Brahmer, J.R.; Ricciuti, B.; et al. Multisystem Immune-Related Adverse Events Associated With Immune Checkpoint Inhibitors for Treatment of Non–Small Cell Lung Cancer. JAMA Oncol. 2020, 6, 1952–1956. [Google Scholar] [CrossRef]
- Ganesan, S.; Mehnert, J. Biomarkers for Response to Immune Checkpoint Blockade. Annu. Rev. Cancer Biol. 2020, 4, 331–351. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Huang, Z.; Teng, F.; Xing, L.; Yu, J. Predictive biomarkers in PD-1/PD-L1 checkpoint blockade immunotherapy. Cancer Treat. Rev. 2015, 41, 868–876. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Peng, W.; Liu, C.; Xu, C.; Lou, Y.; Chen, J.; Yang, Y.; Yagita, H.; Overwijk, W.W.; Lizée, G.; Radvanyi, L.; et al. PD-1 Blockade Enhances T-cell Migration to Tumors by Elevating IFN-γ Inducible Chemokines. Cancer Res. 2012, 72, 5209–5218. [Google Scholar] [CrossRef] [Green Version]
- Bridge, J.A.; Lee, J.C.; Daud, A.; Wells, J.W.; Bluestone, J.A. Cytokines, Chemokines, and Other Biomarkers of Response for Checkpoint Inhibitor Therapy in Skin Cancer. Front. Med. 2018, 5, 351. [Google Scholar] [CrossRef]
- Rivas-Fuentes, S.; Salgado-Aguayo, A.; Pertuz Belloso, S.; Gorocica Rosete, P.; Alvarado-Vásquez, N.; Aquino-Jarquin, G. Role of Chemokines in Non-Small Cell Lung Cancer: Angiogenesis and Inflammation. J. Cancer 2015, 6, 938–952. [Google Scholar] [CrossRef] [Green Version]
- Ji, S.; Chen, H.; Yang, K.; Zhang, G.; Mao, B.; Hu, Y.; Zhang, H.; Xu, J. Peripheral cytokine levels as predictive biomarkers of benefit from immune checkpoint inhibitors in cancer therapy. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 129, 110457. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, H.; Zhang, T.; Yang, X.; Zhong, J.; Wang, Y.; Chi, Y.; Wu, M.; An, T.; Li, J.; et al. Plasma cytokines interleukin-18 and C-X-C motif chemokine ligand 10 are indicative of the anti-programmed cell death protein-1 treatment response in lung cancer patients. Ann. Transl. Med. 2021, 9, 33. [Google Scholar] [CrossRef]
- Boutsikou, E.; Domvri, K.; Hardavella, G.; Tsiouda, D.; Zarogoulidis, K.; Kontakiotis, T. Tumour necrosis factor, interferon-gamma and interleukins as predictive markers of antiprogrammed cell-death protein-1 treatment in advanced non-small cell lung cancer: A pragmatic approach in clinical practice. Ther. Adv. Med. Oncol. 2018, 10, 1758835918768238. [Google Scholar]
- Montfort, A.; Dufau, C.; Colacios, C.; Andrieu-Abadie, N.; Levade, T.; Filleron, T.; Delord, J.-P.; Ayyoub, M.; Meyer, N.; Ségui, B. Anti-TNF, a magic bullet in cancer immunotherapy? J. Immunother. Cancer 2019, 7, 303. [Google Scholar] [CrossRef]
- Alvarez, M.; Otano, I.; Minute, L.; Ochoa, M.C.; Perez-Ruiz, E.; Melero, I.; Berraondo, P. Impact of prophylactic TNF blockade in the dual PD-1 and CTLA-4 immunotherapy efficacy and toxicity. Cell Stress 2019, 3, 236–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Ruiz, E.; Minute, L.; Otano, I.; Alvarez, M.; Ochoa, M.C.; Belsue, V.; de Andrea, C.; Rodriguez-Ruiz, M.E.; Perez-Gracia, J.L.; Marquez-Rodas, I.; et al. Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature 2019, 569, 428–432. [Google Scholar]
- Bertrand, F.; Montfort, A.; Marcheteau, E.; Imbert, C.; Gilhodes, J.; Filleron, T.; Rochaix, P.; Andrieu-Abadie, N.; Levade, T.; Meyer, N.; et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 2017, 8, 2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [PubMed]
- Mildner, F.; Sopper, S.; Amann, A.; Pircher, A.; Pall, G.; Köck, S.; Naismith, E.; Wolf, D.; Gamerith, G. Systematic review: Soluble immunological biomarkers in advanced non-small-cell lung cancer (NSCLC). Crit. Rev. Oncol. Hematol. 2020, 153, 102948. [Google Scholar] [CrossRef]
- Cresswell, G.D.; Nichol, D.; Spiteri, I.; Tari, H.; Zapata, L.; Heide, T.; Maley, C.C.; Magnani, L.; Schiavon, G.; Ashworth, A.; et al. Mapping the breast cancer metastatic cascade onto ctDNA using genetic and epigenetic clonal tracking. Nat. Commun. 2020, 11, 1446. [Google Scholar]
- Troncone, G.; Gridelli, C. The reproducibility of PD-L1 scoring in lung cancer: Can the pathologists do better? Transl. Lung Cancer Res. 2017, 6, S74–S77. [Google Scholar]
- Wang, C.; Hahn, E.; Slodkowska, E.; Eskander, A.; Enepekides, D.; Higgins, K.; Vesprini, D.; Liu, S.K.; Downes, M.R.; Xu, B. Reproducibility of PD-L1 immunohistochemistry interpretation across various types of genitourinary and head/neck carcinomas, antibody clones, and tissue types. Hum. Pathol. 2018, 82, 131–139. [Google Scholar] [CrossRef]
- Scagliotti, G.V.; Parikh, P.; von Pawel, J.; Biesma, B.; Vansteenkiste, J.; Manegold, C.; Serwatowski, P.; Gatzemeier, U.; Digumarti, R.; Zukin, M.; et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 3543–3551. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Torlakovic, E.; Lim, H.J.; Adam, J.; Barnes, P.; Bigras, G.; Chan, A.W.H.; Cheung, C.C.; Chung, J.-H.; Couture, C.; Fiset, P.O.; et al. “Interchangeability” of PD-L1 immunohistochemistry assays: A meta-analysis of diagnostic accuracy. Mod. Pathol. 2020, 33, 4–17. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.S.K.; Wu, Y.-L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Jørgensen, J.T. PD-L1 expression and efficacy of pembrolizumab as monotherapy in NSCLC. Chin. Clin. Oncol. 2020, 9, 60. [Google Scholar] [CrossRef]
- Morita, M.; Tamiya, M.; Fujimoto, D.; Tamiya, A.; Suzuki, H.; Hirano, K.; Fukuda, Y.; Yokoyama, T.; Kominami, R.; Kanazu, M.; et al. Prediction of patients with a tumor proportion score >50% who do not respond to first-line monotherapy with pembrolizumab. BMC Cancer 2020, 20, 93. [Google Scholar] [CrossRef] [Green Version]
- Freidlin, B.; Korn, E.L. A Problematic Biomarker Trial Design. J. Natl. Cancer Inst. 2021, djab144. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez–Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Updated Analysis of KEYNOTE-024: Pembrolizumab Versus Platinum-Based Chemotherapy for Advanced Non–Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score of 50% or Greater. J. Clin. Oncol. 2019, 37, 537–546. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Five-Year Outcomes With Pembrolizumab Versus Chemotherapy for Metastatic Non-Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score ≥ 50. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 2339–2349. [Google Scholar] [CrossRef]
- KEYTRUDA 25 mg/mL Concentrate for Solution for Infusion—Summary of Product Characteristics (SmPC)—(emc). Available online: https://www.medicines.org.uk/emc/product/2498/smpc#gref (accessed on 15 July 2021).
- Czarska-Thorley, D. Opdivo: Pending EC Decision. Available online: https://www.ema.europa.eu/en/medicines/human/summaries-opinion/opdivo-6 (accessed on 26 September 2021).
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Gadgeel, S.; Rodríguez-Abreu, D.; Speranza, G.; Esteban, E.; Felip, E.; Dómine, M.; Hui, R.; Hochmair, M.J.; Clingan, P.; Powell, S.F.; et al. Updated Analysis From KEYNOTE-189: Pembrolizumab or Placebo Plus Pemetrexed and Platinum for Previously Untreated Metastatic Nonsquamous Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2020, 38, 1505–1517. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Shah, M.A.; Muro, K.; Francois, E.; Adenis, A.; Hsu, C.-H.; Doi, T.; Moriwaki, T.; Kim, S.-B.; Lee, S.-H.; et al. Randomized Phase III KEYNOTE-181 Study of Pembrolizumab Versus Chemotherapy in Advanced Esophageal Cancer. J. Clin. Oncol. 2020, 38, 4138–4148. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Shitara, K.; Özgüroğlu, M.; Bang, Y.-J.; Di Bartolomeo, M.; Mandalà, M.; Ryu, M.-H.; Fornaro, L.; Olesiński, T.; Caglevic, C.; Chung, H.C.; et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): A randomised, open-label, controlled, phase 3 trial. Lancet 2018, 392, 123–133. [Google Scholar] [CrossRef]
- Shitara, K.; Van Cutsem, E.; Bang, Y.-J.; Fuchs, C.; Wyrwicz, L.; Lee, K.-W.; Kudaba, I.; Garrido, M.; Chung, H.C.; Lee, J.; et al. Efficacy and Safety of Pembrolizumab or Pembrolizumab Plus Chemotherapy vs. Chemotherapy Alone for Patients With First-line, Advanced Gastric Cancer: The KEYNOTE-062 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1571–1580. [Google Scholar] [CrossRef]
- Shah, M.A.; Kojima, T.; Hochhauser, D.; Enzinger, P.; Raimbourg, J.; Hollebecque, A.; Lordick, F.; Kim, S.-B.; Tajika, M.; Kim, H.T.; et al. Efficacy and Safety of Pembrolizumab for Heavily Pretreated Patients With Advanced, Metastatic Adenocarcinoma or Squamous Cell Carcinoma of the Esophagus: The Phase 2 KEYNOTE-180 Study. JAMA Oncol. 2019, 5, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-T.; Kang, Y.-K.; Satoh, T.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.-S.; Muro, K.; Kang, W.; Yoshikawa, T.; et al. A phase III study of nivolumab (Nivo) in previously treated advanced gastric or gastric esophageal junction (G/GEJ) cancer (ATTRACTION-2): Three-year update data. J. Clin. Oncol. 2020, 38, 383. [Google Scholar] [CrossRef]
- Kato, K.; Cho, B.C.; Takahashi, M.; Okada, M.; Lin, C.-Y.; Chin, K.; Kadowaki, S.; Ahn, M.-J.; Hamamoto, Y.; Doki, Y.; et al. Nivolumab versus chemotherapy in patients with advanced oesophageal squamous cell carcinoma refractory or intolerant to previous chemotherapy (ATTRACTION-3): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 1506–1517. [Google Scholar] [CrossRef]
- Sun, J.-M.; Shen, L.; Shah, M.A.; Enzinger, P.; Adenis, A.; Doi, T.; Kojima, T.; Metges, J.-P.; Li, Z.; Kim, S.-B.; et al. Pembrolizumab plus chemotherapy versus chemotherapy alone for first-line treatment of advanced oesophageal cancer (KEYNOTE-590): A randomised, placebo-controlled, phase 3 study. Lancet 2021, 398, 759–771. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Bragagnoli, A.C.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef]
- Munari, E.; Zamboni, G.; Lunardi, G.; Marchionni, L.; Marconi, M.; Sommaggio, M.; Brunelli, M.; Martignoni, G.; Netto, G.J.; Hoque, M.O.; et al. PD-L1 Expression Heterogeneity in Non–Small Cell Lung Cancer: Defining Criteria for Harmonization between Biopsy Specimens and Whole Sections. J. Thorac. Oncol. 2018, 13, 1113–1120. [Google Scholar] [CrossRef] [Green Version]
- Casadevall, D.; Clavé, S.; Taus, Á.; Hardy-Werbin, M.; Rocha, P.; Lorenzo, M.; Menéndez, S.; Salido, M.; Albanell, J.; Pijuan, L.; et al. Heterogeneity of Tumor and Immune Cell PD-L1 Expression and Lymphocyte Counts in Surgical NSCLC Samples. Clin. Lung Cancer 2017, 18, 682–691.e5. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Perez-Gracia, J.L.; Felip, E.; Kim, D.-W.; Han, J.-Y.; Molina, J.R.; Kim, J.-H.; Dubos Arvis, C.; Ahn, M.-J.; et al. Use of archival versus newly collected tumor samples for assessing PD-L1 expression and overall survival: An updated analysis of KEYNOTE-010 trial. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Fang, W.; Yu, J.; Chen, N.; Zhan, J.; Ma, Y.; Yang, Y.; Huang, Y.; Zhao, H.; Zhang, L. Expression of programmed death ligand-1 on tumor cells varies pre and post chemotherapy in non-small cell lung cancer. Sci. Rep. 2016, 6, 20090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, C.H.; Koh, J.; Ock, C.-Y.; Kim, M.; Keam, B.; Kim, T.M.; Jeon, Y.K.; Kim, D.-W.; Chung, D.H.; Heo, D.S. Temporal evolution of programmed death-ligand 1 expression in patients with non-small cell lung cancer. Korean J. Intern. Med. 2021, 36, 975–984. [Google Scholar] [CrossRef]
- Shklovskaya, E.; Rizos, H. Spatial and Temporal Changes in PD-L1 Expression in Cancer: The Role of Genetic Drivers, Tumor Microenvironment and Resistance to Therapy. Int. J. Mol. Sci. 2020, 21, 7139. [Google Scholar] [CrossRef] [PubMed]
- Echle, A.; Rindtorff, N.T.; Brinker, T.J.; Luedde, T.; Pearson, A.T.; Kather, J.N. Deep learning in cancer pathology: A new generation of clinical biomarkers. Br. J. Cancer 2021, 124, 686–696. [Google Scholar] [CrossRef]
- Goodman, A.M.; Castro, A.; Pyke, R.M.; Okamura, R.; Kato, S.; Riviere, P.; Frampton, G.; Sokol, E.; Zhang, X.; Ball, E.D.; et al. MHC-I genotype and tumor mutational burden predict response to immunotherapy. Genome Med. 2020, 12, 45. [Google Scholar] [CrossRef]
- Ramos-Paradas, J.; Hernández-Prieto, S.; Lora, D.; Sanchez, E.; Rosado, A.; Caniego-Casas, T.; Carrizo, N.; Enguita, A.B.; Muñoz-Jimenez, M.T.; Rodriguez, B.; et al. Tumor mutational burden assessment in non-small-cell lung cancer samples: Results from the TMB2 harmonization project comparing three NGS panels. J. Immunother. Cancer 2021, 9, e001904. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Zhou, K.I.; Peterson, B.; Serritella, A.; Thomas, J.; Reizine, N.; Moya, S.; Tan, C.; Wang, Y.; Catenacci, D.V.T. Spatial and Temporal Heterogeneity of PD-L1 Expression and Tumor Mutational Burden in Gastroesophageal Adenocarcinoma at Baseline Diagnosis and after Chemotherapy. Clin. Cancer Res. 2020, 26, 6453–6463. [Google Scholar] [CrossRef]
- Kazdal, D.; Endris, V.; Allgäuer, M.; Kriegsmann, M.; Leichsenring, J.; Volckmar, A.-L.; Harms, A.; Kirchner, M.; Kriegsmann, K.; Neumann, O.; et al. Spatial and Temporal Heterogeneity of Panel-Based Tumor Mutational Burden in Pulmonary Adenocarcinoma: Separating Biology From Technical Artifacts. J. Thorac. Oncol. 2019, 14, 1935–1947. [Google Scholar] [CrossRef]
- Wang, Z.; Duan, J.; Cai, S.; Han, M.; Dong, H.; Zhao, J.; Zhu, B.; Wang, S.; Zhuo, M.; Sun, J.; et al. Assessment of Blood Tumor Mutational Burden as a Potential Biomarker for Immunotherapy in Patients With Non–Small Cell Lung Cancer With Use of a Next-Generation Sequencing Cancer Gene Panel. JAMA Oncol. 2019, 5, 696–702. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Socinski, M.; Velcheti, V.; Mekhail, T.; Chae, Y.K.; Leal, T.A.; Dowell, J.E.; Tsai, M.L.; Dakhil, C.S.; Stella, P.; Shen, V.; et al. Final efficacy results from B-F1RST, a prospective phase II trial evaluating blood-based tumour mutational burden (bTMB) as a predictive biomarker for atezolizumab (atezo) in 1L non-small cell lung cancer (NSCLC). Ann. Oncol. 2019, 30, v919–v920. [Google Scholar] [CrossRef]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Bakir, M.A.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Litchfield, K.; Reading, J.L.; Puttick, C.; Thakkar, K.; Abbosh, C.; Bentham, R.; Watkins, T.B.K.; Rosenthal, R.; Biswas, D.; Rowan, A.; et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021, 184, 596–614.e14. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yang, C.; He, N.; Zhao, G.; Wang, J.; Yang, Y. Integration of the Tumor Mutational Burden and Tumor Heterogeneity Identify an Immunological Subtype of Melanoma With Favorable Survival. Front. Oncol. 2020, 10, 2435. [Google Scholar] [CrossRef] [PubMed]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Stelloo, E.; Jansen, A.M.L.; Osse, E.M.; Nout, R.A.; Creutzberg, C.L.; Ruano, D.; Church, D.N.; Morreau, H.; Smit, V.T.H.B.M.; van Wezel, T.; et al. Practical guidance for mismatch repair-deficiency testing in endometrial cancer. Ann. Oncol. 2017, 28, 96–102. [Google Scholar] [CrossRef]
- Smyth, E.C.; Wotherspoon, A.; Peckitt, C.; Gonzalez, D.; Hulkki-Wilson, S.; Eltahir, Z.; Fassan, M.; Rugge, M.; Valeri, N.; Okines, A.; et al. Mismatch Repair Deficiency, Microsatellite Instability, and Survival. JAMA Oncol. 2017, 3, 1197–1203. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Imai, K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Semin. Oncol. 2019, 46, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Bonneville, R.; Krook, M.A.; Chen, H.-Z.; Smith, A.; Samorodnitsky, E.; Wing, M.R.; Reeser, J.W.; Roychowdhury, S. Detection of Microsatellite Instability Biomarkers via Next-Generation Sequencing. Methods Mol. Biol. 2020, 2055, 119–132. [Google Scholar]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Tieng, F.Y.F.; Abu, N.; Lee, L.-H.; Ab Mutalib, N.-S. Microsatellite Instability in Colorectal Cancer Liquid Biopsy—Current Updates on Its Potential in Non-Invasive Detection, Prognosis and as a Predictive Marker. Diagnostics 2021, 11, 544. [Google Scholar] [CrossRef]
- Zhuge, L.; Huang, B.; Xie, J.; Gao, Z.; Zheng, D.; Zheng, S.; Xiang, J.; Zhang, J. Immunoscore Signature Predicts Postoperative Survival and Adjuvant Chemotherapeutic Benefits in Esophageal Squamous Cell Carcinoma. Cancer Manag. Res. 2020, 12, 12885–12894. [Google Scholar] [CrossRef]
- Nassif, E.F.; Mlecnik, B.; Thibault, C.; Auvray, M.; Bruni, D.; Colau, A.; Compérat, E.; Bindea, G.; Catteau, A.; Fugon, A.; et al. The Immunoscore in Localized Urothelial Carcinoma Treated with Neoadjuvant Chemotherapy: Clinical Significance for Pathologic Responses and Overall Survival. Cancers 2021, 13, 494. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bifulco, C.; Bindea, G.; Marliot, F.; Lugli, A.; Lee, J.J.; Zlobec, I.; Rau, T.T.; Berger, M.D.; Nagtegaal, I.D.; et al. Multicenter International Society for Immunotherapy of Cancer Study of the Consensus Immunoscore for the Prediction of Survival and Response to Chemotherapy in Stage III Colon Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 3638–3651. [Google Scholar] [CrossRef]
- Federation Francophone de Cancerologie Digestive. Pembrolizumab in Combination with Xelox Bevacizumab in Patients with Microsatellite Stable Mestatic Colorectal Cancer and a High Immune Infiltrate: A Proof of Concept Study. 2020 October Report; No.: NCT04262687. Available online: https://clinicaltrials.gov/ct2/show/NCT04262687 (accessed on 4 August 2021).
- Frontiers|Correlation between PD-L2 Expression and Clinical Outcome in Solid Cancer Patients: A Meta-Analysis|Oncology. Available online: https://www.frontiersin.org/articles/10.3389/fonc.2019.00047/full (accessed on 30 December 2021).
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.M.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef] [Green Version]
- Banna, G.L.; Cantale, O.; Bersanelli, M.; Del Re, M.; Friedlaender, A.; Cortellini, A.; Addeo, A. Are anti-PD1 and anti-PD-L1 alike? The non-small-cell lung cancer paradigm. Oncol. Rev. 2020, 14, 490. [Google Scholar] [CrossRef] [PubMed]
- Solinas, C.; Aiello, M.; Rozali, E.; Lambertini, M.; Willard-Gallo, K.; Migliori, E. Programmed Cell Death-Ligand 2: A Neglected But Important Target in the Immune Response to Cancer? Transl. Oncol. 2020, 13, 100811. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [Green Version]
- Lecocq, Q.; Keyaerts, M.; Devoogdt, N.; Breckpot, K. The Next-Generation Immune Checkpoint LAG-3 and Its Therapeutic Potential in Oncology: Third Time’s a Charm. Int. J. Mol. Sci. 2021, 22, 75. [Google Scholar] [CrossRef]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef] [Green Version]
- Graydon, C.G.; Mohideen, S.; Fowke, K.R. LAG3’s Enigmatic Mechanism of Action. Front. Immunol. 2021, 11, 3444. [Google Scholar] [CrossRef]
- Xu, F.; Liu, J.; Liu, D.; Liu, B.; Wang, M.; Hu, Z.; Du, X.; Tang, L.; He, F. LSECtin Expressed on Melanoma Cells Promotes Tumor Progression by Inhibiting Antitumor T-cell Responses. Cancer Res. 2014, 74, 3418–3428. [Google Scholar] [CrossRef] [Green Version]
- Saleh, R.R.; Peinado, P.; Fuentes-Antrás, J.; Pérez-Segura, P.; Pandiella, A.; Amir, E.; Ocaña, A. Prognostic Value of Lymphocyte-Activation Gene 3 (LAG3) in Cancer: A Meta-Analysis. Front. Oncol. 2019, 9, 1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipson, E.J. Relatlimab (RELA) Plus Nivolumab (NIVO) versus NIVO in First-Line Advanced Melanoma: Primary Phase III Results from RELATIVITY-047 (CA224-047). Available online: https://meetinglibrary.asco.org/record/201596/abstract (accessed on 23 May 2021).
- ASCO 2021: LAG-3 Is Now a Validated Target in Melanoma—Clinical Trials Arena. Available online: https://www.clinicaltrialsarena.com/comment/asco-2021-validated-target-melanoma/ (accessed on 31 December 2021).
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Khair, D.O.; Bax, H.J.; Mele, S.; Crescioli, S.; Pellizzari, G.; Khiabany, A.; Nakamura, M.; Harris, R.J.; French, E.; Hoffmann, R.M.; et al. Combining Immune Checkpoint Inhibitors: Established and Emerging Targets and Strategies to Improve Outcomes in Melanoma. Front. Immunol. 2019, 10, 453. [Google Scholar] [CrossRef] [Green Version]
- Compugen Ltd A Phase 1a/1b Study of COM701 as Monotherapy and in Combination with an Anti-PD-1 Antibody in Subjects with Advanced Solid Tumors. 2021 October Report; No.: NCT03667716. Available online: https://clinicaltrials.gov/ct2/show/NCT03667716 (accessed on 29 December 2021).
- Kelderman, S.; Heemskerk, B.; van Tinteren, H.; van den Brom, R.R.H.; Hospers, G.A.P.; van den Eertwegh, A.J.M.; Kapiteijn, E.W.; de Groot, J.W.B.; Soetekouw, P.; Jansen, R.L.; et al. Lactate dehydrogenase as a selection criterion for ipilimumab treatment in metastatic melanoma. Cancer Immunol. Immunother. 2014, 63, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, S.; Zhang, S.; Liu, Y.; Ma, L.; Zhu, J.; Xin, Y.; Wang, Y.; Yang, C.; Cheng, Y. Systemic immune-inflammation index, neutrophil-to-lymphocyte ratio, platelet-to-lymphocyte ratio can predict clinical outcomes in patients with metastatic non-small-cell lung cancer treated with nivolumab. J. Clin. Lab. Anal. 2019, 33, e22964. [Google Scholar] [CrossRef]
- Capone, M.; Giannarelli, D.; Mallardo, D.; Madonna, G.; Festino, L.; Grimaldi, A.M.; Vanella, V.; Simeone, E.; Paone, M.; Palmieri, G.; et al. Baseline neutrophil-to-lymphocyte ratio (NLR) and derived NLR could predict overall survival in patients with advanced melanoma treated with nivolumab. J. Immunother. Cancer 2018, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, P.F.; Ascierto, P.A.; Pigozzo, J.; Del Vecchio, M.; Maio, M.; Antonini Cappellini, G.C.; Guidoboni, M.; Queirolo, P.; Savoia, P.; Mandalà, M.; et al. Baseline neutrophils and derived neutrophil-to-lymphocyte ratio: Prognostic relevance in metastatic melanoma patients receiving ipilimumab. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Han, X.; Zha, H.; Tao, H.; Li, X.; Yuan, F.; Chen, G.; Wang, L.; Ma, J.; Hu, Y. Systemic Immune-Inflammation Index and Changes of Neutrophil-Lymphocyte Ratio as Prognostic Biomarkers for Patients With Pancreatic Cancer Treated With Immune Checkpoint Blockade. Front. Oncol. 2021, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- McQuade, J.L.; Daniel, C.R.; Hess, K.R.; Mak, C.; Wang, D.Y.; Rai, R.R.; Park, J.J.; Haydu, L.E.; Spencer, C.; Wongchenko, M.; et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: A retrospective, multicohort analysis. Lancet Oncol. 2018, 19, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.T.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R.; et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 2019, 25, 141–151. [Google Scholar] [CrossRef]
- Chehrazi-Raffle, A.; Meza, L.; Alcantara, M.; Dizman, N.; Bergerot, P.; Salgia, N.; Hsu, J.; Ruel, N.; Salgia, S.; Malhotra, J.; et al. Circulating cytokines associated with clinical response to systemic therapy in metastatic renal cell carcinoma. J. Immunother. Cancer 2021, 9, e002009. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tsoi, J.; Onyshchenko, M.; Abril-Rodriguez, G.; Ross-Macdonald, P.; Wind-Rotolo, M.; Champhekar, A.; Medina, E.; Torrejon, D.Y.; Shin, D.S.; et al. Conserved Interferon-γ Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 2020, 38, 500–515.e3. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef] [Green Version]
- Frangieh, C.J.; Melms, J.C.; Thakore, P.I.; Geiger-Schuller, K.R.; Ho, P.; Luoma, A.M.; Cleary, B.; Jerby-Arnon, L.; Malu, S.; Cuoco, M.S.; et al. Multimodal pooled Perturb-CITE-seq screens in patient models define mechanisms of cancer immune evasion. Nat. Genet. 2021, 53, 332–341. [Google Scholar] [CrossRef]
- Hoekstra, M.E.; Vijver, S.V.; Schumacher, T.N. Modulation of the tumor micro-environment by CD8+ T cell-derived cytokines. Curr. Opin. Immunol. 2021, 69, 65–71. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J. Interleukin-23/Th17 pathways and inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 1090–1100. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Zahoor, H.; Lin, Y.; Malhotra, U.; Sander, C.; Butterfield, L.H.; Kirkwood, J.M. Baseline circulating IL-17 predicts toxicity while TGF-β1 and IL-10 are prognostic of relapse in ipilimumab neoadjuvant therapy of melanoma. J. Immunother. Cancer 2015, 3, 39. [Google Scholar] [CrossRef] [Green Version]
- Nonomura, Y.; Otsuka, A.; Nakashima, C.; Seidel, J.A.; Kitoh, A.; Dainichi, T.; Nakajima, S.; Sawada, Y.; Matsushita, S.; Aoki, M.; et al. Peripheral blood Th9 cells are a possible pharmacodynamic biomarker of nivolumab treatment efficacy in metastatic melanoma patients. Oncoimmunology 2016, 5, e1248327. [Google Scholar] [CrossRef]
- Feun, L.G.; Li, Y.-Y.; Wu, C.; Wangpaichitr, M.; Jones, P.D.; Richman, S.P.; Madrazo, B.; Kwon, D.; Garcia-Buitrago, M.; Martin, P.; et al. Phase 2 study of pembrolizumab and circulating biomarkers to predict anticancer response in advanced, unresectable hepatocellular carcinoma. Cancer 2019, 125, 3603–3614. [Google Scholar] [CrossRef]
- Lim, S.Y.; Lee, J.H.; Gide, T.N.; Menzies, A.M.; Guminski, A.; Carlino, M.S.; Breen, E.J.; Yang, J.Y.H.; Ghazanfar, S.; Kefford, R.F.; et al. Circulating Cytokines Predict Immune-Related Toxicity in Melanoma Patients Receiving Anti-PD-1–Based Immunotherapy. Clin. Cancer Res. 2019, 25, 1557–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.-B.; McSweeney, P.; et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. Available online: https://www.nejm.org/doi/10.1056/NEJMoa1601202?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub++0www.ncbi.nlm.nih.gov (accessed on 29 June 2020).
- Schoenfeld, J.D.; Nishino, M.; Severgnini, M.; Manos, M.; Mak, R.H.; Hodi, F.S. Pneumonitis resulting from radiation and immune checkpoint blockade illustrates characteristic clinical, radiologic and circulating biomarker features. J. Immunother. Cancer 2019, 7, 112. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Zheng, J.; Zhu, J.; Xie, C.; Zhang, X.; Han, X.; Song, B.; Ma, Y.; Liu, J. PD-L1 gene polymorphism and high level of plasma soluble PD-L1 protein may be associated with non-small cell lung cancer. Int. J. Biol. Markers 2015, 30, e364–e368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gao, J.; Li, Y.; Nie, J.; Dai, L.; Hu, W.; Chen, X.; Han, J.; Ma, X.; Tian, G.; et al. Circulating PD-L1 in NSCLC patients and the correlation between the level of PD-L1 expression and the clinical characteristics. Thorac. Cancer 2015, 6, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Bu, Z.; Liu, X.; Zhang, L.; Li, Z.; Wu, A.; Wu, X.; Cheng, X.; Xing, X.; Du, H.; et al. Level of circulating PD-L1 expression in patients with advanced gastric cancer and its clinical implications. Chin. J. Cancer Res. 2014, 26, 104–111. [Google Scholar]
- Wei, W.; Xu, B.; Wang, Y.; Wu, C.; Jiang, J.; Wu, C. Prognostic significance of circulating soluble programmed death ligand-1 in patients with solid tumors: A meta-analysis. Medicine 2018, 97, e9617. [Google Scholar] [CrossRef]
- Ng, K.W.; Attig, J.; Young, G.R.; Ottina, E.; Papamichos, S.I.; Kotsianidis, I.; Kassiotis, G. Soluble PD-L1 generated by endogenous retroelement exaptation is a receptor antagonist. eLife 2019, 8, e50256. [Google Scholar] [CrossRef]
- Okuma, Y.; Wakui, H.; Utsumi, H.; Sagawa, Y.; Hosomi, Y.; Kuwano, K.; Homma, S. Soluble Programmed Cell Death Ligand 1 as a Novel Biomarker for Nivolumab Therapy for Non-Small-cell Lung Cancer. Available online: https://pubmed.ncbi.nlm.nih.gov/29859759/ (accessed on 7 February 2021).
- Costantini, A.; Julie, C.; Dumenil, C.; Hélias-Rodzewicz, Z.; Tisserand, J.; Dumoulin, J.; Giraud, V.; Labrune, S.; Chinet, T.; Emile, J.-F.; et al. Predictive role of plasmatic biomarkers in advanced non-small cell lung cancer treated by nivolumab. Oncoimmunology 2018, 7, e1452581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Jilisihan, B.; Wang, W.; Tang, Y.; Keyoumu, S. Soluble LAG3 acts as a potential prognostic marker of gastric cancer and its positive correlation with CD8+T cell frequency and secretion of IL-12 and INF-γ in peripheral blood. Cancer Biomark. Sect. Dis. Markers 2018, 23, 341–351. [Google Scholar] [CrossRef]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Bullock, K.; Gurjao, C.; Braun, D.; Shukla, S.A.; Bossé, D.; Lalani, A.-K.A.; Gopal, S.; Jin, C.; Horak, C.; et al. Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat. Commun. 2019, 10, 4346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Eynde, B.J.; van Baren, N.; Baurain, J.-F. Is There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annu. Rev. Cancer Biol. 2020, 4, 241–256. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Afzal, M.Z.; Mercado, R.R.; Shirai, K. Efficacy of metformin in combination with immune checkpoint inhibitors (anti-PD-1/anti-CTLA-4) in metastatic malignant melanoma. J. Immunother. Cancer 2018, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Rusdi, N.K.; Pawitan, J.A. Cancer Immunotherapy and Flow Cytometry in Immunotherapy Monitoring. Biomed. Pharmacol. J. 2019, 12, 1587–1593. [Google Scholar] [CrossRef]
- Cossarizza, A.; Chang, H.-D.; Radbruch, A.; Acs, A.; Adam, D.; Adam-Klages, S.; Agace, W.W.; Aghaeepour, N.; Akdis, M.; Allez, M.; et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur. J. Immunol. 2019, 49, 1457–1973. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Guo, J.; Cai, Z.; Li, B.; Sun, L.; Shen, Y.; Wang, S.; Wang, Z.; Wang, Z.; Wang, Y.; et al. Th9 Cell Differentiation and Its Dual Effects in Tumor Development. Front. Immunol. 2020, 11, 1026. [Google Scholar] [CrossRef]
- Mengos, A.E.; Gastineau, D.A.; Gustafson, M.P. The CD14+HLA-DRlo/neg Monocyte: An Immunosuppressive Phenotype That Restrains Responses to Cancer Immunotherapy. Front. Immunol. 2019, 10, 1026. [Google Scholar] [CrossRef] [Green Version]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrieta, O.; Montes-Servín, E.; Hernandez-Martinez, J.-M.; Cardona, A.F.; Casas-Ruiz, E.; Crispín, J.C.; Motola, D.; Flores-Estrada, D.; Barrera, L. Expression of PD-1/PD-L1 and PD-L2 in peripheral T-cells from non-small cell lung cancer patients. Oncotarget 2017, 8, 101994–102005. [Google Scholar] [CrossRef] [Green Version]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T.; et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, M.C.; Duong, C.P.M.; Gopalakrishnan, V.; Iebba, V.; Chen, W.-S.; Derosa, L.; Khan, M.A.W.; Cogdill, A.P.; White, M.G.; Wong, M.C.; et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat. Med. 2021, 27, 1432–1441. [Google Scholar] [CrossRef]
- Goldberg, S.B.; Narayan, A.; Kole, A.J.; Decker, R.H.; Teysir, J.; Carriero, N.J.; Lee, A.; Nemati, R.; Nath, S.K.; Mane, S.M.; et al. Early Assessment of Lung Cancer Immunotherapy Response via Circulating Tumor DNA. Clin. Cancer Res. 2018, 24, 1872–1880. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef]
- Chen, Y.; Seeruttun, S.R.; Wu, X.; Wang, Z. Maximum Somatic Allele Frequency in Combination With Blood-Based Tumor Mutational Burden to Predict the Efficacy of Atezolizumab in Advanced Non-small Cell Lung Cancer: A Pooled Analysis of the Randomized POPLAR and OAK Studies. Front. Oncol. 2019, 9, 1432. [Google Scholar] [CrossRef]
- Roh, W.; Chen, P.-L.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Gopalakrishnan, V.; Wang, F.; Cooper, Z.A.; Reddy, S.M.; et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 2017, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Schabath, M.B.; Welsh, E.A.; Fulp, W.J.; Chen, L.; Teer, J.K.; Thompson, Z.J.; Engel, B.E.; Xie, M.; Berglund, A.E.; Creelan, B.C.; et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene 2016, 35, 3209–3216. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.-Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assoun, S.; Theou-Anton, N.; Nguenang, M.; Cazes, A.; Danel, C.; Abbar, B.; Pluvy, J.; Gounant, V.; Khalil, A.; Namour, C.; et al. Association of TP53 mutations with response and longer survival under immune checkpoint inhibitors in advanced non-small-cell lung cancer. Lung Cancer Amst. Neth. 2019, 132, 65–71. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; Bosco, A.; Millward, M.J.; Small, M.; Nowak, A.K.; Lake, R.A. Dynamic versus static biomarkers in cancer immune checkpoint blockade: Unravelling complexity. Nat. Rev. Drug Discov. 2017, 16, 264–272. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, T.J.; Ring, B.Z.; Seitz, R.S.; Hout, D.R.; Schweitzer, B.L. A novel immuno-oncology algorithm measuring tumor microenvironment to predict response to immunotherapies. Heliyon 2021, 7, e06438. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021, 39, 346–360.e7. [Google Scholar] [CrossRef]
- Hayashi, H.; Sugawara, S.; Fukuda, Y.; Fujimoto, D.; Miura, S.; Ota, K.; Ozawa, Y.; Hara, S.; Tanizaki, J.; Azuma, K.; et al. A Randomized Phase 2 Study Comparing Nivolumab with Carboplatin-Pemetrexed for EGFR-Mutated NSCLC with Resistance to EGFR Tyrosine Kinase Inhibitors (WJOG8515L). Clin. Cancer Res. 2021. Available online: https://clincancerres.aacrjournals.org/content/early/2021/12/16/1078-0432.CCR-21-3194.full-text+.full.pdf (accessed on 29 June 2020).
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.-O.; Bracarda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef]
- Wang, L.; Saci, A.; Szabo, P.M.; Chasalow, S.D.; Castillo-Martin, M.; Domingo-Domenech, J.; Siefker-Radtke, A.; Sharma, P.; Sfakianos, J.P.; Gong, Y.; et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat. Commun. 2018, 9, 3503. [Google Scholar] [CrossRef]
- Restifo, N.P.; Marincola, F.M.; Kawakami, Y.; Taubenberger, J.; Yannelli, J.R.; Rosenberg, S.A. Loss of Functional Beta2-Microglobulin in Metastatic Melanomas From Five Patients Receiving Immunotherapy. J. Natl. Cancer Inst. 1996, 88, 100–108. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell–Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Kidman, J.; Principe, N.; Watson, M.; Lassmann, T.; Holt, R.A.; Nowak, A.K.; Lesterhuis, W.J.; Lake, R.A.; Chee, J. Characteristics of TCR Repertoire Associated With Successful Immune Checkpoint Therapy Responses. Front. Immunol. 2020, 11, 587014. [Google Scholar] [CrossRef]
- Oh, D.Y.; Cham, J.; Zhang, L.; Fong, G.; Kwek, S.S.; Klinger, M.; Faham, M.; Fong, L. Immune Toxicities Elicted by CTLA-4 Blockade in Cancer Patients Are Associated with Early Diversification of the T-cell Repertoire. Cancer Res. 2017, 77, 1322–1330. [Google Scholar] [CrossRef] [Green Version]
- Subudhi, S.K.; Aparicio, A.; Gao, J.; Zurita, A.J.; Araujo, J.C.; Logothetis, C.J.; Tahir, S.A.; Korivi, B.R.; Slack, R.S.; Vence, L.; et al. Clonal expansion of CD8 T cells in the systemic circulation precedes development of ipilimumab-induced toxicities. Proc. Natl. Acad. Sci. USA 2016, 113, 11919–11924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardinale, A.; De Luca, C.D.; Locatelli, F.; Velardi, E. Thymic Function and T-Cell Receptor Repertoire Diversity: Implications for Patient Response to Checkpoint Blockade Immunotherapy. Front. Immunol. 2021, 12, 4983. [Google Scholar] [CrossRef]
- Chaput, N.; Lepage, P.; Coutzac, C.; Soularue, E.; Le Roux, K.; Monot, C.; Boselli, L.; Routier, E.; Cassard, L.; Collins, M.; et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Hakozaki, T.; Richard, C.; Elkrief, A.; Hosomi, Y.; Benlaïfaoui, M.; Mimpen, I.; Terrisse, S.; Derosa, L.; Zitvogel, L.; Routy, B.; et al. The Gut Microbiome Associates with Immune Checkpoint Inhibition Outcomes in Patients with Advanced Non–Small Cell Lung Cancer. Cancer Immunol. Res. 2020, 8, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Dong, H.; Xia, L.; Yang, Y.; Zhu, Y.; Shen, Y.; Zheng, H.; Yao, C.; Wang, Y.; Lu, S. The Diversity of Gut Microbiome is Associated With Favorable Responses to Anti-Programmed Death 1 Immunotherapy in Chinese Patients With NSCLC. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2019, 14, 1378–1389. [Google Scholar] [CrossRef] [PubMed]
- Abu-Sbeih, H.; Herrera, L.N.; Tang, T.; Altan, M.; Chaftari, A.-M.P.; Okhuysen, P.C.; Jenq, R.R.; Wang, Y. Impact of antibiotic therapy on the development and response to treatment of immune checkpoint inhibitor-mediated diarrhea and colitis. J. Immunother. Cancer 2019, 7, 242. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wiesnoski, D.H.; Helmink, B.A.; Gopalakrishnan, V.; Choi, K.; DuPont, H.L.; Jiang, Z.-D.; Abu-Sbeih, H.; Sanchez, C.A.; Chang, C.-C.; et al. Fecal Microbiota Transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat. Med. 2018, 24, 1804–1808. [Google Scholar] [CrossRef]
- Sims, T.T.; El Alam, M.B.; Karpinets, T.V.; Dorta-Estremera, S.; Hegde, V.L.; Nookala, S.; Yoshida-Court, K.; Wu, X.; Biegert, G.W.G.; Delgado Medrano, A.Y.; et al. Gut microbiome diversity is an independent predictor of survival in cervical cancer patients receiving chemoradiation. Commun. Biol. 2021, 4, 1–10. [Google Scholar] [CrossRef]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489. [Google Scholar] [CrossRef]
- Shui, L.; Yang, X.; Li, J.; Yi, C.; Sun, Q.; Zhu, H. Gut Microbiome as a Potential Factor for Modulating Resistance to Cancer Immunotherapy. Front. Immunol. 2020, 10, 2989. [Google Scholar] [CrossRef] [Green Version]
- Routy, B.; Chatelier, E.L.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2020, 371, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.-M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zheng, N.; Luo, Q.; Jiang, L.; He, B.; Yuan, X.; Shen, L. Probiotics Lactobacillus reuteri Abrogates Immune Checkpoint Blockade-Associated Colitis by Inhibiting Group 3 Innate Lymphoid Cells. Front. Immunol. 2019, 10, 1235. [Google Scholar] [CrossRef] [PubMed]
- Draper, L.A.; Ryan, F.J.; Dalmasso, M.; Casey, P.G.; McCann, A.; Velayudhan, V.; Ross, R.P.; Hill, C. Autochthonous faecal viral transfer (FVT) impacts the murine microbiome after antibiotic perturbation. BMC Biol. 2020, 18, 173. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Weiner, B.; Ford, C.B.; Sellman, B.R.; Hammond, S.A.; Freeman, D.J.; Dennis, P.; Soria, J.-C.; Wortman, J.R.; Henn, M.R. Intervention strategies for microbial therapeutics in cancer immunotherapy. Immuno-Oncol. Technol. 2020, 6, 9–17. [Google Scholar] [CrossRef]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef] [Green Version]
- Clooney, A.G.; Eckenberger, J.; Laserna-Mendieta, E.; Sexton, K.A.; Bernstein, M.T.; Vagianos, K.; Sargent, M.; Ryan, F.J.; Moran, C.; Sheehan, D.; et al. Ranking microbiome variance in inflammatory bowel disease: A large longitudinal intercontinental study. Gut 2021, 70, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Malla, M.A.; Dubey, A.; Kumar, A.; Yadav, S.; Hashem, A.; Abd_Allah, E.F. Exploring the Human Microbiome: The Potential Future Role of Next-Generation Sequencing in Disease Diagnosis and Treatment. Front. Immunol. 2019, 9, 2868. [Google Scholar] [CrossRef] [PubMed]
- Gharaibeh, R.Z.; Jobin, C. Microbiota and cancer immunotherapy: In search of microbial signals. Gut 2019, 68, 385–388. [Google Scholar] [CrossRef]
- Chatterjee, S.; Lesniak, W.G.; Miller, M.S.; Lisok, A.; Sikorska, E.; Wharram, B.; Kumar, D.; Gabrielson, M.; Pomper, M.G.; Gabelli, S.B.; et al. Rapid PD-L1 detection in tumors with PET using a highly specific peptide. Biochem. Biophys. Res. Commun. 2017, 483, 258–263. [Google Scholar] [CrossRef] [Green Version]
- Puhr, H.C.; Ilhan-Mutlu, A. New emerging targets in cancer immunotherapy: The role of LAG3. ESMO Open 2019, 4, e000482. [Google Scholar] [CrossRef] [Green Version]
- Altman, D.G.; McShane, L.M.; Sauerbrei, W.; Taube, S.E. Reporting recommendations for tumor marker prognostic studies (REMARK): Explanation and elaboration. BMC Med. 2012, 10, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TRIAL ARM | PD-L1 TPS (Number of Patients Receiving Pembrolizumab) | Overall Response Rate (95% CI) (%) | Median PFS in Months (95% CI) | PFS HR Compared to Chemo Alone (95% CI, p-Value) | Median OS in Months (95% CI) | OS HR Compared to Chemo Alone (95% CI, p-Value) | Approval |
|---|---|---|---|---|---|---|---|
| KEYNOTE-024 Single Agent Pembrolizumab (Pre-treated AC and SCC [25,30,31,32] | ≥50% (154) | 44.8 | 10.3 | 0.5 (0.37–0.68, <0.001) | 26.3 (95% CI 18.3–40.4) | 0.62 (0.48–0.81, 0.002) | Ireland, EMA [33], FDA |
| KEYNOTE-042 Single Agent Pembrolizumab (First Line AC and SCC) [26] | All Patients (636) | 16.7 | 0.81 (0.71–0.93, 0.0036) | FDA | |||
| KEYNOTE 042 [26] | ≥50% (298) | 39 (34–45) | 7.1 (5.9–9) | 0.81 (0.67–0.99, 0.017) | 20 (15–24.9) | 0.69 (0.56–0.85, 0.003) | Ireland, EMA, FDA |
| KEYNOTE 042 [26] | ≥20% (412) | 33 (29–38) | 6.2 (5.1–7.8) | 0.94 (0.8–1.11,) | 17.7 (15.3–22.1) | 0.77 (0.64–0.92, 0.002) | FDA |
| KEYNOTE 042 [26] | ≥1% (636) | 27 (24 -31) | 5.4 (4.3–6.2) | 1.07 (0.94–1.21) | 16.7 (13.9–19.7) | 0.81 (0.71–0.93, 0.0018) | FDA |
| KEYNOTE 042 [26] | 1–49% (338) | 13.4 (10.7–18.2) | 0.92 (0.77–1.11,) | FDA | |||
| Pembrolizumab and Chemotherapy (First Line AC) KEYNOTE 189 [34,35] | All Patients (410) | 48 (43.1–53) | 9 (8.1–9.9) | 0.48 (0.4–0.58) | 22 (19.5–25.2) | 0.56 (0.45–0.70) | Ireland, EMA, FDA |
| KEYNOTE 189 [34,35] | ≥50% (132) | 62.1 (53.3–70.4) | 11.1 (9.1–14.4) | 0.36 (0.26 -0.51) | NR (20.4–NR) | 0.59 (0.39–0.86) | |
| KEYNOTE 189 [34,35] | 1–49% (128) | 49.2 (40.3–58.2) | 9.2 (7.8–13.1) | 0.51 (0.36–0.73) | 21.8 (17.7–25.9) | 0.62 (0.42–0.92) | |
| KEYNOTE 189 [34,35] | <1% (127) | 32.3 (24.3–41.2) | 6.2 (4.9–8.1) | 0.64 (0.47–0.89) | 17.2 (13.8–22.8) | 0.52 (0.36–0.74) | |
| Pembrolizumab and Chemotherapy (First Line SCC) KEYNOTE-407 [36] | All Patients (278) | 57.9% (51.9–63.8) | 6.4 (6.2–8.3) | 0.56 (0.45–0.70; <0.001) | 15.9 (13.2–NE) | 0.64; (0.49–0.85; <0.001) | Ireland, EMA, FDA |
| ≥50% (73) | 60.3 (48.1–71.5) | 8.0 (6.1–10.3) | 0.37 (0.24–0.58) | NR (11.3–NE) | 0.64 (0.37–1.10) | ||
| 1–49% (103) | 49.5 (39.5–59.5) | 7.2 (6.0–11.4) | 0.56 (0.39–0.8) | 14.0 (12.8–NE) | 0.57 (0.36–0.90) | ||
| ≥1% (183) | 0.49 (0.38–0.65) | ||||||
| <1% (95) | 63.2 (52.6–72.8) | 6.3 (6.1–6.5) | 0.68 (0.47–0.98) | 15.9 (13.1–NE) | 0.61 (0.38–0.98) |
| TRIAL ARM | PD-L1 CPS (No. of Patients Receiving Pembrolizumab or Nivolumab) | Overall Response Rate (95% CI) (%) | Median PFS in Months (95% CI) | PFS HR Compared to Chemo Alone (95% CI, p-Value) | Median OS in Months (95% CI) | OS HR Compared to Chemo Alone (95% CI, p-Value) | Approval |
|---|---|---|---|---|---|---|---|
| KEYNOTE-061 (pre-treated gastric and GOJ cancer Taxol vs. Pembro) [39] | All patients (296) | 11 | 1.6 | 1.34 (1.12–1.60) | 6.7 (5.4–8.9) | 0.94 (0.79–1.12) | |
| KEYNOTE-061 | CPS ≥ 1 (196) | 16 | 1.5 | 1.27 1.03–1.57) | 9.1 | 0.82, 0.66–1.03; one-sided p = 0.0421 | FDA (gastric cancer 3rd line) |
| KEYNOTE-061 | CPS ≤ 1 (99) | 2 | |||||
| KEYNOTE-061 (post-hoc analysis) | CPS ≥ 10 (53) | 24.5 | FDA (gastric cancer of GOJ cancer 2nd line) | ||||
| KEYNOTE-062 (Pembro alone vs. Pembro + Chemo vs. Chemo alone first line gastric AC) [40] | Pembro Alone (256) | 14.8 | 10.6 (7.7–13.8) | 0.91 (99.2% CI 0.69–1.18) | |||
| Pembro Alone CPS > 1 | 2 (1.5–2.8) | 1.66 (1.37–2.01) | 0.91 (0.74–1.1) | ||||
| Pembro Alone CPS ≥ 10 | 23 | 2.9 (1.6–5.4) | 1.10 (0.79–1.51) | 17.4 (9.1–23.1) | 0.69 (0.49–0.97) | ||
| Pembro + Chemo (250) | 37.2 | 12.5 (10.8–13.9) | 0.85 (0.7–1.03) | ||||
| Pembro + Chemo CPS ≥ 1 | |||||||
| Pembro + Chemo CPS ≥ 10 | 6.9 (5.7–7.3) | 0.84 (0.7–1.02) | 12.3 (9.5–14.8) | 0.85 (0.62–1.17, 0.16) | |||
| KEYNOTE-180 (Pre-treated Oesophageal AC and SCC) [41] | 9.9 (5.2–16.7) | ||||||
| CPS < 10 (63) | 6 (2–16) | 2.0 (1.9–2.1) | 5.4 (3.9–6.3) | ||||
| CPS ≥ 10 (58) | 14 (6–25) | 2.0 (1.9–2.2) | 6.3 (4.4–9.3) | ||||
| SCC CPS ≥ 10 (35) | 20 | FDA | |||||
| KEYNOTE-181 (pre-treated AC and SCC Pembro vs. chemo) [37] | All Patients (314) | 13.1 (9.5–17.3) | 2.1 (2.1–2.2) | 1.11 (0.94–1.31) | 7.1 | 0.89 (0.75–1.05, 0.0560) | |
| CPS ≥ 10 | 21.5 (14.1–30.5) | 2.6 (2.1–4.1) | 0.73 (0.54 to 0.97) | 9.3 (6.6–12.5) | 0.69 (0.52–0.93, 0.0074) | FDA | |
| SCC | 16.7 (11.8–22.6) | 2.2 (2.1–3.2) | 0.92 (0.75–1.13) | 8.2 | 0.78 (0.63–0.96, 0.0095) | ||
| SCC CPS < 10 | 11.9 | 2.1 (2.1–2.4) | 7.3 (5.7–9.2) | ||||
| AC CPS < 10 | 3.3 | 2.1 (1.9–2.1) | 5.1 (4.1–7.1) | ||||
| ATTRACTION-2 (pretreated gastric or GOJ AC, Nivo vs. Placebo) [42] | N/A (493 received Nivo) | 11.9 | 1.61 (1.54–2.30) | 0.60 (0.49–0.75, < 0.0001) | 5.26 (4.60–6.37) | 0.62 (0.51–0.76, p < 0.0001) | FDA |
| ATTRACTION-3 (pre-treated oesophageal SCC, Nivo vs. placebo) [43] | N/A (210 received Nivo) | 10.9 (9.2–13.3) | 0.77 (0.62–0.96, 0.019) | FDA, EMA | |||
| KEYNOTE -590 (Advanced first line oesophageal or GOJ cancer, chemo and Pembro, 73.5% SCC, 25.5% AC) [44] | All patients (373) | 45.0 (39.9–50.2) | 6.3 (6.2–6.9) | 0.65 (0.55–0.76; p < 0.0001). | 12.4 (10.5, 14.0) | 0.73 (0.62–0.86, <0.0001) | FDA |
| SCC CPS ≥ 10 (143) | 7.3 (6.2–8.2) | 0.53 (0.40–0.69) | 13.9 (11.1–17.7) | 0.57 (0.43–0.75, <0.0001) | FDA, | ||
| All SCC (274) | 7.5 | 12.6 (10.2–14.3) | 0.72 (0.60–0.88, 0.0006) | FDA | |||
| All CPS ≥ 10 (186) | 7.5 (6.2–8.2) | 13.5 (11.1–15.6) | 0·62 (0.49–0.78, p < 0.0001) | FDA, | |||
| AC CPS ≥ 10 (43) | 8.0 (6.0–8.3) | 0.49 (0.30–0.81) | 12.1 (9.6–18.7) | 0.83 (0.52–1.34) | FDA, EMA | ||
| AC CPS < 10 (54) | 6.3 (5.6–8.3) | 0.76 (0.49–1.19) | 12.7 (8.1–16.1) | 0.66 (0.42–1.04) | |||
| CheckMate 649 (Nivo + chemo vs. chemo alone in first line gastric, GOJ, oesophageal AC) [45] | All patients (603) | 58 (54–62) | 7.7 (7.1–8.5) | 0.77 (0.68–0.87) | 13.8 (12.6–14.6) | 0.8 (0.68–0.94, <0.0002) | FDA |
| CPS ≥ 5 (473) | 60 (55–65) | 7.7 (7.0–9.1) | 0·68 (98% CI 0·56–0·81, <0·0001) | 14.4 (13.1–16.2) | 0.71 (98.4% CI 0·59–0·86, <0.0001) | FDA, EMA | |
| CPS ≥ 1 (641) | 60 | 7.5 (7.0–8.4) | 0.74 (0.65–0.85) | 14.1 (11.6–15) | 0.77 (99.3% CI 0.064–0.92, <0.0001) | FDA | |
| CPS < 5 | 55 | 7.5 | 0.93 (0.76–1.12) | 12.4 | 0.94 (0.78–1.13) | FDA | |
| CPS < 1 (140) | 51 | 8.7 | 0.93 (0.69–1.26) | 13.8 | 0.79 (0.7–0.89) | FDA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Healey Bird, B.; Nally, K.; Ronan, K.; Clarke, G.; Amu, S.; Almeida, A.S.; Flavin, R.; Finn, S. Cancer Immunotherapy with Immune Checkpoint Inhibitors-Biomarkers of Response and Toxicity; Current Limitations and Future Promise. Diagnostics 2022, 12, 124. https://doi.org/10.3390/diagnostics12010124
Healey Bird B, Nally K, Ronan K, Clarke G, Amu S, Almeida AS, Flavin R, Finn S. Cancer Immunotherapy with Immune Checkpoint Inhibitors-Biomarkers of Response and Toxicity; Current Limitations and Future Promise. Diagnostics. 2022; 12(1):124. https://doi.org/10.3390/diagnostics12010124
Chicago/Turabian StyleHealey Bird, Brian, Ken Nally, Karine Ronan, Gerard Clarke, Sylvie Amu, Ana S. Almeida, Richard Flavin, and Stephen Finn. 2022. "Cancer Immunotherapy with Immune Checkpoint Inhibitors-Biomarkers of Response and Toxicity; Current Limitations and Future Promise" Diagnostics 12, no. 1: 124. https://doi.org/10.3390/diagnostics12010124