
Next-Generation Molecular Investigations in Lysosomal Diseases: Clinical Integration of a Comprehensive Targeted Panel

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. NGS Sequencing
- (i)
- The bcl2fastq conversion software (Illumina, v2.20) was used for reads demultiplexing and generation of Fastq files. Sequenced reads were mapped to the human reference sequence (GRCh37, Hg19) using the Burrows–Wheeler Aligner (BWA v.0.7.17). Read duplicates were marked with Picard tools (v2.18.0), local realignments around indels, base-quality-score recalibration and variant calling were performed with the Genome Analysis Toolkit (GATK 4.0.6.0). Single-nucleotide variants and small indels were identified with the GATK HaplotypeCaller (v4.0.6.0), VarScan2 (v2.4.3) and Vardict (v1.5.1). Variants were then annotated with SnpEff (v.4.2) and Alamut-batch (v.1.12).
- (ii)
3. Results
3.1. Quality Metrics
3.2. Panel Performances for the Detection of SNVs and Indels
3.3. Panel Performances for the Detection of CNVs
3.4. Clinical Utility Assessment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coutinho, M.F.; Alves, S. From rare to common and back again: 60years of lysosomal dysfunction. Mol. Genet. Metab. 2016, 117, 53–65. [Google Scholar] [CrossRef]
- de Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef]
- Schultz, M.L.; Tecedor, L.; Chang, M.; Davidson, B.L. Clarifying lysosomal storage diseases. Trends Neurosci. 2011, 34, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Tebani, A.; Afonso, C.; Marret, S.; Bekri, S. Omics-based strategies in precision medicine: Toward a Paradigm shift in inborn errors of metabolism investigations. Int. J. Mol. Sci. 2016, 17, 1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudrié-Arnaud, B.; Marguet, F.; Patrier, S.; Martinovic, J.; Louillet, F.; Broux, F.; Charbonnier, F.; Dranguet, H.; Coutant, S.; Vezain, M.; et al. Metabolic causes of nonimmune hydrops fetalis: A next-generation sequencing panel as a first-line investigation. Clin. Chim. Acta. 2018, 481, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tebani, A.; Abily-Donval, L.; Afonso, C.; Marret, S.; Bekri, S. Clinical metabolomics: The new metabolic window for inborn errors of metabolism investigations in the post-genomic era. Int. J. Mol. Sci. 2016, 17, 1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Encarnação, M.; Coutinho, M.F.; Silva, L.; Ribeiro, D.; Ouesleti, S.; Campos, T.; Santos, H.; Martins, E.; Cardoso, M.T.; Vilarinho, L.; et al. Assessing lysosomal disorders in the NGS era: Identification of novel rare variants. Int. J. Mol. Sci. 2020, 21, 6355. [Google Scholar] [CrossRef] [PubMed]
- Gheldof, A.; Seneca, S.; Stouffs, K.; Lissens, W.; Jansen, A.; Laeremans, H.; Verloo, P.; Schoonjans, A.S.; Meuwissen, M.; Barca, D.; et al. Clinical implementation of gene panel testing for lysosomal storage diseases. Mol. Genet. Genom. Med. 2019, 7, e00527. [Google Scholar] [CrossRef]
- Málaga, D.R.; Brusius-Facchin, A.C.; Siebert, M.; Pasqualim, G.; Saraiva-Pereira, M.L.; Souza, C.F.M.; Schwartz, I.V.D.; Matte, U.; Giugliani, R. Sensitivity, advantages, limitations, and clinical utility of targeted next-generation sequencing panels for the diagnosis of selected lysosomal storage disorders. Genet. Mol. Biol. 2019, 42, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, G.; García-Seisdedos, D.; Ciubotariu, C.; Piris-Villaespesa, M.; Gandía, M.; Martín-Moro, F.; Gutiérrez-Solana, L.G.; Morado, M.; López-Jiménez, J.; Sánchez-Herranz, A.; et al. Early detection of lysosomal diseases by screening of cases of idiopathic splenomegaly and/or thrombocytopenia with a next-generation sequencing gene panel. JIMD Rep. 2020, 51, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Hrebícek, M.; Mrázová, L.; Seyrantepe, V.; Durand, S.; Roslin, N.M.; Nosková, L.; Hartmannová, H.; Ivánek, R.; Cízkova, A.; Poupetová, H.; et al. Mutations in TMEM76 * cause mucopolysaccharidosis IIIC (Sanfilippo C syndrome). Am. J. Hum. Genet. 2006, 79, 807–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backenroth, D.; Homsy, J.; Murillo, L.R.; Glessner, J.; Lin, E.; Brueckner, M.; Lifton, R.; Goldmuntz, E.; Chung, W.K.; Shen, Y. CANOES: Detecting rare copy number variants from whole exome sequencing data. Nucleic Acids Res. 2014, 42, e97. [Google Scholar] [CrossRef] [PubMed]
- Muller, E.; Brault, B.; Holmes, A.; Legros, A.; Jeannot, E.; Campitelli, M.; Rousselin, A.; Goardon, N.; Frébourg, T.; Krieger, S.; et al. Genetic profiles of cervical tumors by high-throughput sequencing for personalized medical care. Cancer Med. 2015, 4, 1484–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, D.L.; Schröder, J.; Penington, J.S.; Do, H.; Molania, R.; Dobrovic, A.; Speed, T.P.; Papenfuss, A.T. GRIDSS: Sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 2017, 27, 2050–2060. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef] [Green Version]
- Leman, R.; Gaildrat, P.; Le Gac, G.; Ka, C.; Fichou, Y.; Audrezet, M.P.; Caux-Moncoutier, V.; Caputo, S.M.; Boutry-Kryza, N.; Léone, M.; et al. Novel diagnostic tool for prediction of variant spliceogenicity derived from a set of 395 combined in silico/in vitro studies: An international collaborative effort. Nucleic Acids Res. 2018, 46, 7913–7923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartegni, L.; Wang, J.; Zhu, Z.; Zhang, M.Q.; Krainer, A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Quenez, O.; Cassinari, K.; Coutant, S.; Lecoquierre, F.; Le Guennec, K.; Rousseau, S.; Richard, A.C.; Vasseur, S.; Bouvignies, E.; Bou, J.; et al. Detection of copy-number variations from NGS data using read depth information: A diagnostic performance evaluation. Eur. J. Hum. Genet. 2020. [Google Scholar] [CrossRef]
- Naureckiene, S.; Sleat, D.E.; Lackland, H.; Fensom, A.; Vanier, M.T.; Wattiaux, R.; Jadot, M.; Lobel, P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2000, 290, 2298–2301. [Google Scholar] [CrossRef]
- Sleat, D.E.; Donnelly, R.J.; Lackland, H.; Liu, C.G.; Sohar, I.; Pullarkat, R.K.; Lobel, P. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science 1997, 277, 1802–1805. [Google Scholar] [CrossRef] [PubMed]
- Sleat, D.E.; Gin, R.M.; Sohar, I.; Wisniewski, K.; Sklower-Brooks, S.; Pullarkat, R.K.; Palmer, D.N.; Lerner, T.J.; Boustany, R.M.; Uldall, P.; et al. Mutational analysis of the defective protease in classic late-infantile neuronal ceroid lipofuscinosis, a neurodegenerative lysosomal storage disorder. Am. J. Hum. Genet. 1999, 64, 1511–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maegawa, G.H.; Tropak, M.; Buttner, J.; Stockley, T.; Kok, F.; Clarke, J.T.; Mahuran, D.J. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J. Biol. Chem. 2007, 282, 9150–9161. [Google Scholar] [CrossRef] [Green Version]
- Huie, M.L.; Chen, A.S.; Tsujino, S.; Shanske, S.; DiMauro, S.; Engel, A.G.; Hirschhorn, R. Aberrant splicing in adult onset glycogen storage disease type II (GSDII): Molecular identification of an IVS1 (−13T⟶G) mutation in a majority of patients and a novel IVS10 (+1GT⟶CT) mutation. Hum. Mol. Genet. 1994, 3, 2231–2236. [Google Scholar] [CrossRef]
- Wan, L.; Lee, C.C.; Hsu, C.M.; Hwu, W.L.; Yang, C.C.; Tsai, C.H.; Tsai, F.J. Identification of eight novel mutations of the acid alpha-glucosidase gene causing the infantile or juvenile form of glycogen storage disease type II. J. Neurol. 2008, 255, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Matos, L.; Alves, S. From bedside to cell biology: A century of history on lysosomal dysfunction. Gene 2015, 555, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.B.; Winchester, B. Lysosomal Storage Disorders—A Practial Guide, 1st ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2012; pp. 20–28. [Google Scholar]
- Argmann, C.A.; Houten, S.M.; Zhu, J.; Schadt, E.E. A next generation multiscale view of inborn errors of metabolism. Cell Metab. 2016, 23, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Olkhovych, N.V.; Gorovenko, N.G. Determination of frequencies of alleles, associated with the pseudodeficiency of lysosomal hydrolases, in population of Ukraine. Ukr. Biochem. J. 2016, 88, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.; Ajayi, T.; Specchio, N.; de Los Reyes, E.; Gissen, P.; Ballon, D.; Dyke, J.P.; Cahan, H.; Slasor, P.; Jacoby, D.; et al. Study of intraventricular cerliponase alfa for CLN2 disease. N. Engl. J. Med. 2018, 378, 1898–1907. [Google Scholar] [CrossRef] [PubMed]


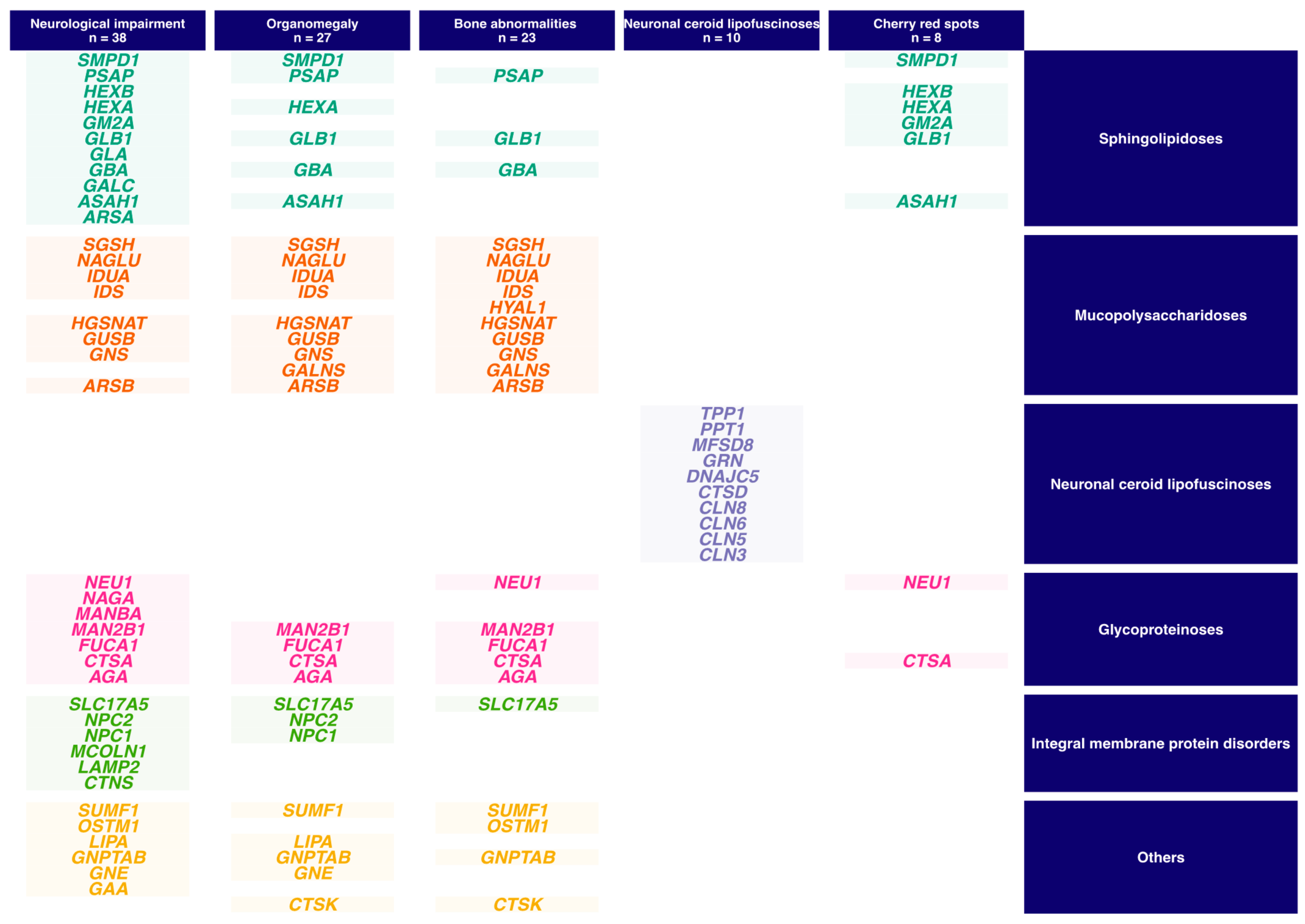{kind=link}
| Disease | Inheritance | Gene | NM_ |
|---|---|---|---|
| α-glucosidase deficiency | AR | GAA | NM_000152.3 |
| α-mannosidase deficiency | AR | MAN2B1 | NM_000528.3 |
| Aspartylglucosaminidase deficiency | AR | AGA | NM_000027.3 |
| β-mannosidase deficiency | AR | MANBA | NM_005908.3 |
| α-fucosidase deficiency | AR | FUCA1 | NM_000147.4 |
| Cathepsin A deficiency | AR | CTSA | NM_000308.2 |
| α-N-acetylgalactosaminidase deficiency | AR | NAGA | NM_000262.2 |
| α-neuraminidase deficiency | AR | NEU1 | NM_000434.3 |
| Cystinosin deficiency | AR | CTNS | NM_004937.2 |
| Lysosome-associated membrane protein 2 deficiency | XL | LAMP2 | NM_002294.2 |
| Niemann-Pick disease type C1 | AR | NPC1 | NM_000271.4 |
| Niemann-Pick disease type C2 | AR | NPC2 | NM_006432.3 |
| Sialin deficiency | AR | SLC17A5 | NM_012434.4 |
| Mucolipin 1 deficiency | AR | MCOLN1 | NM_020533.2 |
| Lysosomal acid lipase deficiency | AR | LIPA | NM_000235.2 |
| Cathepsin K deficiency | AR | CTSK | NM_000396.3 |
| UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase deficiency | AR | GNE | NM_005476.5 |
| UDP-N-acetylglucosamine-1-phosphotransferase α/β subunit deficiency | AR | GNPTAB | NM_024312.4 |
| α-iduronidase deficiency | AR | IDUA | NM_000203.3 |
| Iduronate sulfatase deficiency | XLR | IDS | NM_000202.5 |
| Heparan N-sulfatase deficiency | AR | SGSH | NM_000199.3 |
| N-acetylglucosaminidase deficiency | AR | NAGLU | NM_000263.3 |
| Heparan-α-glucosaminide N-acetyltransferase deficiency | AR | HGSNAT | NM_152419.2 |
| N-acetylglucosamine 6-sulfatase deficiency | AR | GNS | NM_002076.3 |
| N-acetylgalactosamine 6-sulfatase deficiency | AR | GALNS | NM_000512.4 |
| Hyaluronidase deficiency | AR | HYAL1 | NM_153281.1 |
| N-acetylgalactosamine 4-sulfatase deficiency | AR | ARSB | NM_000046.3 |
| β-glucuronidase deficiency | AR | GUSB | NM_000181.3 |
| Palmitoyl-protein thioesterase 1 deficiency | AR | PPT1 | NM_000310.3 |
| Cathepsin D deficiency | AR | CTSD | NM_001909.4 |
| Progranulin deficiency | AD, AR | GRN | NM_002087.2 |
| Tripeptidyl-peptidase 1 deficiency | AR | TPP1 | NM_000391.3 |
| CLN3 disease | AR | CLN3 | NM_001042432.1 |
| CLN4 disease | AD | DNAJC5 | NM_025219.2 |
| CLN5 disease | AR | CLN5 | NM_006493.2 |
| CLN6 disease | AR | CLN6 | NM_017882.2 |
| CLN7 disease | AR | MFSD8 | NM_152778.2 |
| CLN8 disease | AR | CLN8 | NM_018941.3 |
| Osteopetrosis | AR | OSTM1 | NM_014028.3 |
| Formyl-glycine generating enzyme deficiency | AR | SUMF1 | NM_182760.3 |
| GM2 activator protein deficiency | AR | GM2A | NM_000405.4 |
| Arylsulfatase A deficiency | AR | ARSA | NM_000487.5 |
| Acid ceramidase deficiency, inflammatory phenotype | AR | ASAH1 | NM_177924.3 |
| α-Galactosidase A deficiency | XL | GLA | NM_000169,2 |
| Glucocerebrosidase deficiency | AR | GBA | NM_001005741.2 |
| β-galactosylceramidase deficiency | AR | GALC | NM_000153.3 |
| Acid sphingomyelinase deficiency | AR | SMPD1 | NM_000543.4 |
| β-hexosaminidase β-subunit deficiency | AR | HEXB | NM_000521.3 |
| β-hexosaminidase α-subunit deficiency | AR | HEXA | NM_000520.4 |
| β-galactosidase deficiency, GM1 gangliosidosis phenotype | AR | GLB1 | NM_000404.2 |
| Atypical Gaucher disease due to saposin C deficiency | AR | PSAP | NM_002778.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sudrié-Arnaud, B.; Snanoudj, S.; Dabaj, I.; Dranguet, H.; Abily-Donval, L.; Lebas, A.; Vezain, M.; Héron, B.; Marie, I.; Duval-Arnould, M.; et al. Next-Generation Molecular Investigations in Lysosomal Diseases: Clinical Integration of a Comprehensive Targeted Panel. Diagnostics 2021, 11, 294. https://doi.org/10.3390/diagnostics11020294
Sudrié-Arnaud B, Snanoudj S, Dabaj I, Dranguet H, Abily-Donval L, Lebas A, Vezain M, Héron B, Marie I, Duval-Arnould M, et al. Next-Generation Molecular Investigations in Lysosomal Diseases: Clinical Integration of a Comprehensive Targeted Panel. Diagnostics. 2021; 11(2):294. https://doi.org/10.3390/diagnostics11020294
Chicago/Turabian StyleSudrié-Arnaud, Bénédicte, Sarah Snanoudj, Ivana Dabaj, Hélène Dranguet, Lenaig Abily-Donval, Axel Lebas, Myriam Vezain, Bénédicte Héron, Isabelle Marie, Marc Duval-Arnould, and et al. 2021. "Next-Generation Molecular Investigations in Lysosomal Diseases: Clinical Integration of a Comprehensive Targeted Panel" Diagnostics 11, no. 2: 294. https://doi.org/10.3390/diagnostics11020294