
Epidemiology of Mucopolysaccharidoses Update



Abstract
:1. Introduction
2. Updated Epidemiology of MPS in the Seven Countries
2.1. Prevalence of MPS in Brazil
2.2. Prevalence of MPS in Colombia (Cundinamarca and Boyacá)
2.3. Prevalence of MPS in Malaysia
2.4. Prevalence of MPS in Mexico
2.5. Prevalence of MPS in Pakistan
2.6. Prevalence of MPS in Taiwan
2.7. Prevalence of MPS in Turkey
2.8. Prevalence of MPS in Other Countries
2.8.1. Japan
2.8.2. Switzerland
2.8.3. Saudi Arabia
2.8.4. South Korea
2.8.5. China
2.8.6. India
2.8.7. Tunisia
2.8.8. Australia
2.8.9. British Columbia (Canada)
2.8.10. Czech Republic
2.8.11. Denmark
2.8.12. Norway
2.8.13. Sweden
2.8.14. Estonia
2.8.15. Germany
2.8.16. The Netherlands
2.8.17. Northern Ireland
2.8.18. Poland
2.8.19. Portugal
2.8.20. The United States
3. Mutations
3.1. MPS I Mutations
3.2. MPS II Mutations
3.3. MPS III Mutations
3.3.1. MPS IIIA
3.3.2. MPS IIIB
3.3.3. MPS IIIC
3.3.4. MPS IIID
3.4. MPS IV Mutations
3.4.1. MPS IVA
3.4.2. MPS IVB
3.5. MPS VI Mutations
3.6. MPS VII Mutations
3.7. MPS IX Mutations
4. Frequency of MPS with Newborn Screening
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CS | Chondroitin Sulfate |
| DS | Dermatan Sulfate |
| DBS | Dried Blood Spots |
| ERT | Enzyme Replacement Therapy |
| GAG | Glycosaminoglycans |
| GALNS | N-Acetylgalactosamine-6-Sulfate Sulfatase |
| GLB1 | ß-Galactosidase |
| GNS | N-Acetylglucosamine 6-Sulfatase |
| G4S | N-Acetylgalactosamine-4-sulfatase |
| GUSB | ß-D-Glucuronidase |
| HS | Heparan Sulfate |
| HGSNAT | α-Glucosaminidase |
| HSCT | Homepoietic Stem Cell Transplantation |
| I2S | Iduronate-2-Sulfatase |
| IDUA | α-L-iduronidase |
| KS | Keratan Sulfate |
| MPS | Mucopolysaccharidoses |
| NAGLU | α-N-Acetylglucosaminidase |
| NBS | Newborn Screening |
| SGHS | Heparan-N-Sulfatase |
| SRT | Substrate Reduction Therapy |
References
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; Orii, T.; et al. Epidemiology of Mucopolisaccharidoses. Mol. Genet. Metab. 2018, 121, 227–240. [Google Scholar] [CrossRef]
- Kubaski, F.; De Oliveira Poswar, F.; Michelin-Tirelli, K.; Burin, M.G.; Rojas-Málaga, D.; Brusius-Facchin, A.C.; Leistner-Segal, S.; Giugliani, R. Diagnosis of Mucopolysaccharidoses. Diagnostics 2020, 10, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nørmark, M.B.; Kjaer, N.; Lund, A.M. Prevalence of Mucopolysaccharidosis Types I, II, and VI in the Pediatric and Adult Population with Carpal Tunnel Syndrome (CTS). Retrospective and Prospective Analysis of Patients Treated for CTS. JIMD Rep. 2016, 4, 113–116. [Google Scholar]
- Taylor, K.R.; Gallo, R.L. Glycosaminoglycans and Their Proteoglycans: Host-associated Molecular Patterns for Initiation and Modulation of Inflammation. FASEB J. 2006, 20, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Lee, C.L.; Lo, Y.T.; Wang, T.J.; Huang, S.F.; Chen, T.L.; Wang, Y.S.; Niu, D.M.; Chuang, C.K.; Lin, S.P. The Relationships between Urinary Glycosaminoglycan Levels and Phenotypes of Mucopolysaccharidoses. Mol. Genet. Genom. Med. 2018, 6, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Federhen, A.; Pasqualim, G.; de Freitas, T.F.; Gonzalez, E.A.; Trapp, F.; Matte, U.; Giugliani, R. Estimated Birth Prevalence of Mucopolysaccharidoses in Brazil. Am. J. Med. Genet. Part A 2019, 182, 469–483. [Google Scholar] [CrossRef]
- Singh, R.; Chopra, S.; Graham, C.; Langer, M.; Ng, R.; Ullal, A.J.; Pamula, V.K. Emerging Approaches for Fluorescence-Based Newborn Screening of Mucopolysaccharidoses. Diagnostics 2020, 10, 294. [Google Scholar] [CrossRef]
- Martins, C.; De Medeiros, P.F.V.; Wood, J.; Lacerda, L.; Geraghty, M.T.; Giugliani, R.; Pshezhetsky, A.V.; de Medeiros, P.F.V.; Leistner-Segal, S.; Dridi, L.; et al. Molecular Characterization of a Large Group of Mucopolysaccharidosis Type IIIC Patients Reveals the Evolutionary History of the Disease. Hum. Mutat. 2019, 40, 1084–1100. [Google Scholar] [CrossRef]
- Poletto, E.; Pasqualim, G.; Giugliani, R.; Matte, U.; Baldo, G. Worldwide Distribution of Common IDUA Pathogenic Variants. Clin. Genet. 2018, 94, 95–102. [Google Scholar] [CrossRef]
- Lehman, T.J.A.; Miller, N.; Norquist, B.; Underhill, L.; Keutzer, J. Diagnosis of the Mucopolysaccharidoses. Rheumatology 2011, 50, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Qubbaj, W.; Al-Aqeel, A.I.; Al-Hassnan, Z.; Al-Duraihim, A.; Awartani, K.; Al-Rejjal, R.; Coskun, S. Preimplantation Genetic Diagnosis of Morquio Disease. Prenat. Diagn. 2008, 28, 900–903. [Google Scholar] [CrossRef]
- Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Brooks, B.; Varshaver, I.; Avitzour, M.; Margalioth, E.J.; Levy-Lahad, E.; Elstein, D.; Epsztejn-Litman, S.; et al. Preventing Mucopolysaccharidosis Type II (Hunter Syndrome): PGD and Establishing a Hunter (46, XX) Stem Cell Line. Prenat. Diagn. 2011, 31, 853–860. [Google Scholar] [CrossRef]
- Stapleton, M.; Arunkumar, N.; Kubaski, F.; Mason, R.W.; Tadao, O.; Tomatsu, S. Clinical Presentation and Diagnosis of Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 125, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Vairo, F.; Federhen, A.; Baldo, G.; Riegel, M.; Burin, M.; Leistner-Segal, S.; Giugliani, R. Diagnostic and Treatment Strategies in Mucopolysaccharidosis VI. Appl. Clin. Genet. 2015, 8, 245–255. [Google Scholar] [PubMed] [Green Version]
- Sawamoto, K.; Chen, H.H.; Alméciga-Díaz, C.J.; Mason, R.W.; Tomatsu, S. Gene Therapy for Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 123, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, K.; Stapleton, M.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J.; Losada, J.C.; Suarez, D.A.; Tomatsu, S. Therapeutic Options for Mucopolysaccharidoses: Current and Emerging Treatments; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; Volume 79, ISBN 4026501901147. [Google Scholar]
- Cheema, H.A.; Malik, H.S.; Hashmi, M.A.; Fayyaz, Z.; Mushtaq, I.; Shahzadi, N. Mucopolysaccharidoses—Clinical Spectrum and Frequency of Different Types. J. Coll. Physicians Surg. 2017, 27, 80–83. [Google Scholar]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biol. Blood Marrow Transplant. 2019, 25, e226–e246. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.; Caseiro, C.; Lemos, M.; Lopes, L.; Fontes, A.; Ribeiro, H.; Pinto, E.; Silva, E.; Rocha, S.; Marcão, A.; et al. Prevalence of Lysosomal Storage Diseases in Portugal. Eur. J. Hum. Genet. 2004, 12, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of Lysosomal Storage Disorders. J. Am. Med. Assoc. 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Poorthuis, B.J.H.M.; Wevers, R.A.; Kleijer, W.J.; Groener, J.E.M.; De Jong, J.G.N.; Van Weely, S.; Niezen-Koning, K.E.; Van Diggelen, O.P. The Frequency of Lysosomal Storage Diseases in The Netherlands. Hum. Genet. 1999, 105, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Hèreon, B.; Mikaeloff, Y.; Froissart, R.; Caridade, G.; Caillaud, C.; Levade, T.; Chabrol, B.; Valayannopoulos, V.; Michelakakis, H.; Zafeiriou, D.; et al. Incidence and Natural History of Mucopolysaccharidosis Type III in France and Comparison with United Kingdom and Greece. Am. J. Med. Genet. Part A 2010, 155, 58–68. [Google Scholar] [CrossRef]
- Lin, H.Y.; Lin, S.P.; Chuang, C.K.; Niu, D.M.; Chen, M.R.; Tsai, F.J.; Chao, M.C.; Chiu, P.C.; Lin, S.J.; Tsai, L.P.; et al. Incidence of the Mucopolysaccharidoses in Taiwan, 1984-2004. Am. J. Med. Genet. Part. A 2009, 149, 960–964. [Google Scholar] [CrossRef]
- Gómez, A.M.; García-Robles, R.; Suárez-Obando, F. Estimation of the Mucopolysaccharidoses Frequencies and Cluster Analysis in the Colombian Provinces of Cundinamarca and Boyacá. Biomedica 2012, 32, 602–609. [Google Scholar]
- Josahkian, J.A.; Trapp, F.B.; Burin, M.G.; Michelin-Tirelli, K.; de Magalhães, A.P.P.S.; Fernanda Medeiros, S.; Bender, F.; De Mari, J.F.; Facchin, B.; Leistner-Segal, S.; et al. Updated Incidence and Relative Frequency of Mucopolysaccharidoses Across Brazilian Regions. Genet. Mol. Biol. 2020, 1–56. [Google Scholar]
- Ardila, A.U.; Fajardo, A.A. Selective Screening of 32940 Colombian Patients for the Detection of Lysosomal Metabolic Disorders: Memories of 22 Years of Research (1995–2016). In Proceedings of the 13th International Congress of Inborn Errors of Metabolism—ICIEM, Rio De Janeiro, Brazil, 5–8 September 2017; Volume 5, p. 340, Abstract No: 757. [Google Scholar]
- Ngu, L. Diagnosis and Management of Patients with Mucopolysaccharidoses in Malaysia. J. Mucopolysaccharidosis Rare Dis. 2018, 4, 11–14. [Google Scholar]
- Ngu, L.H.; Ong Peitee, W.; Leong, H.Y.; Chew, H.B. Case Report of Treatment Experience with Idursulfase Beta (Hunterase) in an Adolescent Patient with MPS II. Mol. Genet. Metab. Rep. 2017, 12, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Omar, A.; Jalil, J.A.; Shakrin, N.M.; Ngu, L.H.; Yunus, Z.M. Selective Screening for Detection of Mucopolysaccharidoses in Malaysia; A Two-Year Study (2014–2016). Mol. Genet. Metab. Rep. 2019, 19, 100469. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Ruvalcaba, S.D.C.; Brambila-Tapia, A.J.L.; Juárez-Osuna, J.A.; Da Silva-José, T.D.; García-Ortiz, J.E. Biochemical Diagnosis of Mucopolysaccharidosis in a Mexican Reference Center. Genet. Mol. Biol. 2020, 43, e20180347. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chuang, C.K.; Lee, C.L.; Tu, R.Y.; Lo, Y.T.; Chiu, P.C.; Niu, D.M.; Fang, Y.Y.; Chen, T.L.; Tsai, F.J.; et al. Mucopolysaccharidosis III in Taiwan: Natural History, Clinical and Molecular Characteristics of 28 Patients Diagnosed during a 21-Year Period. Am. J. Med. Genet. Part A 2018, 176, 1799–1809. [Google Scholar] [CrossRef]
- Lin, H.Y.; Lee, C.L.; Chang, C.Y.; Chiu, P.C.; Chien, Y.H.; Niu, D.M.; Tsai, F.J.; Hwu, W.L.; Lin, S.J.; Lin, J.L.; et al. Survival and Diagnostic Age of 175 Taiwanese Patients with Mucopolysaccharidoses (1985–2019). Orphanet. J. Rare Dis. 2020, 15, 1–11. [Google Scholar] [CrossRef]
- Teke Kısa, P.; Köse, E.; Ateşoğlu, M.; Arslan, N. Evaluation of Demographic and Clinical Characteristics of Patients with Mucopolysaccharidosis. J. Pediatr. Res. 2017, 4, 59–62. [Google Scholar] [CrossRef]
- El Moustafa, K.; Sivri, S.; Karahan, S.; Coşkun, T.; Akbıyık, F.; Lay, İ. Screening for Mucopolysaccharidoses in the Turkish Population: Analytical and Clinical Performance of an Age-Range Specific, Dye-Based, Urinary Glycosaminoglycan Assay. Clin. Chim. Acta 2017, 464, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Church, H.; Petty, J.; Righart, J.; Parkes, O.; Egerton, C.; Savage, W.; Tylee, K. The Incidence of Mucopolysaccharidoses and Related Disorders in the Turkish Population: A 3 Year Study. Mol. Genet. Metab. 2013, 108, S30. [Google Scholar] [CrossRef]
- Moammar, H.; Cheriyan, G.; Mathew, R.; Al-Sannaa, N. Incidence and Patterns of Inborn Errors of Metabolism in the Eastern Province of Saudi Arabia, 1983–2008. Ann. Saudi Med. 2010, 30, 271. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.Y.; Sohn, Y.B.; Jin, D.K. An Overview of Korean Patients with Mucopolysaccharidosis and Collaboration through the Asia Pacific MPS Network. Intractable Rare Dis. Res. 2014, 3, 79–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Qiu, W.; Ye, J.; Han, L.; Gu, X.; Zhang, H. Demographic Characteristics and Distribution of Lysosomal Storage Disorder Subtypes in Eastern China. J. Hum. Genet. 2016, 61, 345–349. [Google Scholar] [CrossRef]
- Sheth, J.; Mistri, M.; Sheth, F.; Shah, R.; Bavdekar, A.; Godbole, K.; Nanavaty, N.; Datar, C.; Kamate, M.; Oza, N.; et al. Burden of Lysosomal Storage Disorders in India: Experience of 387 Affected Children from a Single Diagnostic Facility. In JIMD Reports–Volume 12; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Ben Turkia, H.; Tebib, N.; Azzouz, H.; Abdelmoula, M.S.; Ben Chehida, A.; Chemli, J.; Monastiri, K.; Chaabouni, M.; Sanhagi, H.; Zouari, B.; et al. Incidence of Mucopolysaccharidoses in Tunisia. Tunis Med. 2009, 87, 782–785. [Google Scholar]
- Nelson, J.; Crowhurst, J.; Carey, B.; Greed, L. Incidence of the Mucopolysaccharidoses in Western Australia. Am. J. Med. Genet. 2003, 123 A, 310–313. [Google Scholar] [CrossRef]
- Lowry, R.B.; Applegarth, D.A.; Toone, J.R.; MacDonald, E.; Thunem, N.Y. An Update on the Frequency of Mucopolysaccharide Syndromes in British Columbia. Hum. Genet. 1990, 85, 389–390. [Google Scholar] [CrossRef]
- Applegarth, D.A.; Toone, J.R.; Lowry, R.B. Incidence of Inborn Errors of Metabolism in British Columbia, 1969–1996. Pediatrics 2000, 105, e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowry, R.B.; Renwick, D.H. Relative Frequency of the Hurler and Hunter Syndromes. New Engl. J. Med. 1971, 284, 221–222. [Google Scholar]
- Poupětová, H.; Ledvinová, J.; Berná, L.; Dvořáková, L.; Kožich, V.; Elleder, M. The Birth Prevalence of Lysosomal Storage Disorders in the Czech Republic: Comparison with Data in Different Populations. J. Inherit. Metab. Dis. 2010, 33, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Malm, G.; Lund, A.M.; Månsson, J.E.; Heiberg, A. Mucopolysaccharidoses in the Scandinavian Countries: Incidence and Prevalence. Acta Paediatr. Int. J. Paediatr. 2008, 97, 1577–1581. [Google Scholar] [CrossRef]
- Krabbi, K.; Joost, K.; Zordania, R.; Talvik, I.; Rein, R.; Huijmans, J.G.M.; Verheijen, F.V.; Õunap, K. The Live-Birth Prevalence of Mucopolysaccharidoses in Estonia. Genet. Test. Mol. Biomarkers 2012, 16, 846–849. [Google Scholar] [CrossRef]
- Baehner, F.; Schmiedeskamp, C.; Krummenauer, F.; Miebach, E.; Bajbouj, M.; Whybra, C.; Kohlschütter, A.; Kampmann, C.; Beck, M. Cumulative Incidence Rates of the Mucopolysaccharidoses in Germany. J. Inherit. Metab. Dis. 2005, 28, 1011–1017. [Google Scholar] [CrossRef]
- Nelson, J. Incidence of the Mucopolysaccharidoses in Northern Ireland. Hum. Genet. 1997, 101, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Jurecka, A.; Ługowska, A.; Golda, A.; Czartoryska, B.; Tylki-Szymańska, A. Prevalence Rates of Mucopolysaccharidoses in Poland. J. Appl. Genet. 2015, 56, 205–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemper, A.R. Newborn Screening for Mucopolysaccharidosis Type 1 (MPS I): A Systematic Review of Evidence; Report of Final Findings. Final Version 1.1; Duke University: Durham, NC, USA, 2015; pp. 3–60. [Google Scholar]
- Scott, C.R.; Elliott, S.; Buroker, N.; Thomas, L.I.; Keutzer, J.; Glass, M.; Gelb, M.H.; Turecek, F. Identification of Infants at Risk for Developing Fabry, Pompe or Mucopolysaccharidosis-I from Newborn Blood Spots by Tandem Mass Spectrometry. J. Pediatr. 2013, 163, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puckett, Y.; Bui, E.; Zelicoff, A.; Montano, A. Epidemiology of Mucopolysaccharidoses (MPS) in the United States: Challenges and Opportunities. Mol. Genet. Metab. 2017, 120, S111. [Google Scholar] [CrossRef]
- Zahoor, M.Y.; Cheema, H.A.; Ijaz, S.; Anjum, M.N.; Ramzan, K.; Bhinder, M.A. Mapping of IDUA Gene Variants in Pakistani Patients with Mucopolysaccharidosis Type 1. J. Pediatr. Endocrinol. Metab. 2019, 32, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kamranjam, M.; Alaei, M. Mutation Analysis of the IDUA Gene in Iranian Patients with Mucopolysaccharidosis Type 1: Identification of Four Novel Mutations. Genet. Test. Mol. Biomark. 2019, 23, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Atçeken, N.; Özgül, R.K.; Yücel Yilmaz, D.; Tokatli, A.; Coşkun, T.; Sivri, H.S.; Dursun, A.; Karaca, M. Evaluation and Identification of IDUA Gene Mutations in Turkish Patients with Mucopolysaccharidosis Type I. Turk. J. Med. Sci. 2016, 46, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Bunge, S.; Kleijer, W.J.; Steglich, C.; Beck, M.; Zuther, C.; Morris, C.P.; Schwinger, E.; Hopwood, J.J.; Scott, H.S.; Gal, A. Mucopolysaccharidosis Type I: Identification of 8 Novel Mutations and Determination of the Frequency of the Two Common α-L-Iduronidase Mutations (W402X and Q70X) among European Patients. Hum. Mol. Genet. 1994, 3, 861–866. [Google Scholar] [CrossRef]
- Voskoboeva, E.Y.; Krasnopolskaya, X.D.; Mirenburg, T.V.; Weber, B.; Hopwood, J.J. Molecular Genetics of Mucopolysaccharidosis Type I: Mutation Analysis among the Patients of the Former Soviet Union. Mol. Genet. Metab. 1998, 65, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Bertola, F.; Filocamo, M.; Casati, G.; Mort, M.; Rosano, C.; Tylki-Szymanska, A.; Tüysüz, B.; Gabrielli, O.; Grossi, S.; Scarpa, M.; et al. IDUA Mutational Profiling of a Cohort of 102 European Patients with Mucopolysaccharidosis Type I: Identification and Characterization of 35 Novel α-L-Iduronidase (IDUA) Alleles. Hum. Mutat. 2011, 32, 2189–2210. [Google Scholar] [CrossRef] [Green Version]
- Alif, N.; Hess, K.; Straczek, J.; Sebbar, S.; N’Bou, A.; Nabet, P.; Dousset, B. Mucopolysaccharidosis Type I: Characterization of a Common Mutation That Causes Hurler Syndrome in Moroccan Subjects. Ann. Hum. Genet. 1999, 63, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chkioua, L.; Khedhiri, S.; Jaidane, Z.; Ferchichi, S.; Habib, S.; Froissart, R.; Bonnet, V.; Chaabouni, M.; Dandana, A.; Jrad, T.; et al. La Mucopolysaccharidose de Type I: Identification Des Mutations Du Gène Alpha-L-Iduronidase Dans Des Familles Tunisiennes. Arch. Pediatr. 2007, 14, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Chkioua, L.; Khedhiri, S.; Kassab, A.; Bibi, A.; Ferchichi, S.; Froissart, R.; Vianey-Saban, C.; Laradi, S.; Miled, A. Molecular Analysis of Mucopolysaccharidosis Type I in Tunisia: Identification of Novel Mutation and Eight Novel Polymorphisms. Diagn. Pathol. 2011, 6, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Laradi, S.; Tukel, T.; Erazo, M.; Shabbeer, J.; Chkioua, L.; Khedhiri, S.; Ferchichi, S.; Chaabouni, M.; Miled, A.; Desnick, R.J. Mucopolysaccharidosis I: α-L-Iduronidase Mutations in Three Tunisian Families. J. Inherit. Metab. Dis. 2005, 28, 1019–1026. [Google Scholar] [CrossRef]
- Tebani, A.; Zanoutene-Cheriet, L.; Adjtoutah, Z.; Abily-Donval, L.; Brasse-Lagnel, C.; Laquerrière, A.; Marret, S.; Benabdellah, A.C.; Bekri, S. Clinical and Molecular Characterization of Patients with Mucopolysaccharidosis Type I in an Algerian Series. Int. J. Mol. Sci. 2016, 17, 743. [Google Scholar] [CrossRef] [Green Version]
- Venturi, N.; Rovelli, A.; Parini, R.; Menni, F.; Brambillasca, F.; Bertagnolio, F.; Uziel, G.; Gatti, R.; Filocamo, M.; Donati, M.A.; et al. Molecular Analysis of 30 Mucopolysaccharidosis Type I Patients: Evaluation of the Mutational Spectrum in Italian Population and Identification of 13 Novel Mutations. Hum. Mutat. 2002, 20, 231. [Google Scholar] [CrossRef]
- Yamagishi, A.; Tomatsu, S.; Fukuda, S.; Uchiyama, A.; Shimozawa, N.; Suzuki, Y.; Kondo, N.; Sukegawa, K.; Orii, T. Mucopolysaccharidosis Type I: Identification of Common Mutations That Cause Hurler and Scheie Syndromes in Japanese Populations. Hum. Mutat. 1996, 7, 23–29. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, W.; Shi, H.; Qiu, Z.; Meng, Y.; Yao, F.; Wei, M. Mucopolysaccharidosis I Mutations in Chinese Patients: Identification of 27 Novel Mutations and 6 Cases Involving Prenatal Diagnosis. Clin. Genet. 2012, 81, 443–452. [Google Scholar] [CrossRef]
- Kwak, M.J.; Huh, R.; Kim, J.; Park, H.D.; Cho, S.Y.; Jin, D.K. Report of 5 Novel Mutations of the α-Liduronidase Gene and Comparison of Korean Mutations in Relation with Those of Japan or China in Patients with Mucopolysaccharidosis I. BMC Med. Genet. 2016, 17, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azab, B.; Dardas, Z.; Hamarsheh, M.; Alsalem, M.; Kilani, Z.; Kilani, F.; Awidi, A.; Jafar, H.; Amr, S. Novel Frameshift Variant in the IDUA Gene Underlies Mucopolysaccharidoses Type I in a Consanguineous Yemeni Pedigree. Mol. Genet. Metab. Rep. 2017, 12, 76–79. [Google Scholar] [CrossRef]
- Gul, R.; Firasat, S.; Hussain, M.; Afshan, K.; Nawaz, D. IDUA Gene Mutations in Mucopolysaccharidosis Type-1 Patients from Two Pakistani Inbred Families. Congenit. Anom. 2019, 60, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Chkioua, L.; Boudabous, H.; Jaballi, I.; Grissa, O.; Ben Turkia, H.; Tebib, N.; Laradi, S. Novel Splice Site IDUA Gene Mutation in Tunisian Pedigrees with Hurler Syndrome. Diagn. Pathol. 2018, 13, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taghikhani, M.; Khatami, S.; Abdi, M.; Hakhamaneshi, M.S.; Alaei, M.R.; Zamanfar, D.; Vakili, R. Mutation Analysis and Clinical Characterization of Iranian Patients with Mucopolysaccharidosis Type I. J. Clin. Lab. Anal. 2019, 33, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Tu, R.Y.; Chern, S.R.; Lo, Y.T.; Fran, S.; Wei, F.J.; Huang, S.F.; Tsai, S.Y.; Chang, Y.H.; Lee, C.L.; et al. Identification and Functional Characterization of IDS Gene Mutations Underlying Taiwanese Hunter Syndrome (Mucopolysaccharidosis Type Ii). Int. J. Mol. Sci. 2020, 21, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.P.; Chang, J.H.; Lee-Chen, G.J.; Lin, D.S.; Lin, H.Y.; Chuang, C.K. Detection of Hunter Syndrome (Mucopolysaccharidosis Type II) in Taiwanese: Biochemical and Linkage Studies of the Iduronate-2-Sulfatase Gene Defects in MPS II Patients and Carriers. Clin. Chim. Acta 2006, 369, 29–34. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Zhang, X.; Wang, Y.; Qiu, W.; Ye, J.; Han, L.; Gao, X.; Gu, X. Analysis of the IDS Gene in 38 Patients with Hunter Syndrome: The c.879G>A (p.Gln293Gln) Synonymous Variation in a Female Create Exonic Splicing. PLoS ONE 2011, 6, e22951. [Google Scholar] [CrossRef] [Green Version]
- Kadali, S.; Mohammad, S.; Akella, N.; Rama, R.; Vijaya, D.; Bodiga, L.; Naushad, S.M.; Radha Rama Devi, A.; Bodiga, V.L. Biochemical, Machine Learning and Molecular Approaches for the Differential Diagnosis of Mucopolysaccharidoses. Mol. Cell. Biochem. 2019, 458, 27–37. [Google Scholar] [CrossRef]
- Kato, T.; Kato, Z.; Kuratsubo, I.; Tanaka, N.; Ishigami, T.; Kajihara, J.I.; Sukegawa-Hayasaka, K.; Orii, K.; Isogai, K.; Fukao, T.; et al. Mutational and Structural Analysis of Japanese Patients with Mucopolysaccharidosis Type II. J. Hum. Genet. 2005, 50, 395–402. [Google Scholar] [CrossRef]
- Li, X.Y.; Shi, X.Y.; Ju, J.; Hu, X.H.; Yang, X.F.; Zou, L.P. A Novel Iduronate 2-Sulfatase Mutation in a Chinese Family with Mucopolysaccharidosis Type II. World J. Pediatr. 2012, 8, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.B.; Ki, C.S.; Kim, C.H.; Ko, A.R.; Yook, Y.J.; Lee, S.J.; Kim, S.J.; Park, S.W.; Yeau, S.; Kwon, E.K.; et al. Identification of 11 Novel Mutations in 49 Korean Patients with Mucopolysaccharidosis Type II. Clin. Genet. 2012, 81, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Pollard, L.M.; Jones, J.R.; Wood, T.C. Molecular Characterization of 355 Mucopolysaccharidosis Patients Reveals 104 Novel Mutations. J. Inherit. Metab. Dis. 2013, 36, 179–187. [Google Scholar] [CrossRef]
- Narayanan, D.L.; Srivastava, P.; Mandal, K.; Gambhir, P.S.; Phadke, S.R. Hunter Syndrome in Northern India: Clinical Features and Mutation Spectrum. Indian Pediatr. 2016, 53, 134–136. [Google Scholar] [CrossRef] [PubMed]
- Montfort, M.; Vilageliu, L.; Garcia-Giralt, N.; Guidi, S.; Coll, M.J.; Chabás, A.; Grinberg, D. Mutation 1091delC Is Highly Prevalent in Spanish Sanfilippo Syndrome Type A Patients. Hum. Mutat. 1998, 12, 274–279. [Google Scholar] [CrossRef]
- Bunge, S.; Ince, H.; Steglich, C.; Kleijer, W.J.; Beck, M.; Zaremba, J.; van Diggelen, O.P.; Weber, B.; Hopwood, J.J.; Gal, A. Identification of 16 Sulfamidase Gene Mutations Including the Common R74C in Patients with Mucopolysaccharidosis Type IIIA (Sanfilippo A). Hum. Mutat. 1997, 10, 479–485. [Google Scholar] [CrossRef]
- Rady, P.L.; Surendran, S.; Vu, A.T.; Hawkins, J.C.; Michals-Matalon, K.; Tyring, S.K.; Merren, J.; Kumar, A.K.; Matalon, R. Founder Mutation R245H of Sanfilippo Syndrome Type A in the Cayman Islands. Genet. Test. 2002, 6, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, H.; Kumar, D.T.; Doss, C.G.P.; Zayed, H. Bioinformatics Classification of Mutations in Patients with Mucopolysaccharidosis IIIA. Metab. Brain Dis. 2019, 34, 1577–1594. [Google Scholar] [CrossRef] [Green Version]
- Beesley, C.E.; Young, E.P.; Vellodi, A.; Winchester, B.G. Mutational Analysis of Sanfilippo Syndrome Type A (MPS IIIA): Identification of 13 Novel Mutations. J. Med. Genet. 2000, 37, 704–707. [Google Scholar] [CrossRef] [Green Version]
- Di Natale, P.; Balzano, N.; Esposito, S.; Villani, G.R.D. Identification of Molecular Defects in Italian Sanfilippo a Patients Including 13 Novel Mutations. Hum. Mutat. 1998, 11, 313–320. [Google Scholar] [CrossRef]
- Weber, B.; Van De Kamp, J.J.P.; Kleijer, W.J.; Guo, X.H.; Blanch, L.; Van Diggelen, O.P.; Wevers, R.; Poorthuis, B.J.H.M.; Hopwood, J.J. Identification of a Common Mutation (R245H) in Sanfilippo A Patients from the Netherlands. J. Inherit. Metab. Dis. 1998, 21, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Beesley, C.; Moraitou, M.; Winchester, B.; Schulpis, K.; Dimitriou, E.; Michelakakis, H. Sanfilippo B Syndrome: Molecular Defects in Greek Patients. Clin. Genet. 2004, 65, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Beesley, C.E.; Young, E.P.; Vellodi, A.; Winchester, B.G. Identification of 12 Novel Mutations in the α-N-Acetylglucosaminidase Gene in 14 Patients with Sanfilippo Syndrome Type B (Mucopolysaccharidosis Type IIIB). J. Med. Genet. 1998, 35, 910–914. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, E.E.A.; Fateen, E.M. Identification of Three Novel Homozygous NAGLU Mutations in Egyptian Patients with Sanfilippo Syndrome B. Meta Gene 2019, 21, 100580. [Google Scholar] [CrossRef]
- Schmidtchen, A.; Greenberg, D.; Zhao, H.G.; Li, H.H.; Huang, Y.; Tieu, P.; Zhao, H.Z.; Cheng, S.; Zhao, Z.; Whitley, C.B.; et al. NAGLU Mutations Underlying Sanfilippo Syndrome Type B. Am. J. Hum. Genet. 1998, 62, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierzynowska, K.; Mański, A.; Limanówka, M.; Wierzba, J.; Gaffke, L.; Anikiej, P.; Węgrzyn, G. Untypically Mild Phenotype of a Patient Suffering from Sanfilippo Syndrome B with the c.638C>T/c.889C>T (p.Pro213Leu/p.Arg297Ter) Mutations in the NAGLU Gene. Mol. Genet. Genom. Med. 2020, 8, 1–6. [Google Scholar] [CrossRef]
- Hettiarachchi, D.; Nethikumara, N.; Pathirana, B.A.P.S.; Weththasigha, K.; Dissanayake, W.D.N.; Dissanayake, V.H.W. A Novel Mutation in the NAGLU Gene Associated with Sanfilippo Syndrome Type B (Mucopolysaccharidosis III B). Clin. Case Rep. 2018, 6, 1051–1054. [Google Scholar] [CrossRef] [PubMed]
- Ouesleti, S.; Coutinho, M.F.; Ribeiro, I.; Miled, A.; Mosbahi, D.S.; Alves, S.; Mosbahi, S.; Alves, S. Update of the Spectrum of Mucopolysaccharidoses Type III in Tunisia: Identification of Three Novel Mutations and in Silico Structural Analysis of the Missense Mutations. World J. Pediatr. 2017, 13, 374–380. [Google Scholar] [CrossRef]
- Shafaat, M.; Hashemi, M.; Majd, A.; Abiri, M.; Zeinali, S. Genetic Testing of Mucopolysaccharidoses Disease Using Multiplex PCR- Based Panels of STR Markers: In Silico Analysis of Novel Mutations. Metab. Brain Dis. 2019, 34, 1447–1455. [Google Scholar] [CrossRef]
- Tanaka, T.; Suzuki, T.; Nakamura, J.; Kawamura, Y.; Sadahiro, S.; Kijima, H.; Yamamoto, I.; Vo, N.; Yamaguchi, M.; Irino, Y.; et al. Huaier Regulates Cell Fate by the Rescue of Disrupted Transcription Control in the Hippo Signaling Pathway. Arch. Clin. Biomed. Res. 2017, 01, 179–199. [Google Scholar] [CrossRef]
- Zeng, Q.; Fan, Y.; Wang, L.; Huang, Z.; Gu, X.; Yu, Y. Molecular Defects Identified by Whole Exome Sequencing in a Child with Atypical Mucopolysaccharidosis IIIB. J. Pediatr. Endocrinol. Metab. 2017, 30, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Feldhammer, M.; Durand, S.; Mrázová, L.; Boucher, R.M.; Laframboise, R.; Steinfeld, R.; Wraith, J.E.; Michelakakis, H.; Van Diggelen, O.P.; Hřebíček, M.; et al. Sanfilippo Syndrome Type C: Mutation Spectrum in the Heparan Sulfate Acetyl-CoA: α-Glucosaminide N-Acetyltransferase (HGSNAT) Gene. Hum. Mutat. 2009, 30, 918–925. [Google Scholar] [CrossRef]
- Fedele, A.O.; Filocamo, M.; Di Rocco, M.; Sersale, G.; Lübke, T.; di Natale, P.; Cosma, M.P.; Ballabio, A. Mutational Analysis of the HGSNAT Gene in Italian Patients with Mucopolysaccharidosis IIIC (Sanfilippo C Syndrome). Mutation in Brief #959. Online. Hum. Mutat. 2007, 28, 523. [Google Scholar] [PubMed]
- Velasco, H.M.; Sanchez, Y.; Martin, A.M.; Umaña, L.A. Natural History of Sanfilippo Syndrome Type C in Boyacá, Colombia: A Neurogenetic Description. J. Child. Neurol. 2017, 32, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Huh, H.J.; Seo, J.Y.; Cho, S.Y.; Ki, C.S.; Lee, S.Y.; Kim, J.W.; Park, H.D.; Jin, D.K. The First Korean Case of Mucopolysaccharidosis IIIC (Sanfilippo Syndrome Type C) Confirmed by Biochemical and Molecular Investigation. Ann. Lab. Med. 2013, 33, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Hřebíček, M.; Mrázová, L.; Seyrantepe, V.; Durand, S.; Roslin, N.M.; Nosková, L.; Hartmannová, H.; Ivánek, R.; Čížková, A.; Poupétová, H.; et al. Mutations in TMEM76* Cause Mucopolysaccharidosis IIIC (Sanfilippo C Syndrome). Am. J. Hum. Genet. 2006, 79, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Ruijter, G.J.G.; Valstar, M.J.; van de Kamp, J.M.; van der Helm, R.M.; Durand, S.; van Diggelen, O.P.; Wevers, R.A.; Poorthuis, B.J.; Pshezhetsky, A.V.; Wijburg, F.A. Clinical and Genetic Spectrum of Sanfilippo Type C (MPS IIIC) Disease in The Netherlands. Mol. Genet. Metab. 2008, 93, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Valstar, J.M.; Bertoli-Avella, A.M.; Wessels, M.W.; Ruijter, G.J.G.; de Graaf, B.; Olmer, R.; Elfferich, P.; Neijs, S.; Kariminejad, R.; Ezgü, F.S.; et al. Mucopolysaccharidosis Type IIID: 12 New Patients and 15 Novel Mutations. Hum. Mutat. 2010, 31, E1348–E1360. [Google Scholar]
- Al Kaissi, A.; Hofstaetter, J.; Weigel, G.; Grill, F.; Ganger, R.; Kircher, S.G. The Constellation of Skeletal Deformities in a Family with Mixed Types of Mucopolysaccharidoses. Medicine 2016, 95, e4561. [Google Scholar] [CrossRef] [PubMed]
- Mok, A.; Cao, H.; Hegele, R.A. Genomic Basis of Mucopolysaccharidosis Type IIID ( MIM 252940 ) Revealed by Sequencing of GNS Encoding N -Acetylglucosamine-6-Sulfatase. Genomics 2003, 81, 1–5. [Google Scholar] [CrossRef]
- Jansen, A.C.M.; Cao, H.; Kaplan, P.; Silver, K.; Leonard, G.; De Meirleir, L.; Lissens, W.; Liebaers, I.; Veilleux, M.; Andermann, F.; et al. Sanfilippo Syndrome Type D. Arch. Neurol. 2007, 64, 1629–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidchol, A.M.; Dalal, A.; Shah, H.; Suryanarayana, S.; Nampoothiri, S.; Kabra, M.; Gupta, N.; Danda, S.; Gowrishankar, K.; Phadke, S.R.; et al. GALNS Mutations in Indian Patients with Mucopolysaccharidosis IVA. Am. J. Med. Genet. Part A 2014, 164, 2793–2801. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Montaño, A.M.; Nishioka, T.; Gutierrez, M.A.; Peña, O.M.; Trandafirescu, G.G.; Lopez, P.; Yamaguchi, S.; Noguchi, A.; Orii, T. Mutation and Polymorphism Spectrum of the GALNS Gene in Mucopolysaccharidosis IVA (Morquio A). Hum. Mutat. 2005, 26, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Bochernitsan, A.N.; Brusius-Facchin, A.C.; Couto, R.R.; Kubaski, F.; dos Santos Lopes, S.S.; Gondim, C.E.; de Medeiros, P.F.V.; de Souza, C.F.M.; Giugliani, R.; Leistner-Segal, S. Spectrum of GALNS Mutations and Haplotype Study in Brazilian Patients with Mucopolysaccharidosis Type IVA. Meta Gene 2018, 16, 77–84. [Google Scholar] [CrossRef]
- Tomatsu, S.; Fukuda, S.; Cooper, A.; Wraith, J.E.; Rezvi, G.M.M.; Yamagishi, A.; Yamada, N.; Kato, Z.; Isogai, K.; Sukegawa, K.; et al. Mucopolysaccharidosis IVA: Identification of a Common Missense Mutation I113F in the N-Acetylgalactosamine-6-Sulfate Sulfatase Gene. Am. J. Hum. Genet. 1995, 57, 556–563. [Google Scholar]
- Yamada, N.; Fukuda, S.; Tomatsu, S.; Muller, V.; Hopwood, J.J.; Nelson, J.; Kato, Z.; Yamagishi, A.; Sukegawa, K.; Kondo, N.; et al. Molecular Heterogeneity in Mucopolysaccharidosis IVA in Australia and Northern Ireland: Nine Novel Mutations Including T312S, a Common Allele That Confers a Mild Phenotype. Hum. Mutat. 1998, 11, 202–208. [Google Scholar] [CrossRef]
- Tomatsu, S.; Filocamo, M.; Orii, K.O.; Sly, W.S.; Gutierrez, M.A.; Nishioka, T.; Serrato, O.P.; Natale, P.; Montaño, A.M.; Yamaguchi, S.; et al. Mucopolysaccharidosis IVA (Morquio A): Identification of Novel Common Mutations in the N-Acetylgalactosamine-6-Sulfate Sulfatase (GALNS) Gene in Italian Patients. Hum. Mutat. 2004, 24, 187–188. [Google Scholar] [CrossRef]
- Terzioglu, M.; Tokatli, A.; Coskun, T.; Emre, S. Molecular Analysis of Turkish Mucopolysaccharidosis IVA (Morquio A) Patients: Identification of Novel Mutations in the N-Acetylgalactosamine-6-Sulfate Sulfatase (GALNS) Gene. Hum. Mutat. 2002, 20, 477–478. [Google Scholar] [CrossRef] [PubMed]
- Morrone, A.; Caciotti, A.; Atwood, R.; Davidson, K.; Du, C.; Francis-Lyon, P.; Harmatz, P.; Mealiffe, M.; Mooney, S.; Oron, T.R.; et al. Morquio a Syndrome-Associated Mutations: A Review of Alterations in the GALNS Gene and a New Locus-Specific Database. Hum. Mutat. 2014, 35, 1271–1279. [Google Scholar] [CrossRef] [Green Version]
- Ishii, N.; Oohira, T.; Oshima, A.; Sakuraba, H.; Endo, F.; Matsuda, I.; Sukegawa, K.; Orii, T.; Suzuki, Y. Clinical and Molecular Analysis of a Japanese Boy with Morquio B Disease. Clin. Genet. 1995, 48, 103–108. [Google Scholar] [CrossRef]
- Oshima, A.; Yoshida, K.; Shimmoto, M.; Fukuhara, Y.; Sakuraba, H.; Suzuki, Y. Human β-Galactosidase Gene Mutations in Morquio B Disease. Am. J. Hum. Genet. 1991, 49, 1091–1093. [Google Scholar]
- Abumansour, I.S.; Yuskiv, N.; Paschke, E.; Stockler-Ipsiroglu, S. Morquio-B Disease: Clinical and Genetic Characteristics of a Distinct GLB1-Related Dysostosis Multiplex. JIMD Rep. 2020, 51, 30–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschke, E.; Milos, I.; Kreimer-Erlacher, H.; Hoefler, G.; Beck, M.; Hoeltzenbein, M.; Kleijer, W.; Levade, T.; Michelakakis, H.; Radeva, B.; et al. Mutation Analyses in 17 Patients with Deficiency in Acid β-Galactosidase: Three Novel Point Mutations and High Correlation of Mutation W273L with Morquio Disease Type B. Hum. Genet. 2001, 109, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, R.; Chabás, A.; Coll, M.J.; Miranda, C.S.; Vilageliu, L.; Grinberg, D. Twenty-One Novel Mutations in the GLB1 Gene Identified in a Large Group of GM1-Gangliosidosis and Morquio B Patients: Possible Common Origin for the Prevalent p.R59H Mutation among Gypsies. Hum. Mutat. 2006, 27, 1060. [Google Scholar] [CrossRef]
- Gucev, Z.S.; Tasic, V.; Jancevska, A.; Zafirovski, G.; Kremensky, I.; Sinigerska, I.; Nanba, E.; Higaki, K.; Gucev, F.; Suzuki, Y. Novel β-Galactosidase Gene Mutation p.W273R in a Woman with Mucopolysaccharidosis Type IVB (Morquio B) and Lack of Response to in Vitro Chaperone Treatment of Her Skin Fibroblasts. Am. J. Med. Genet. Part. A 2008, 146, 1736–1740. [Google Scholar] [CrossRef]
- Al-Sannaa, N.A.; Al-Abdulwahed, H.Y.; Al-Majed, S.I. The Clinical and Genetic Spectrum of Maroteaux-Lamy Syndrome (Mucopolysaccharidosis VI ) in the Eastern Province of Saudi Arabia. J. Community Genet. 2018, 9, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Aminzadeh, M.; Malekpour, N.; Ghandil, P. Identification of Arylsulfatase B Gene Mutations and Clinical Presentations of Iranian Patients with Mucopolysaccharidosis VI. Gene 2019, 706, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Dursun, A.; Karaca, M.; Alehan, D.; Turan-Dizdar, H.; Aksoy, C.; Arslan, U.; Sivri, H.S.; Kılıç, M.; Dursun, A.; Coşkun, T.; et al. Genotypic-Phenotypic Features and Enzyme Replacement Therapy Outcome in Patients with Mucopolysaccharidosis VI from Turkey. Am. J. Med. Genet. Part. A 2017, 173, 2954–2967. [Google Scholar]
- Lin, H.; Chen, M.; Chuang, C.; Chen, C.; Lin, D.; Chien, Y.; Ke, Y.; Tsai, F.; Pan, H. Enzyme Replacement Therapy for Mucopolysaccharidosis VI—Experience in Taiwan. J. Inherit. Metab. Dis. 2010, 3, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Lin, S.; Wang, C.; Hwu, W.; Chuang, C.; Lin, S.; Tsai, Y.; Chen, C.; Tsai, F. Clinica Chimica Acta Genetic Analysis of Mucopolysaccharidosis Type VI in Taiwanese Patients. Clin. Chim. Acta 2008, 394, 89–93. [Google Scholar] [CrossRef]
- Furujo, M.; Kubo, T.; Kosuga, M.; Okuyama, T. Enzyme Replacement Therapy Attenuates Disease Progression in Two Japanese Siblings with Mucopolysaccharidosis Type VI. Mol. Genet. Metab. 2011, 104, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Ke, Y.; Chou, I.; Wang, C.; Tsai, F. Deletion of Exon 4 in the N-Acetylgalactosamine-4-Sulfatase Gene in a Taiwanese Patient with Mucopolysaccharidosis Type VI. Tohoku J. Exp. Med. 2015, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Costa-Motta, F.M.; Bender, F.; Acosta, A.; Abé-Sandes, K.; Machado, T.; Bomfim, T.; Sorte, T.B.; da Silva, D.; Bittles, A.; Giugliani, R.; et al. A Community-Based Study of Mucopolysaccharidosis Type VI in Brazil: The Influence of Founder Effect, Endogamy and Consanguinity. Hum. Hered. 2014, 4, 189–196. [Google Scholar] [CrossRef]
- Hançer, V.; Büyükdoğan, M.; Babameto-Laku, A. A Novel Pathological ARSB Mutation (c. 870G > A; p. Trp290stop) in Mucopolysaccharidosis Type VI Patients. Mol. Syndromol. 2019, 10, 272–275. [Google Scholar] [CrossRef]
- Chen, H.; Kao, S.; Lin, H.; Huang, Y.; Kumar, A.B. Taiwan National Newborn Screening Program by Tandem Mass. J. Pediatr. 2018, 205, 1–7. [Google Scholar]
- Coutinho, M.F.; Encarnaç, M.; Matos, L.; Silva, L.; Ribeiro, D.; Santos, J.I.; João Prata, M.; Vilarinho, L. Molecular Characterization of a Novel Splicing Mutation Underlying Mucopolysaccharidosis (MPS) Type VI—Indirect Proof of Principle on Its Pathogenicity. Diagnostics 2020, 10, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malekpour, N.; Vakili, R.; Hamzehloie, T. Mutational Analysis of ARSB Gene in Mucopolysaccharidosis Type VI: Identification of Three Novel Mutations in Iranian Patients. Iran J. Basic Med. Sci. 2018, 21, 950–956. [Google Scholar] [PubMed]
- Mosallanejad, A.; Alaei, M.; Ghaffari, S.; Rafati, M.; Saneifard, H. Non-Progressive Nonimmune Hydrops Fetalis Caused by a Novel Mutation in GUSB Gene. Iran. J. Child. Neurol. 2020, 14, 101–106. [Google Scholar] [PubMed]
- Guffon, N.; Froissart, R.; Fouilhoux, A. A Rare Late Progression Form of Sly Syndrome Mucopolysaccharidosis. JIMD Rep. 2019, 49, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Pan, J.; Linpeng, S.; Li, Z.; Tan, H.; Wu, L. Identification of Five Novel Mutations Causing Rare Lysosomal Storage Diseases. Med. Sci. Monit. 2019, 25, 7634–7644. [Google Scholar] [CrossRef]
- Furlan, F.; Rovelli, A.; Rigoldi, M.; Filocamo, M.; Tappino, B.; Friday, D.; Gasperini, S.; Mariani, S.; Izzi, C.; Bondioni, M.P.; et al. A New Case Report of Severe Mucopolysaccharidosis Type VII: Diagnosis, Treatment with Haematopoietic Cell Transplantation and Prenatal Diagnosis in a Second Pregnancy 11 Medical and Health Sciences 1114 Paediatrics and Reproductive Medicine. Ital. J. Pediatr. 2018, 44, 1–7. [Google Scholar]
- Kubaski, F.; Brusius, A.C.-F.; Mason, R.W.; Patel, P.; Burin, M.G.; Michelin-Tirelli, K.; Kessler, R.G.; Bender, F.; Leistner-Segal, S.; Moreno, C.A.; et al. Elevation of Glycosaminoglycans in the Amniotic Fluid of a Fetus with Mucopolysaccharidosis VII: Prenatal Diagnosis of an MPS VII Fetus. Prenat. Diagn. 2017, 37, 435–439. [Google Scholar] [CrossRef]
- Fukuda, S.; Tomatsu, S.; Sukegawa, K.; Sasaki, T.; Yamada, Y.; Kuwahara, T.; Okamoto, H.; Ikedo, Y.; Yamaguchi, S.; Orii, T. Molecular Analysis of Mucopolysaccharidosis Type VII. J. Inherit. Metab. Dis. 1991, 14, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Vervoort, R.; Lissens, W.; Hoo, J.J.; Valentine, L.A.; Sly, W.S. β-Glucuronidase P408S, P415L Mutations: Evidence That Both Mutations Combine to Produce an MPS VII Allele in Certain Mexican Patients. Hum. Genet. 1996, 98, 281–284. [Google Scholar] [CrossRef]
- Islam, M.R.; Arreola, M.P.G.; Wong, P.; Tomatsu, S.; Corona, J.S.; Sly, W.S. β-Glucuronidase P408S, P415L Allele in a Mexican Population: Population Screening in Guadalajara and Prenatal Diagnosis. Prenat. Diagn. 1998, 18, 822–825. [Google Scholar] [CrossRef]
- Tomatsu, S.; Fukuda, S.; Sukegawa, K.; Ikedo, Y.; Yamada, S.; Yamada, Y.; Sasaki, T.; Okamoto, H.; Kuwahara, T.; Yamaguchi, S.; et al. Mucopolysaccharidosis Type VII: Characterization of Mutations and Molecular Heterogeneity. Am. J. Hum. Genet. 1991, 48, 89–96. [Google Scholar]
- Tomatsu, S.; Sukegawa, K.; Ikedo, Y.; Fukuda, S.; Yamada, Y.; Sasaki, T.; Okamoto, H.; Kuwabara, T.; Orii, T. Molecular Basis of Mucopolysaccharidosis Type VII: Replacement of Ala619 in β-Glucuronidase with Val. Gene 1990, 89, 283–287. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Dung, V.C.; Grubb, J.; Sly, W. Mutations and Polymorphisms in GUSB Gene in Mucopolysaccharidosis VII (Sly Syndrome). Hum. Mutat. 2009, 30, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vervoort, R.; Rafiqul Islam, M.; Sly, W.S.; Zabot, M.T.; Kleijer, W.J.; Chabas, A.; Fensom, A.; Young, E.P.; Liebaers, I.; Lissens, W. Molecular Analysis of Patients with β-Glucuronidase Deficiency Presenting as Hydrops Fetalis or as Early Mucopolysaccharidosis VII. Am. J. Hum. Genet. 1996, 58, 457–471. [Google Scholar]
- Vervoort, R.; Buist, N.R.M.; Kleijer, W.J.; Wevers, R.; Fryns, J.P.; Liebaers, I.; Lissens, W. Molecular Analysis of the β-Glucuronidase Gene: Novel Mutations in Mucopolysaccharidosis Type VII and Heterogeneity of the Polyadenylation Region. Hum. Genet. 1997, 99, 462–468. [Google Scholar] [CrossRef]
- Vervoort, R.; Lissens, W.; Liebaers, I. Molecular Analysis of a Patient with Hydrops Fetalis Caused by Β-glucuronidase Deficiency, and Evidence for Additional Pseudogenes. Hum. Mutat. 1993, 2, 443–445. [Google Scholar] [CrossRef] [PubMed]
- Imundo, L.; LeDuc, C.A.; Guha, S.; Brown, M.; Perino, G.; Gushulak, L.; Triggs-Raine, B.; Chung, W.K. A Complete Deficiency of Hyaluronoglucosaminidase 1 (HYAL1) Presenting as Familial Juvenile Idiopathic Arthritis. J. Inherit. Metab. Dis. 2011, 34, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Natowicz, M.R.; Short, M.P.; Wang, Y.; Dickersin, G.R.; Gebhardt, M.; Rosenthal, D.; Sims, K.; Rosenberg, A. Clinical and Biochemical Manifestations of Hyaluronidase Deficiency. N. Engl. J. Med. 1996, 335, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Triggs-Raine, B.; Salo, T.J.; Zhang, H.; Wicklow, B.A.; Natowicz, M.R. Mutations in HYAL1, a Member of a Tandemly Distributed Multigene Family Encoding Disparate Hyaluronidase Activities, Cause a Newly Described Lysosomal Disorder, Mucopolysaccharidosis IX. Proc. Natl. Acad. Sci. USA 1999, 96, 6296–6300. [Google Scholar] [CrossRef] [Green Version]
- Kiykim, E.; Barut, K.; Cansever, M.S.; Zeybek, C.A.; Zubarioglu, T.; Aydin, A.; Kasapcopur, O. Screening Mucopolysaccharidosis Type IX in Patients with Juvenile Idiopathic Arthritis. JIMD Rep. 2016, 25, 21–24. [Google Scholar]
- CDC Newborn Screening Portal. Available online: https://www.cdc.gov/newbornscreening/ (accessed on 5 August 2020).
- Maccready, R.A.; Hussey, M.G. Newborn Phenylketonuria Detection Program in Massachusetts. Am. J. Public Health Nations Health 1964, 54, 2075–2081. [Google Scholar] [CrossRef] [Green Version]
- Caggana, M.; Jones, E.A.; Shahied, S.I.; Tanksley, S.; Hermerath, C.A.; Lubin, I.M. Newborn Screening: From Guthrie to Whole Genome Sequencing. Public Health Rep. 2013, 128, 14–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelb, M.H. Newborn Screening for Lysosomal Storage Diseases: Methodologies, Screen Positive Rates, Normalization of Datasets, Second-Tier Tests, and Post-Analysis Tools. Int. J. Neonatal. Screen. 2018, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, M.; Kubaski, F.; Mason, R.W.; Shintaku, H.; Kobayashi, H.; Yamaguchi, S.; Taketani, T.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Newborn Screening for Mucopolysaccharidoses: Measurement of Glycosaminoglycans by LC-MS/MS. Mol. Genet. Metab. Rep. 2020, 22, 100563. [Google Scholar] [CrossRef]
- Huang, T.; Cao, Y.; Zeng, J.; Dong, J.; Sun, X.; Chen, J.; Gao, P. Tandem Mass Spectrometry-Based Newborn Screening Strategy Could Be Used to Facilitate Rapid and Sensitive Lung Cancer Diagnosis. Onco. Targets Ther. 2016, 9, 2479–2487. [Google Scholar]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Waggoner, D.; Tinkle, B.; Braddock, S.R.; Schneider, M.; Grange, D.K.; Nash, C.; Shryock, H.; et al. Newborn Screening for Lysosomal Storage Disorders in Illinois: The Initial 15-Month Experience. J. Pediatr. 2017, 190, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Caggana, M.; Bailey, S.M.; Desnick, R.J.; Estrella, L.; Holzman, I.; Kelly, N.R.; Kornreich, R.; Gabriel, S.; Martin, M.; et al. The New York pilot newborn screening program for lysosomal storage diseases: Report of the First 65,000 Infants. Genet. Med. 2019, 21, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Martínez, J.I.; Limón-Rojas, A.E.; Gaytán-García, M.D.J.; Reyna-Figueroa, J.; Wakida-Kusunoki, G.; del Rocío Delgado-Calvillo, M.; Cantú-Reyna, C.; Cruz-Camino, H.; Cervantes-Barragán, D.E. Newborn Screening for Six Lysosomal Storage Disorders in a Cohort of Mexican Patients: Three-Year Findings from a Screening Program in a Closed Mexican Health System. Mol. Genet. Metab. 2017, 121, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Schaap, T.; Bach, G. Incidence of Mucopolysaccharidoses in Israel: Is Hunter Disease a “Jewish Disease”? Hum. Genet. 1980, 223, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Jurecka, A.; Zakharova, E.; Cimbalistiene, L.; Gusina, N.; Malinova, V.; Rózdzyńska-Swiątkowska, A.; Golda, A.; Kulpanovich, A.; Abdilova, G.K.; Voskoboeva, E.; et al. Mucopolysaccharidosis Type VI in Russia, Kazakhstan, and Central and Eastern Europe. Pediatr. Int. 2014, 56, 520–525. [Google Scholar] [CrossRef]
- Moore, D.; Connock, M.J.; Wraith, E.; Lavery, C. The Prevalence of and Survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie Syndromes in the UK. Orphanet J. Rare Dis. 2008, 3, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Federhen, A.; Burin, M.; Leistner-Segal, S.; Matte, U.; Tirelli, K.; Facchin, A.; Pasqualim, G.; Bender, F.; Rafaelli, C.; Giugliani, R. Minimal Estimated Incidence of MPS I, II, IV-A and VI in Brazil and Comparison to the Rest of the World. J. Inborn Errors Metab. Screen. 2016, 4, 9–10. [Google Scholar]
- Eyskens, F.; Devos, S. Newborn Screening for Lysosomal Storage Disorders in Belgium: The Importance of Sex- and Age-Dependent Reference Ranges Journal. J. Inborn Errors Metab. Screen. 2017, 5. [Google Scholar] [CrossRef]
- Bravo, H.; Neto, E.C.; Schulte, J.; Pereira, J.; Filho, C.S.; Bittencourt, F.; Sebastião, F.; Bender, F.; de Magalhães, A.P.S.; Guidobono, R.; et al. Investigation of Newborns with Abnormal Results in a Newborn Screening Program for Four Lysosomal Storage Diseases in Brazil. Mol. Genet. Metab. Rep. 2017, 12, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Paciotti, S.; Persichetti, E.; Pagliardini, S.; Deganuto, M.; Rosano, C.; Balducci, C.; Codini, M.; Filocamo, M.; Menghini, A.R.; Pagliardini, V.; et al. First Pilot Newborn Screening for Four Lysosomal Storage Diseases in an Italian Region: Identification and Analysis of a Putative Causative Mutation in the GBA Gene. Clin. Chim. Acta 2012, 413, 1827–1831. [Google Scholar] [CrossRef]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn Screening for Lysosomal Storage Disorders by Tandem Mass Spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef]
- Lin, S.P.; Lin, H.Y.; Wang, T.J.; Chang, C.Y.; Lin, C.H.; Huang, S.F.; Tsai, C.C.; Liu, H.L.; Keutzer, J.; Chuang, C.K. A Pilot Newborn Screening Program for Mucopolysaccharidosis Type i in Taiwan. Orphanet J. Rare Dis. 2013, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.C.; Chiang, C.C.; Niu, D.M.; Wang, C.H.; Kao, S.M.; Tsai, F.J.; Huang, Y.H.; Liu, H.C.; Huang, C.K.; Gao, H.J.; et al. Detecting Multiple Lysosomal Storage Diseases by Tandem Mass Spectrometry—A National Newborn Screening Program in Taiwan. Clin. Chim. Acta 2014, 431, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.; Lin, H.; Wang, T.; Huang, Y.; Chan, M.; Liao, H.; Lo, Y.; Wang, L.; Tu, R.; Fang, Y.; et al. Status of Newborn Screening and Follow up Investigations for Mucopolysaccharidoses I and II in Taiwan. Orphanet J. Rare Dis. 2018, 13, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, M.-J.; Liao, H.-C.; Gelb, M.H.; Chuang, C.-K.; Liu, M.-Y.; Chen, H.-J.; Kao, S.-M.; Lin, H.-Y.; Huang, Y.-H.; Kumar, A.B.; et al. Taiwan National Newborn Screening Program by Tandem Mass Spectrometry for Mucopolysaccharidoses Types I, II, and VI. J. Pediatr. 2019, 205, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.-H.H.; Lee, N.C.; Chen, P.W.; Yeh, H.Y.; Gelb, M.H.; Chiu, P.C.; Chu, S.Y.; Lee, C.H.; Lee, A.R.; Hwu, W.L. Newborn Screening for Morquio Disease and Other Lysosomal Storage Diseases: Results from the 8-Plex Assay for 70,000 Newborns. Orphanet J. Rare Dis. 2020, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.; Buroker, N.; Cournoyer, J.J.; Potier, A.M.; Trometer, J.D.; Elbin, C.; Schermer, M.J.; Kantola, J.; Boyce, A.; Turecek, F.; et al. Pilot Study of Newborn Screening for Six Lysosomal Storage Diseases Using Tandem Mass Spectrometry. Mol. Genet. Metab. 2016, 118, 304–309. [Google Scholar] [CrossRef]
- Minter Baerg, M.M.; Stoway, S.D.; Hart, J.; Mott, L.; Peck, D.S.; Nett, S.L.; Eckerman, J.S.; Lacey, J.M.; Turgeon, C.T.; Gavrilov, D.; et al. Precision Newborn Screening for Lysosomal Disorders. Genet. Med. 2018, 20, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, P.V.; Klug, T.; Vermette, L.; Raburn-Miller, J.; Kiesling, J.; Rogers, S. Incidence of 4 Lysosomal Storage Disorders from 4 Years of Newborn Screening. JAMA Pediatr. 2018, 172, 696–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, P.V.; Campbell, C.; Klug, T.; Rogers, S.; Raburn-Miller, J.; Kiesling, J. Lysosomal Storage Disorder Screening Implementation: Findings from the First Six Months of Full Population Pilot Testing in Missouri. J. Pediatr. 2015, 166, 172–177. [Google Scholar] [CrossRef]
- Burton, B.K.; Hoganson, G.E.; Grange, D.K.; Braddock, S.R.; Christensen, K.M.; Hitchins, L.; Hickey, R.; Shao, R.; Basheeruddin, K.; Basheeruddin, K. Newborn Screening for Mucopolysaccharidosis Type II (MPS II) in Illinois: The First Year’s Experience. Mol. Genet. Metab. 2019, 126, S34. [Google Scholar] [CrossRef]
- Scott, C.R.; Elliott, S.; Hong, X.; Huang, J.; Kumar, A.B.; Yi, F.; Pendem, N.; Chennamaneni, N.K.; Gelb, M.H. Newborn Screening for Mucopolysaccharidoses: Results of a Pilot Study with 100,000 Dried Blood Spots. J. Pediatr. 2019, 216, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Lee, C.L.; Lo, Y.T.; Tu, R.Y.; Chang, Y.H.; Chang, C.Y.; Chiu, P.C.; Chang, T.M.; Tsai, W.H.; Niu, D.M.; et al. An At-Risk Population Screening Program for Mucopolysaccharidoses by Measuring Urinary Glycosaminoglycans in Taiwan. Diagnostics 2019, 9, 140. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.B.; Hong, X.; Yi, F.; Wood, T.; Gelb, M.H. Tandem Mass Spectrometry-Based Multiplex Assays for α- Mannosidosis and FucosidosisS Public Access. Mol. Genet. Metab 2019, 127, 207–211. [Google Scholar] [CrossRef]
- Langan, T.J.; Jalal, K.; Barczykowski, A.L.; Carter, R.L.; Stapleton, M.; Orii, K.; Fukao, T.; Kobayashi, H.; Yamaguchi, S.; Tomatsu, S. Development of a Newborn Screening Tool for Mucopolysaccharidosis Type I Based on Bivariate Normal Limits: Using Glycosaminoglycan and Alpha-L-Iduronidase Determinations on Dried Blood Spots to Predict Symptoms. JIMD Rep. 2020, 52, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, Z.M.; Urdaneta, L.; Klein, T.; Fuller, M.; Gelb, M.H. Evaluation of Multiple Methods for Quantification of Glycosaminoglycan Biomarkers in Newborn Dried Blood Spots from Patients with Severe and Attenuated Mucopolysaccharidosis-I. Int. J. Neonatal. Screen. 2020, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, Y.; Fuji, N.; Yamazaki, N.; Hirakiyama, A.; Kamioka, T.; Seo, J.H.; Mashima, R.; Kosuga, M.; Okuyama, T. A Molecular Analysis of the GAA Gene and Clinical Spectrum in 38 Patients with Pompe Disease in Japan. Mol. Genet. Metab. Rep. 2018, 14, 3–9. [Google Scholar] [CrossRef]
- Dʹavanzo, F.; Rigon, L.; Zanetti, A.; Tomanin, R. Mucopolysaccharidosis Type II: One Hundred Years of Research, Diagnosis, and Treatment. Int. J. Mol. Sci. 2020, 21, 1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ru, M.H.; Boelens, J.J.; Das, A.M.; Jones, S.A.; Van Der Lee, J.H.; Mahlaoui, N.; Mengel, E.; Offringa, M.; O’Meara, A.; Parini, R.; et al. Enzyme Replacement Therapy and/or Hematopoietic Stem Cell Transplantation at Diagnosis in Patients with Mucopolysaccharidosis Type I: Results of a European Consensus Procedure. Orphanet J. Rare Dis. 2011, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kubaski, F.; Yabe, H.; Suzuki, Y.; Seto, T.; Hamazaki, T.; Mason, R.W.; Xie, L.; Gunnar, T.; Onsten, H.; Leistner-Segal, S.; et al. Hematopoietic Stem Cell Transplantation for Patients with Mucopolysaccharidosis II HHS Public Access. Biol. Blood Marrow Transpl. 2017, 23, 1795–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubot, P.; Sabourdy, F.; Plat, G.; Jubert, C.; Cancès, C.; Broué, P.; Touati, G.; Levade, T. First Report of a Patient with MPS Type VII, Due to Novel Mutations in GUSB, Who Underwent Enzyme Replacement and Then Hematopoietic Stem Cell Transplantation. Int. J. Mol. Sci. 2019, 20, 5345. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| MPS | Associated Gene | Deficient Enzyme | GAG Accumulation | The Severity of Disease | Treatment Options |
|---|---|---|---|---|---|
| MPS I-H | IDUA | α-L-iduronidase | HS, DS | Severe | ERT, HSCT |
| MPS I-S | IDUA | α-L-iduronidase | DS | Mild | ERT, HSCT |
| MPS I-H/S | IDUA | α-L-iduronidase | DS | Intermediate | ERT, HSCT |
| MPS IIA | IDS | iduronidase-2-sulfatase | HS, DS | Severe | ERT, HSCT |
| MPS IIB | IDS | iduronidase-2-sulfatase | HS, DS | Mild | ERT, HSCT |
| MPS IIIA | SGSH | heparan-N-sulfatase | HS | Variable in the severity | Symptomatic and supportive |
| MPS IIIB | NAGLU | α-N-acetylglucosaminidase | HS | Variable in the severity | Symptomatic and supportive |
| MPS IIIC | HGSNAT | α-glucosaminidase acetyltransferase | HS | Variable in the severity | Symptomatic and supportive |
| MPS IIID | GNS | N-acetylglucosamine-6-sulfatase | HS | Variable in the severity | Symptomatic and supportive |
| MPS IVA | GALNS | N-acetylglucosamine-6- sulfate sulfatase | C6S, KS | Variable in the severity | ERT, HSCT |
| MPS IVB | GLB1 | ß-galactosidase | KS | Mild | ERT, HSCT |
| MPS VI | ARSB | N-acetylglucosamine-4-sulfatase | DS, C4S | Variable in the severity | ERT, HSCT |
| MPS VII | GUSB | ß-glucuronidase | HS, DS, C4S, C6S | Variable in the severity | ERT, HSCT |
| MPS IX | HYAL1 | Hyaluronidase | Hyaluronan | Mild | Symptomatic and supportive |
| Country | Study Period | Total MPS Cases | MPS Types | Combined Prevalence | References | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III (A–D) | IV (A–B) | VI | VII | |||||
| Africa | ||||||||||
| Tunisia | 1970–2005 | 96 | 0.63 | 0.29 | 0.7 | 0.45 | 0.3 | N/A | 2.27 | [40] |
| Asia | ||||||||||
| Japan | 1982–2009 | 467 | 0.23 | 0.84 | 0.26 | 0.15 | 0.03 | 0.02 | 1.53 | [1] |
| Saudi Arabia | 1983–2008 | 28 | 3.62 | N/A | 1.8 | 3.62 | 7.85 | N/A | 16.9 | [36] |
| South Korea | 1994–2013 | 147 | 0.21 | 0.74 | 0.25 | 0.13 | 0.019 | N/A | 1.35 | [37] |
| Taiwan | 1984–2004 | 130 | 0.11 | 1.07 | 0.39 | 0.33 | 0.14 | N/A | 2.04 | [23] |
| Israel | 1967–1975 | 8 | N/A | 1.48 | N/A | N/A | N/A | N/A | N/A | [20] |
| Australia | ||||||||||
| Australia | 1980–1996 | 188 | 1.14 | 0.74 | 1.51 | 0.59 | 0.43 | 0.047 | 4.46 | [20] |
| Western Australia | 1969–1996 | 18 | 0.94 | 0.31 | 1.71 | 0.16 | 0.31 | N/A | 3.43 | [41] |
| Europe | ||||||||||
| Belarus | 1983–2011 | 13 | N/A | N/A | N/A | N/A | 0.37 | N/A | N/A | [17] |
| Czech Republic | 1975–2008 | 119 | 0.72 | 0.43 | 0.91 | 0.73 | 0.05 | 0.02 | 3.72 | [45] |
| Denmark | 1975–2004 | 33 | 0.54 | 0.27 | 0.43 | 0.48 | 0.05 | N/A | 1.77 | [46] |
| Estonia | 1985–2006 | 15 | N/A | 2.16 | 1.62 | N/A | 0.27 | N/A | 4.05 | [47] |
| Estonia | 1983–2011 | 2 | N/A | N/A | N/A | N/A | 0.4 | N/A | N/A | [17] |
| France | 1990–2006 | 128 | N/A | N/A | 0.73 | N/A | N/A | N/A | N/A | [162] |
| Germany | 1980–1995 | 474 | 0.69 | 0.64 | 1.57 | 0.38 | 0.23 | N/A | 3.51 | [48] |
| Greece | 1990–2006 | 20 | N/A | N/A | 0.97 | N/A | N/A | N/A | N/A | [162] |
| Lithuania | 1983–2011 | 8 | N/A | N/A | N/A | N/A | 0.64 | N/A | N/A | [17] |
| Northern Ireland | 1958–1985 | 34 | 1.66 | 0.71 | 0.36 | 1.3 | N/A | N/A | 4 | [49] |
| Norway | 1979–2004 | 45 | 1.85 | 0.13 | 0.27 | 0.76 | 0.07 | N/A | 3.08 | [46] |
| Poland | 1970–2010 | 392 | 0.22 | 0.46 | 0.86 | 0.14 | 0.0132 | N/A | 1.8 | [50] |
| Poland | 1983–2011 | 5 | N/A | N/A | N/A | N/A | 0.036 | N/A | N/A | [17] |
| Portugal | 1982–2001 | 353 | 1.33 | 1.09 | 0.84 | 0.6 | 0.42 | N/A | 4.8 | [163] |
| Sweden | 1975–2004 | 52 | 0.67 | 0.27 | 0.67 | 0.07 | 0.07 | N/A | 1.75 | [46] |
| Switzerland | 1975–2008 | 41 | 0.19 | 0.46 | 0.38 | 0.38 | 0.11 | 0.038 | 1.56 | [1] |
| The Netherlands | 1970–1996 | 331 | 1.19 | 0.67 | 1.89 | 0.36 | 0.15 | 0.24 | 4.5 | [21] |
| The United Kingdom | 1981–2003 | 167 | 1.07 | N/A | N/A | N/A | N/A | N/A | N/A | [22] |
| The United Kingdom | 1990–2006 | 126 | N/A | N/A | 1.21 | N/A | N/A | N/A | N/A | [162] |
| North America | ||||||||||
| British Columbia | 1952–1986 | N/A | 0.69 | 0.9 | 0.3 | 0.46 | N/A | N/A | N/A | [42] |
| British Columbia | 1969–1996 | 20 | 0.58 | 0.1 | 0.29 | 0.39 | 0.29 | 0.29 | 1.94 | [43] |
| The United States | 1995–2005 | N/A | 0.34 | 0.29 | 0.38 | 0.09 | 0.05 | 0.05 | 1.2 | [53] |
| Mexico | 2012–2017 | 198 | 0.19 | 0.15 | 0.17 | 1.1 | 0.17 | 0.23 | 2.23 | [30] |
| South America | ||||||||||
| Brazil | 1994–2012 | 600 | 0.24 | 0.38 | N/A | 0.11 | 0.31 | N/A | 1.04 | [164] |
| Brazil | 1994–2015 | 823 | 0.24 | 0.37 | 0.21 | 0.14 | 0.28 | 0.02 | 1.25 | [6] |
| Brazil | 1982–2019 | 1652 | 0.29 | 0.48 | 0.06 | 0.07 | 0.35 | 0.02 | 1.57 | [25] |
| Colombian province (Cundinamarca and Boyacá)–Colombia | 1998–2007 | 35 | 0.45 | 0.45 | 0.17 | 0.68 | 0.23 | N/A | 1.98 | [165] |
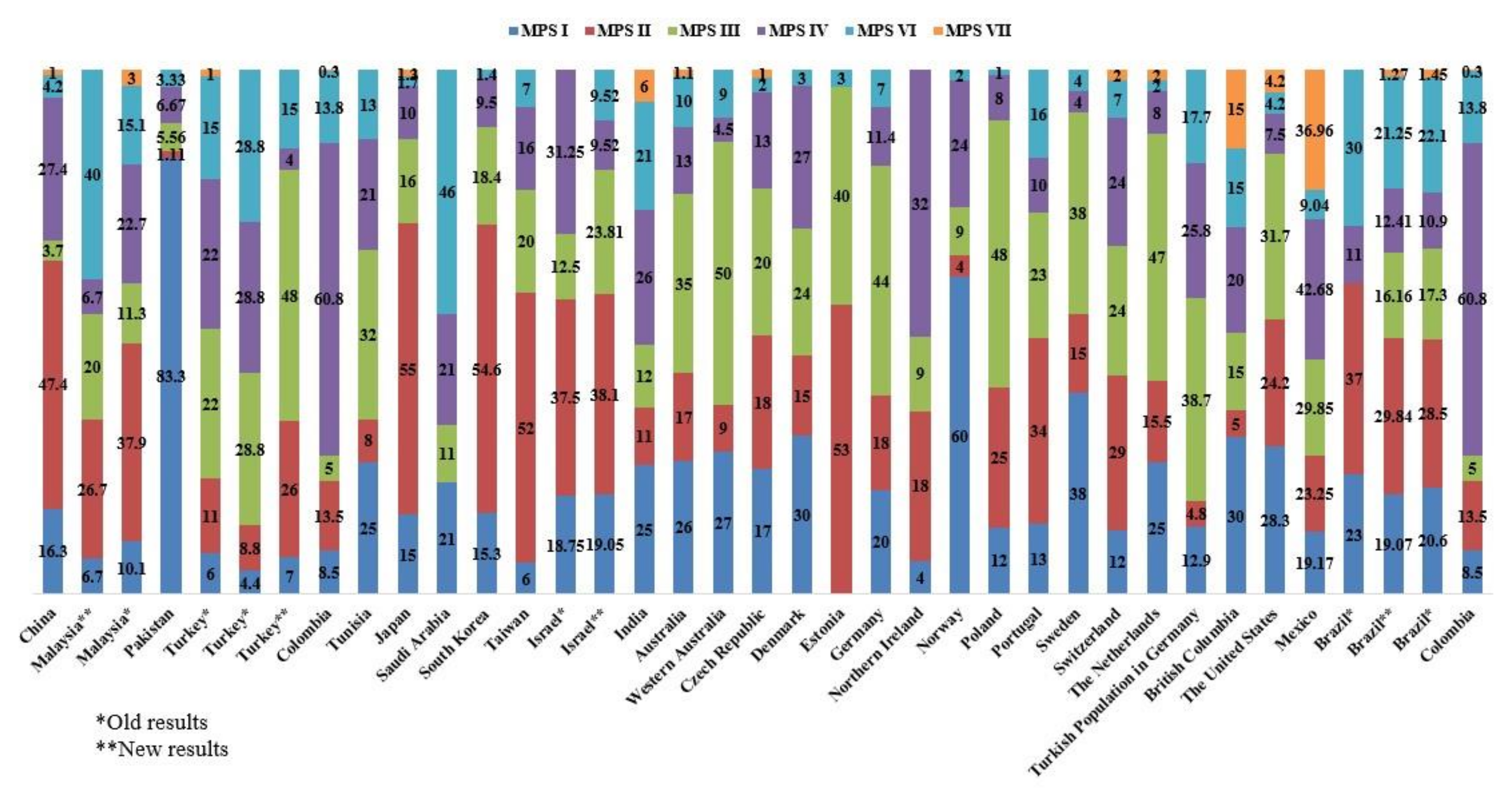
| Study Period | Total MPS Cases | MPS I | MPS II | MPS III | MPS IV | MPS VI | MPS VII | References | |
|---|---|---|---|---|---|---|---|---|---|
| China | 2006–2012 | 190 | 16.3 | 47.4 | 3.7 | 27.4 | 4.2 | 1 | [38] |
| Malaysia | 2014–2016 | 15 | 6.7 | 26.7 | 20 | 6.7 | 40 | N/A | [29] |
| Malaysia | N/A | 79 | 10.1 | 37.9 | 11.3 | 22.7 | 15.1 | 3 | [28] |
| Pakistan | 2013–2015 | 90 | 83.3 | 1.11 | 5.56 | 6.67 | 3.33 | N/A | [17] |
| Turkey | 2009–2011 | 339 | 6 | 11 | 22 | 22 | 15 | 1 | [35] |
| Turkey | N/A | 45 | 4.4 | 8.8 | 28.8 | 28.8 | 28.8 | N/A | [34] |
| Turkey | N/A | 27 | 7 | 26 | 48 | 4 | 15 | N/A | [33] |
| Tunisia | 1970–2005 | 96 | 25 | 8 | 32 | 21 | 13 | N/A | [40] |
| Japan | 1982–2009 | 467 | 15 | 55 | 16 | 10 | 1.7 | 1.3 | [1] |
| Saudi Arabia | 1983–2008 | 28 | 21 | N/A | 11 | 21 | 46 | N/A | [36] |
| South Korea | 1994–2013 | 147 | 15.3 | 54.6 | 18.4 | 9.5 | 1.4 | N/A | [37] |
| Taiwan | 1984–2004 | 130 | 6 | 52 | 20 | 16 | 7 | N/A | [23] |
| Israel | 1973 | 16 | 18.75 | 37.5 | 12.5 | 31.25 | N/A | N/A | [20] |
| Israel | 1974–1979 | 21 | 19.05 | 38.1 | 23.81 | 9.52 | 9.52 | N/A | [20] |
| India | 2002–2012 | 85 | 25 | 11 | 12 | 26 | 21 | 6 | [39] |
| Australia | 1980–1996 | 188 | 26 | 17 | 35 | 13 | 10 | 1.1 | [20] |
| Western Australia | 1969–1996 | 18 | 27 | 9 | 50 | 4.5 | 9 | N/A | [41] |
| Czech Republic | 1975–2008 | 119 | 17 | 18 | 20 | 13 | 2 | 1 | [45] |
| Denmark | 1975–2004 | 33 | 30 | 15 | 24 | 27 | 3 | N/A | [46] |
| Estonia | 1985–2006 | 15 | N/A | 53 | 40 | N/A | 3 | N/A | [47] |
| Germany | 1980–1995 | 474 | 20 | 18 | 44 | 11.4 | 7 | N/A | [48] |
| Northern Ireland | 1958–1985 | 34 | 4 | 18 | 9 | 32 | N/A | N/A | [49] |
| Norway | 1979–2004 | 45 | 60 | 4 | 9 | 24 | 2 | N/A | [46] |
| Poland | 1970–2010 | 392 | 12 | 25 | 48 | 8 | 1 | N/A | [50] |
| Portugal | 1982–2001 | 353 | 13 | 34 | 23 | 10 | 16 | N/A | [19] |
| Sweden | 1975–2004 | 52 | 38 | 15 | 38 | 4 | 4 | N/A | [46] |
| Switzerland | 1975–2008 | 41 | 12 | 29 | 24 | 24 | 7 | 2 | [1] |
| The Netherlands | 1970–1996 | 331 | 25 | 15.5 | 47 | 8 | 2 | 2 | [88] |
| Turkish Population in Germany | 1980–1995 | 62 | 12.9 | 4.8 | 38.7 | 25.8 | 17.7 | N/A | [48] |
| British Columbia | 1969–1996 | 20 | 30 | 5 | 15 | 20 | 15 | 15 | [43] |
| The United States | 1995–2005 | N/A | 28.3 | 24.2 | 31.7 | 7.5 | 4.2 | 4.2 | [53] |
| Mexico | 2012–2017 | 198 | 19.17 | 23.25 | 29.85 | 42.68 | 9.04 | 36.96 | [30] |
| Brazil | 1994–2012 | 600 | 23 | 37 | N/A | 11 | 30 | N/A | [164] |
| Brazil | 1994–2015 | 823 | 20.6 | 28.5 | 17.3 | 10.9 | 22.1 | 1.45 | [6] |
| Brazil | 1982–2019 | 1652 | 19.07 | 29.84 | 16.16 | 12.41 | 21.25 | 1.27 | [25] |
| Colombia | 1995–2016 | 319 | 8.5 | 13.5 | 5 | 60.8 | 13.8 | 0.3 | [26] |
| Country | Years | Number of DBS Screened | Positive Screened Result | Confirmation Method | Positive | Carrier | Pseudodeficiency | Frequency per 100,000 Birth | References | |
|---|---|---|---|---|---|---|---|---|---|---|
| MPS I | Belgium | 2015–2016 | 20,000 | 54 | Liquid chromatography-tandem mass spectrometry | NA | NA | NA | NA | [166] |
| Brazil | No date | 10,567 | 2 | DNA sequence analysis | 0 | 1 | 1 | 0 | [167] | |
| Italy—Umbria | No date | 3403 | 13 | Enzyme assay | 3 | NA | NA | 0 | [168] | |
| North East Italy | 2015-2017 | 44,411 | 13 | Tandem mass spectrometry | 1 | 2 | 5 | 1/44,411 | [169] | |
| Japan | 2012–2015 | 18,222 | 300 | Liquid chromatography-tandem mass spectrometry | 0 | 0 | 0 | 0 | [157] | |
| Mexico | 2012–2016 | 20,018 | 72 | Leukocyte enzyme activity and DNA sequence analysis | 2 | 0 | NA | 9.99 | [161] | |
| Taiwan | 2008–2013 | 35,285 | 58 | DNA sequence analysis | 2 | 1 | NA | 5.67 | [170] | |
| Taiwan | 2012–2013 | 60,473 | 61 | DNA sequence analysis | 0 | NA | NA | 0 | [171] | |
| Taiwan | 2015–2017 | 294,196 | 84 | DNA sequence analysis | 4 | 0 | 0 | 1.35 | [172] | |
| Taiwan | 2015–2017 | 130,237 | 120 | DNA sequence analysis | 5 | 0 | 0 | 3.8 | [173] | |
| Taiwan | 2018–2019 | 73,743 | 178 | UPLC-MS/MS | 1 | NA | NA | 1/73,743 | [174] | |
| USA—Washington | No date | 43,000 | NA | Mass spectrometry and fluorometric assay | 6 | NA | NA | 13.95 | [175] | |
| USA—Washington | No Date | 106,526 | 9 | Tandem mass spectrometry | 3 | 1 | NA | 8.44 (1/35,700) | [52] | |
| USA—Kentucky | 2016–2017 | 55,161 | 76 | Tandem mass spectrometry | 1 | NA | NA | 1.81 | [176] | |
| USA—Illinois | 2014–2016 | 219,973 | 151 | Tandem mass spectrometry | 1 | 5 | 30 | 1/219,973 | [159] | |
| USA—Missouri | 2013–2014 | 174,636 | 70 | Tandem mass spectrometry | 1 | 3 | 25 | 1/174,636 | [51] | |
| USA—Missouri | 2013–2017 | 308 | 133 | Enzyme assay | 2 | NA | 71 | 0.64 | [177] | |
| USA—Missouri | January–June 2013 | 43,701 | 32 | Enzyme assay | 1 | 2 | 7 | 2.28 | [178] | |
| USA—New York | 2015 | 35,816 | 13 | Liquid chromatography-tandem mass spectrometry and DNA sequence analysis | 0 | 4 | 8 | 0 | [160] | |
| USA—New York | No date | 43,000 | 6 | Mass spectrometry and fluorometric assay | NA | 2 | NA | 13.6 | [175] | |
| MPS II | USA—Illinois | 2014 | 93,219 | 9 | Tandem mass spectrometry | 1 | 3 | 5 | NA | [179] |
| USA—Washington | No date | 105,214 | 25 | Liquid chromatography-tandem mass spectrometry and DNA sequence analysis | 1 | 2 | NA | NA | [180] | |
| Taiwan | 2015–2017 | 294,196 | 84 | Leukocyte enzyme assay and DNA sequence analysis | 3 | 0 | 0 | 1.96 | [172] | |
| Taiwan | 2018–2019 | 73,743 | 56 | UPLC-MS/MS | 3 | NA | NA | 1/24,581 | [174] | |
| Taiwan | August to December 2015 | 28,799 | 53 | DNA sequence analysis | 0 | NA | NA | NA | [181] | |
| January 2016–August 2017 | 101,376 | 184 | DNA sequence analysis | 0 | NA | 38 | [181] | |||
| MPS IIIB | USA—Washington | No date | 103,001 | 0 | Liquid chromatography-tandem mass spectrometry and DNA sequence analysis | 0 | 0 | 0 | 0 | [180] |
| Taiwan | 2018–2019 | 73,743 | 14 | UPLC-MS/MS | 3 | NA | NA | 1/24,581 | [174] | |
| MPS IVA | USA—Washington | No date | 106,106 | 8 | Liquid chromatography-tandem mass spectrometry and DNA sequence analysis | 0 | 0 | 0 | 0 | [180] |
| Taiwan | 2018–2019 | 73,743 | 70 | UPLC-MS/MS | 6 | NA | NA | 1/12,291 | [174] | |
| MPS VI | USA—Washington | No date | 103,259 | 4 | Liquid chromatography-tandem mass spectrometry and DNA sequence analysis | 0 | 0 | 0 | 0 | [180] |
| Taiwan | August to December 2015 | 131,075 | 176 | DNA sequence analysis | 0 | NA | NA | 0 | [173] | |
| Taiwan | 2018–2019 | 73,743 | 11 | UPLC-MS/MS | 0 | NA | NA | NA | [174] | |
| MPS VII | USA—Washington | No date | 94,931 | 1 | Liquid chromatography-tandem mass spectrometry and DNA sequence analysis | 0 | 0 | 0 | 0 | [180] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Celik, B.; Tomatsu, S.C.; Tomatsu, S.; Khan, S.A. Epidemiology of Mucopolysaccharidoses Update. Diagnostics 2021, 11, 273. https://doi.org/10.3390/diagnostics11020273
Celik B, Tomatsu SC, Tomatsu S, Khan SA. Epidemiology of Mucopolysaccharidoses Update. Diagnostics. 2021; 11(2):273. https://doi.org/10.3390/diagnostics11020273
Chicago/Turabian StyleCelik, Betul, Saori C. Tomatsu, Shunji Tomatsu, and Shaukat A. Khan. 2021. "Epidemiology of Mucopolysaccharidoses Update" Diagnostics 11, no. 2: 273. https://doi.org/10.3390/diagnostics11020273