
Stereoselective Syntheses of Organophosphorus Compounds

Abstract
:1. Introduction
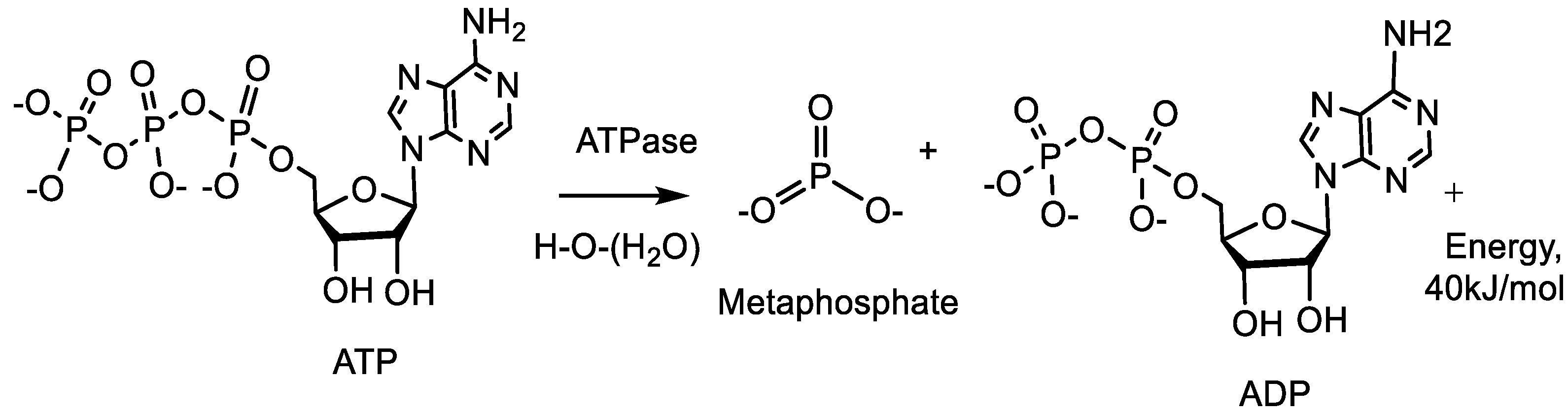
2. Stereochemistry of Organophosphorus Compounds
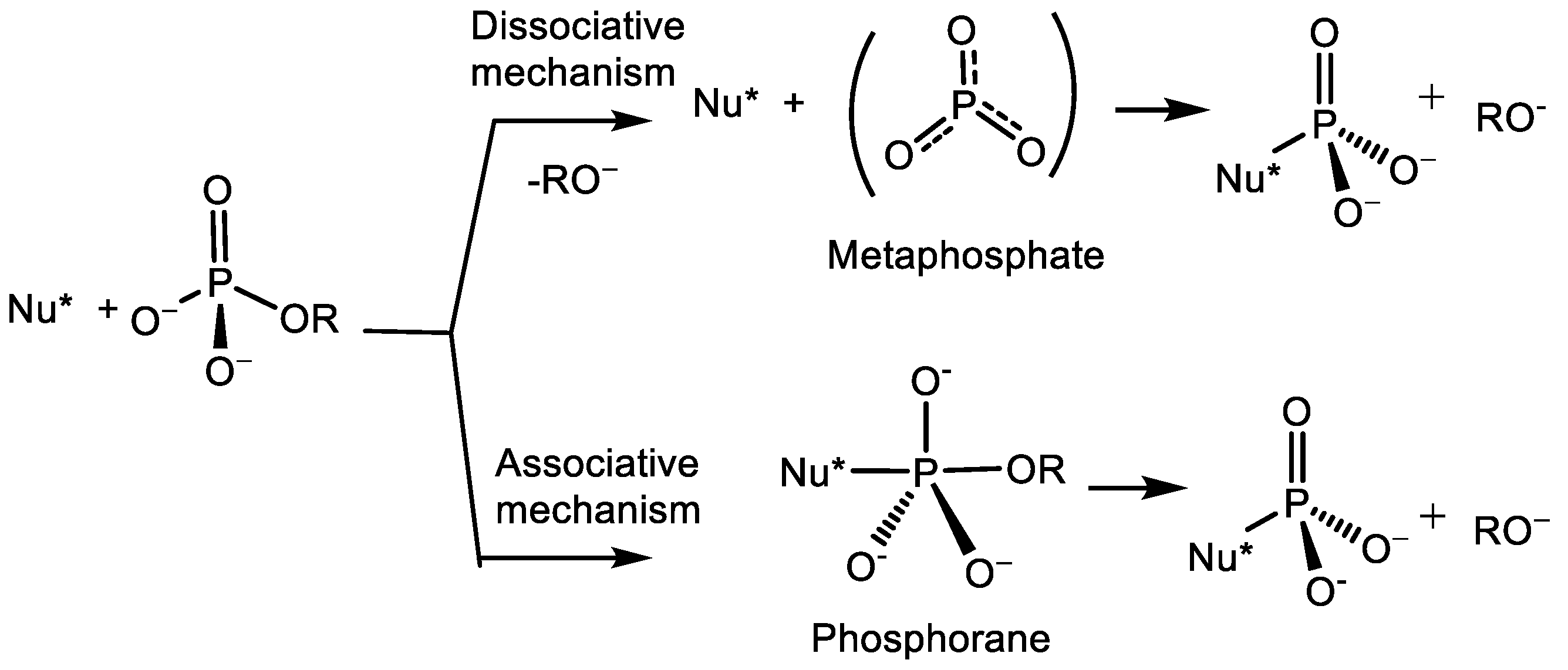
- (a)
- dissociative SN1-type mechanism that consider the formation of a stable metaphosphate ion (PO3), which is attacked by a nucleophile in the subsequent, rate limiting step of reaction;
- (b)
- associative, two-step addition-elimination mechanism through the formation of a phosphorane intermediate.
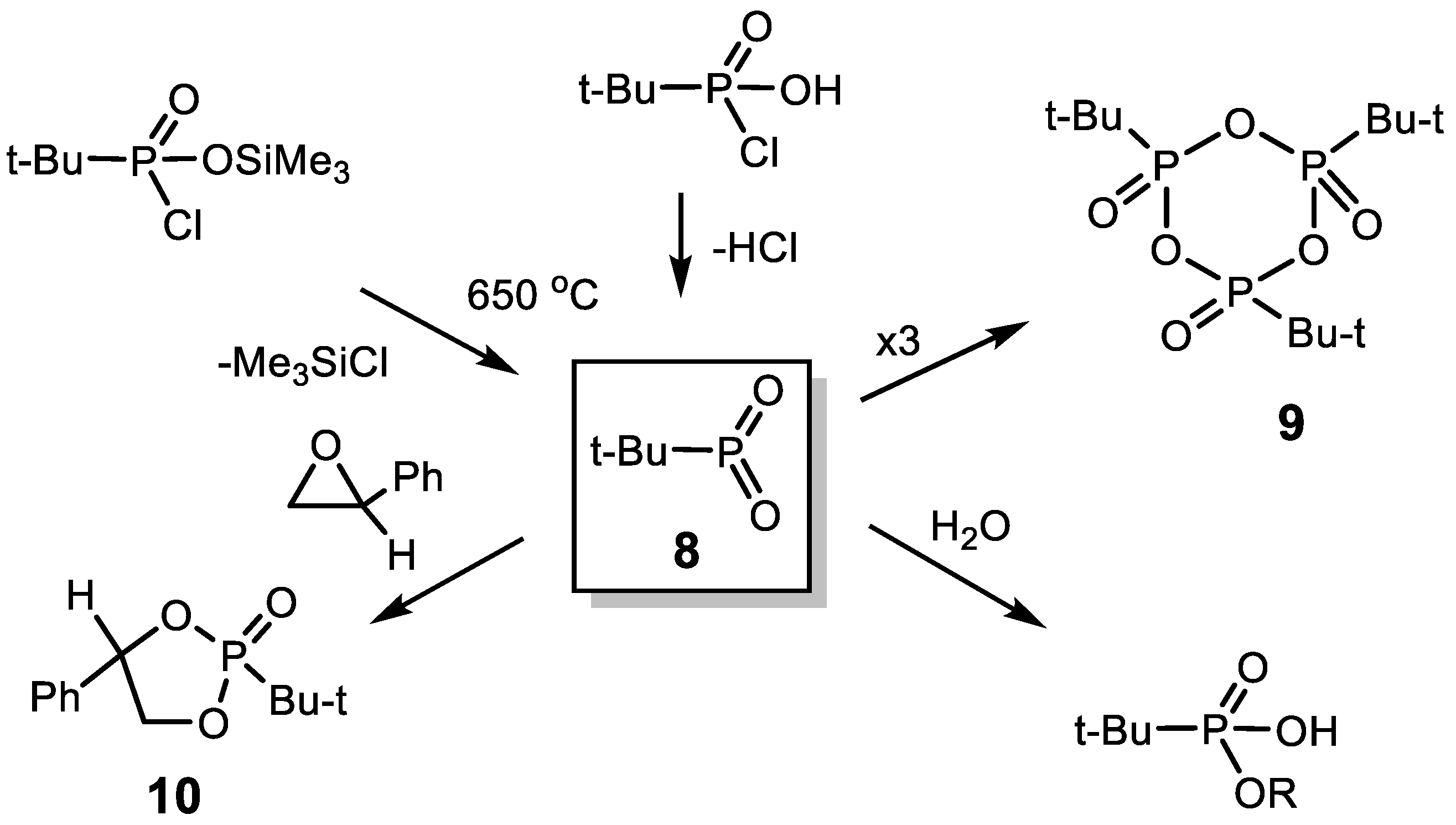
- (1)
- Dioxaphosphorane 8 readily polymerizes to the trimer tert-butyldioxophosphorane 9, which was isolated and its structure confirmed by spectroscopy and elemental analysis.
- (2)
- Compound 8 reacts with styrene oxide to form a [2+3]-cycloaddition product 9, which is a five-membered phosphorus heterocycle, 2-tert-butyl-2-phenyl-1,3,2-dioxophospholane 10, which was obtained as a mixture two diastereomers in a ratio of 1:2 and purified by distillation under reduced pressure.
- (3)
3. Methods for the Synthesis of Chiral Organophosphorus Compounds
3.1. Asymmetric Catalysis
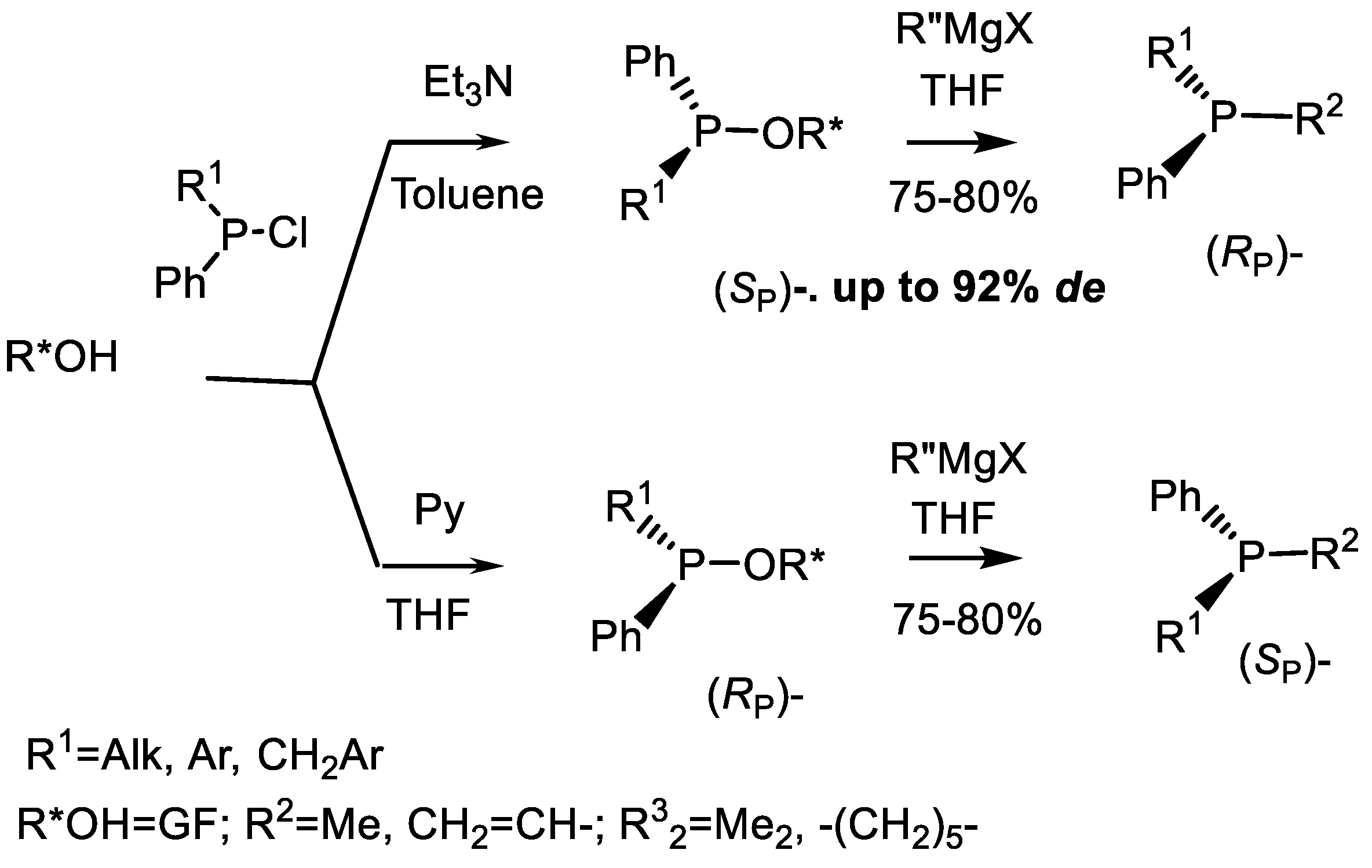
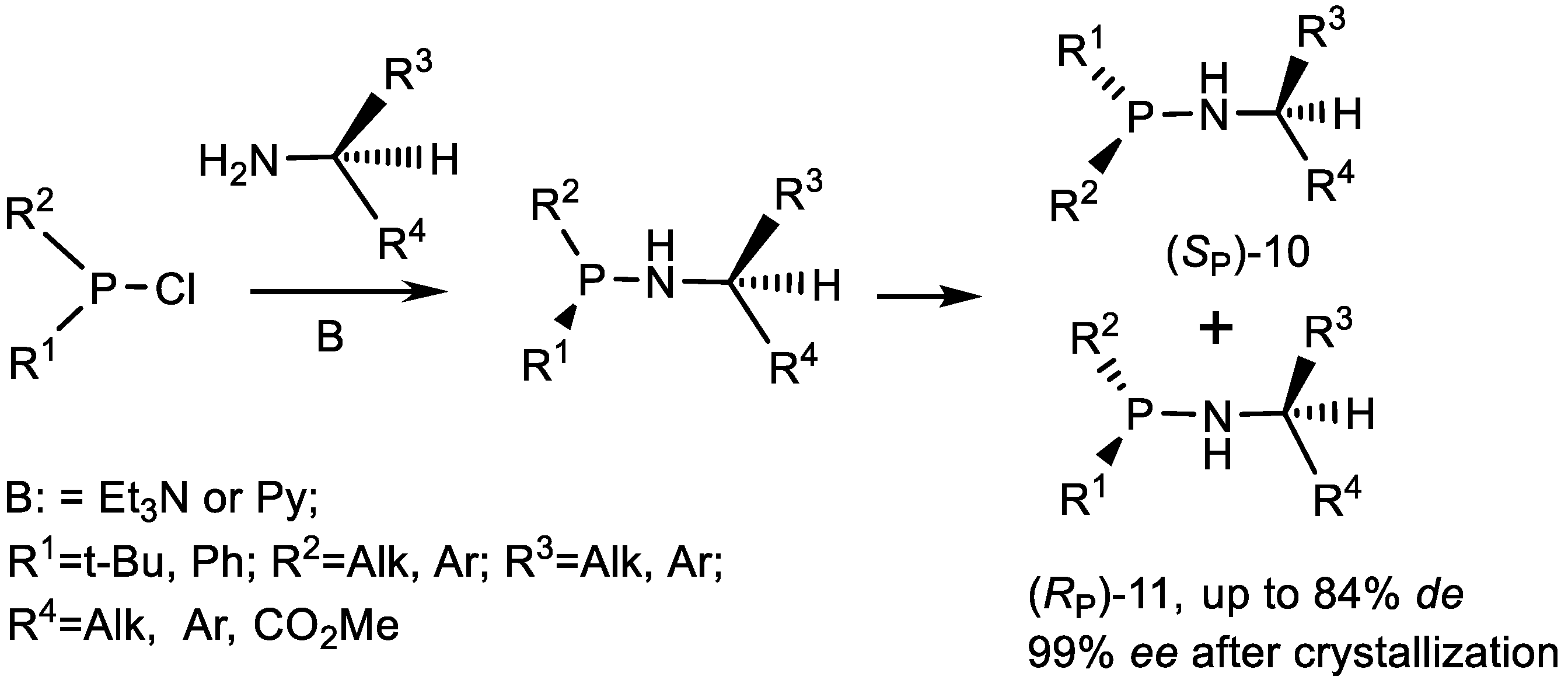
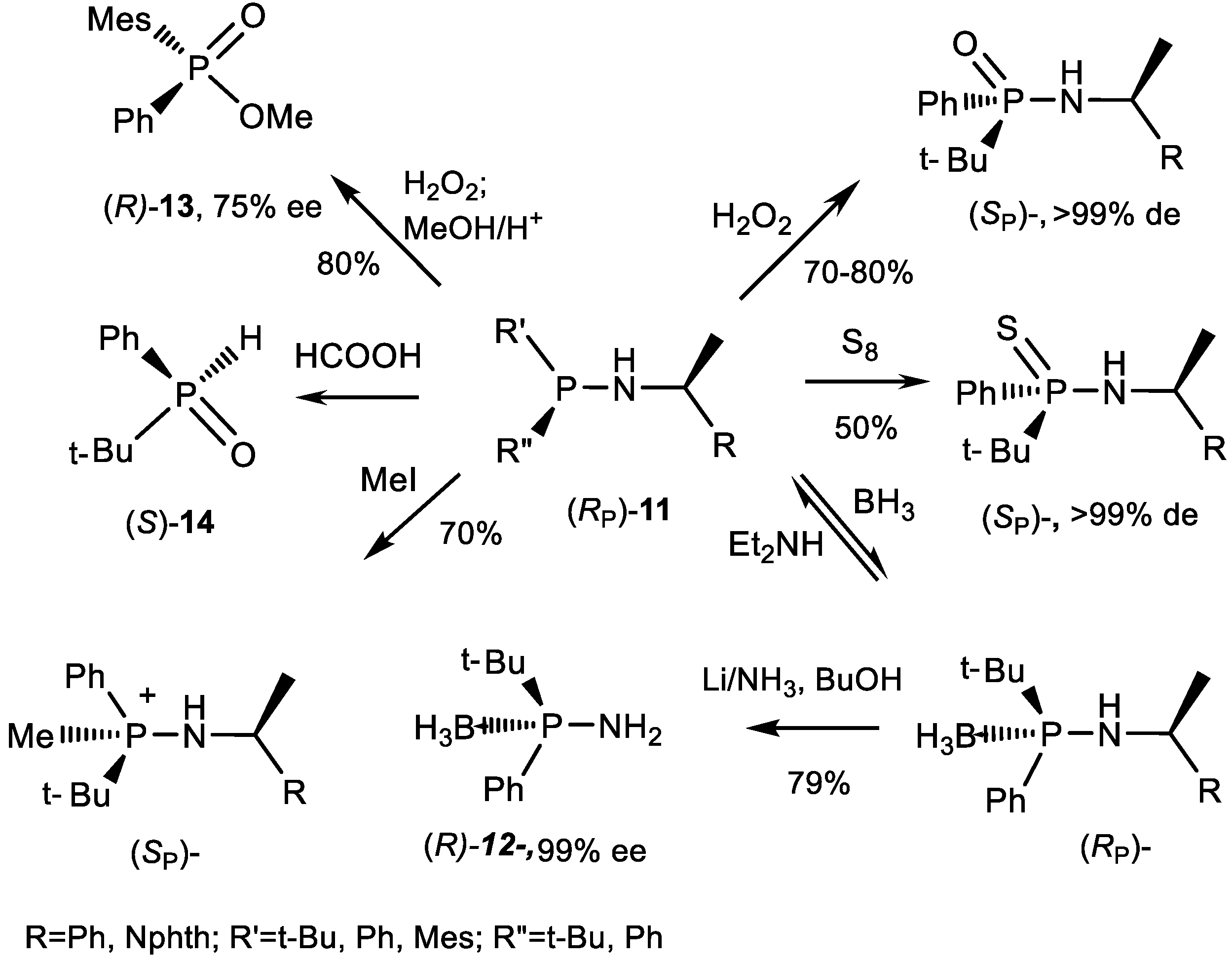
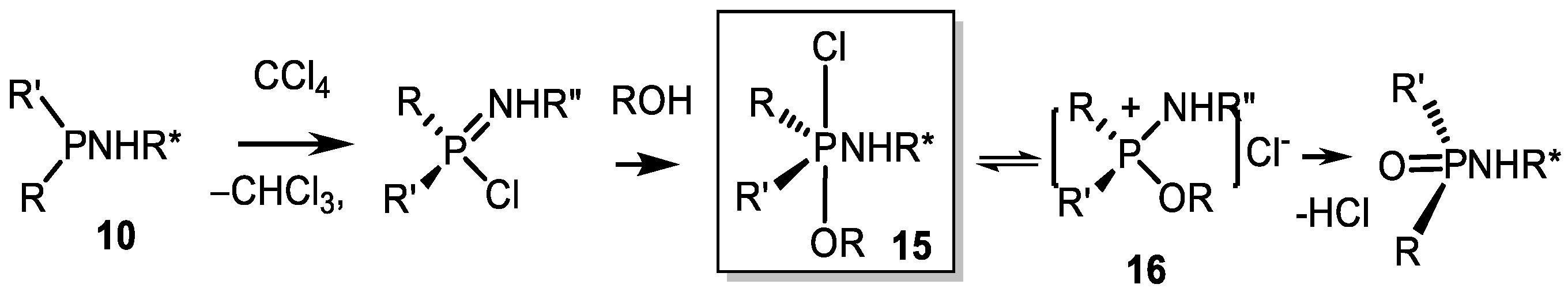
3.2. Diastereoselective Substitution Reactions
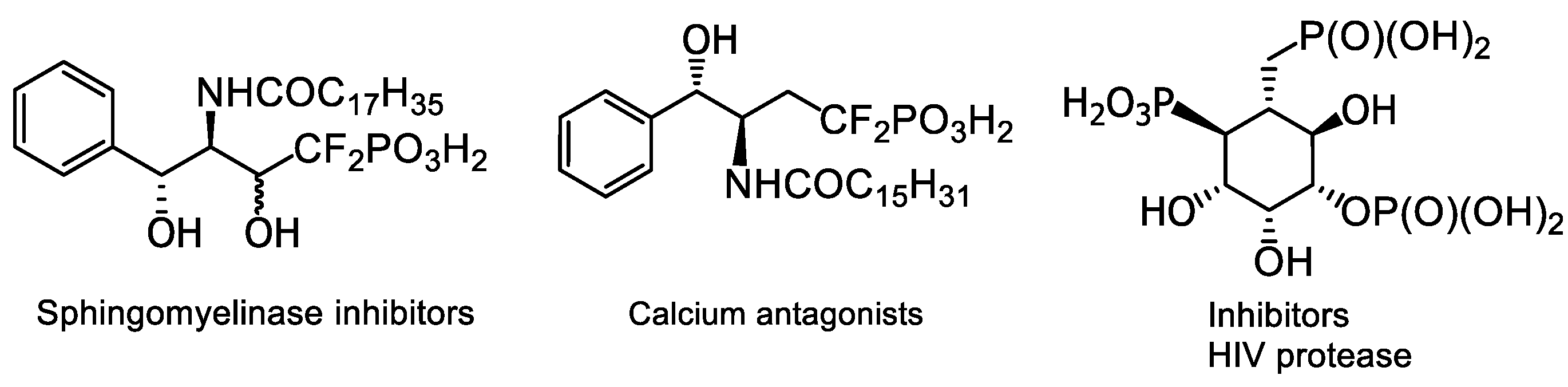
4. Phosphorus Analogues of Natural Compounds
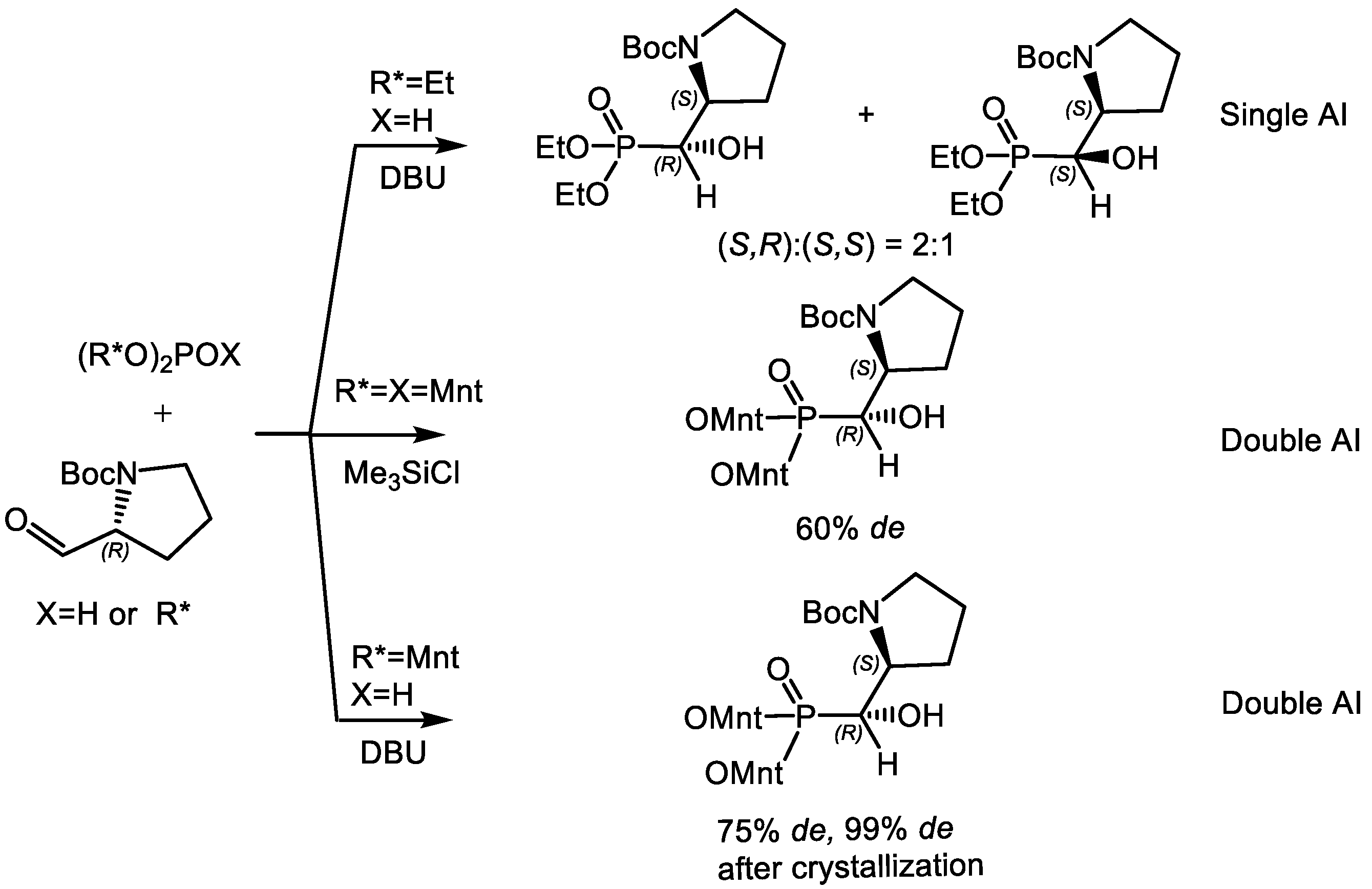
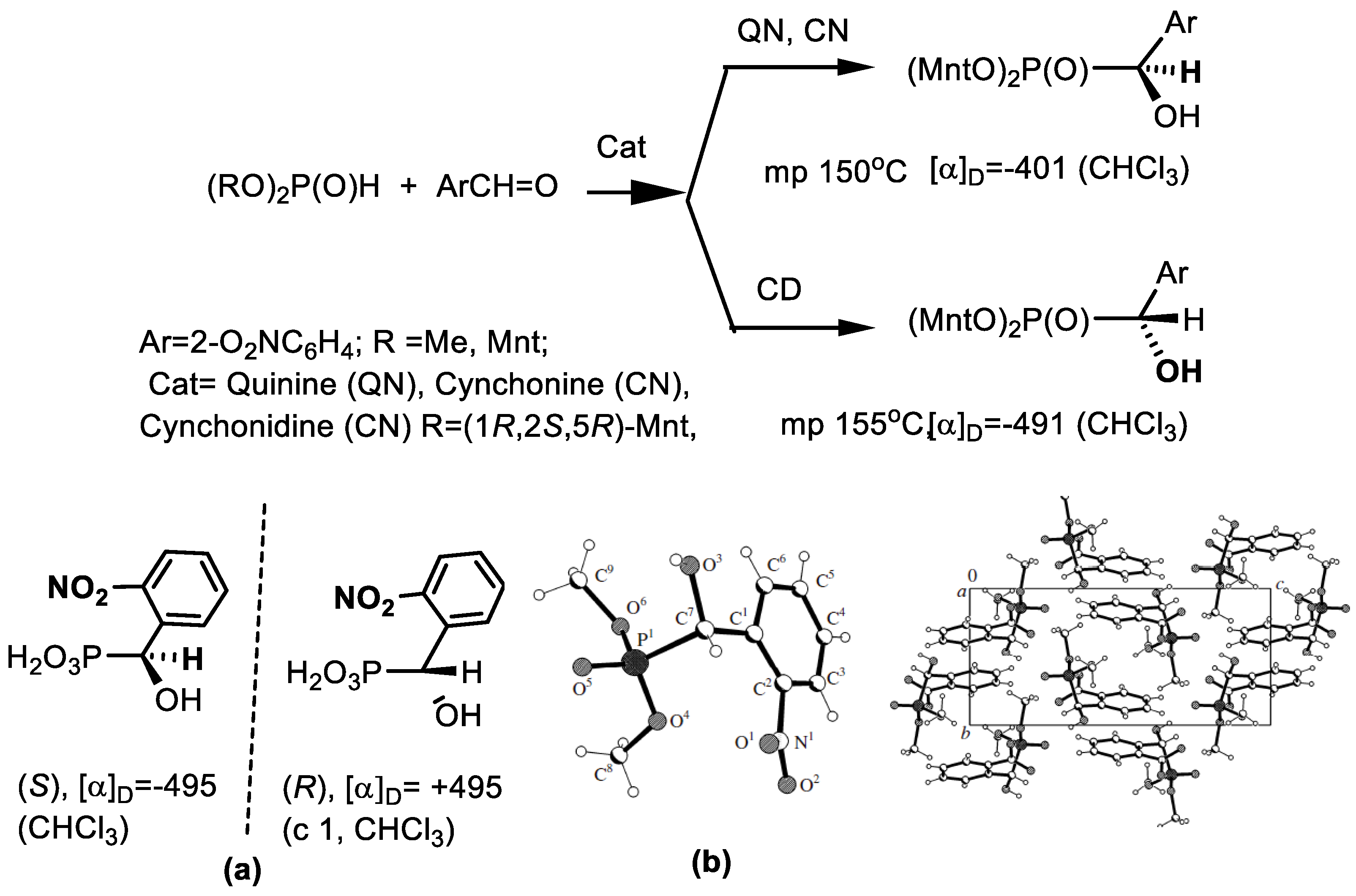
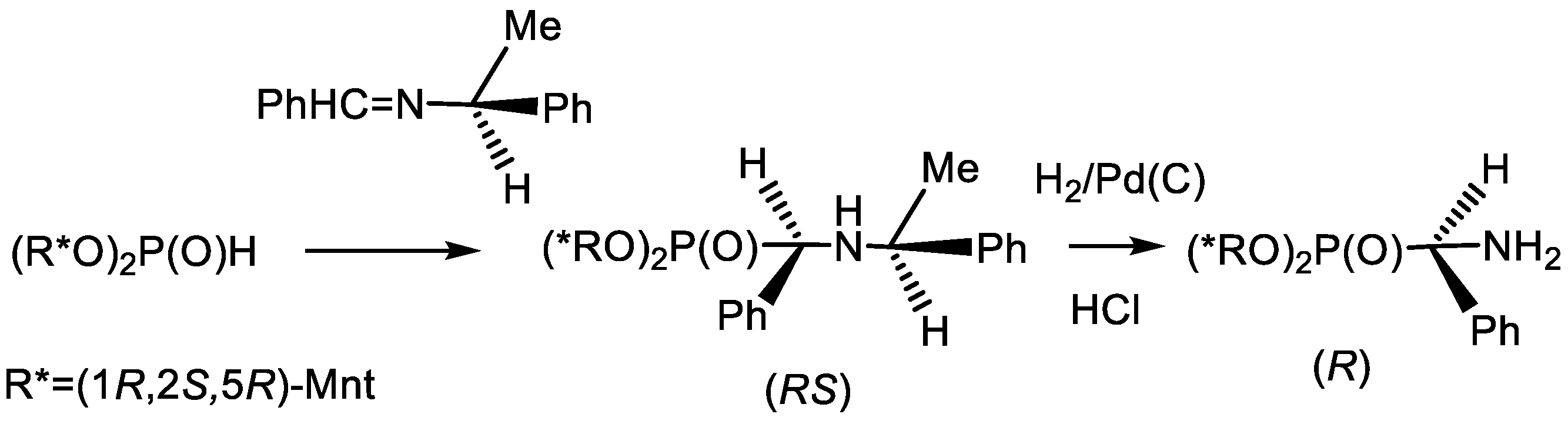
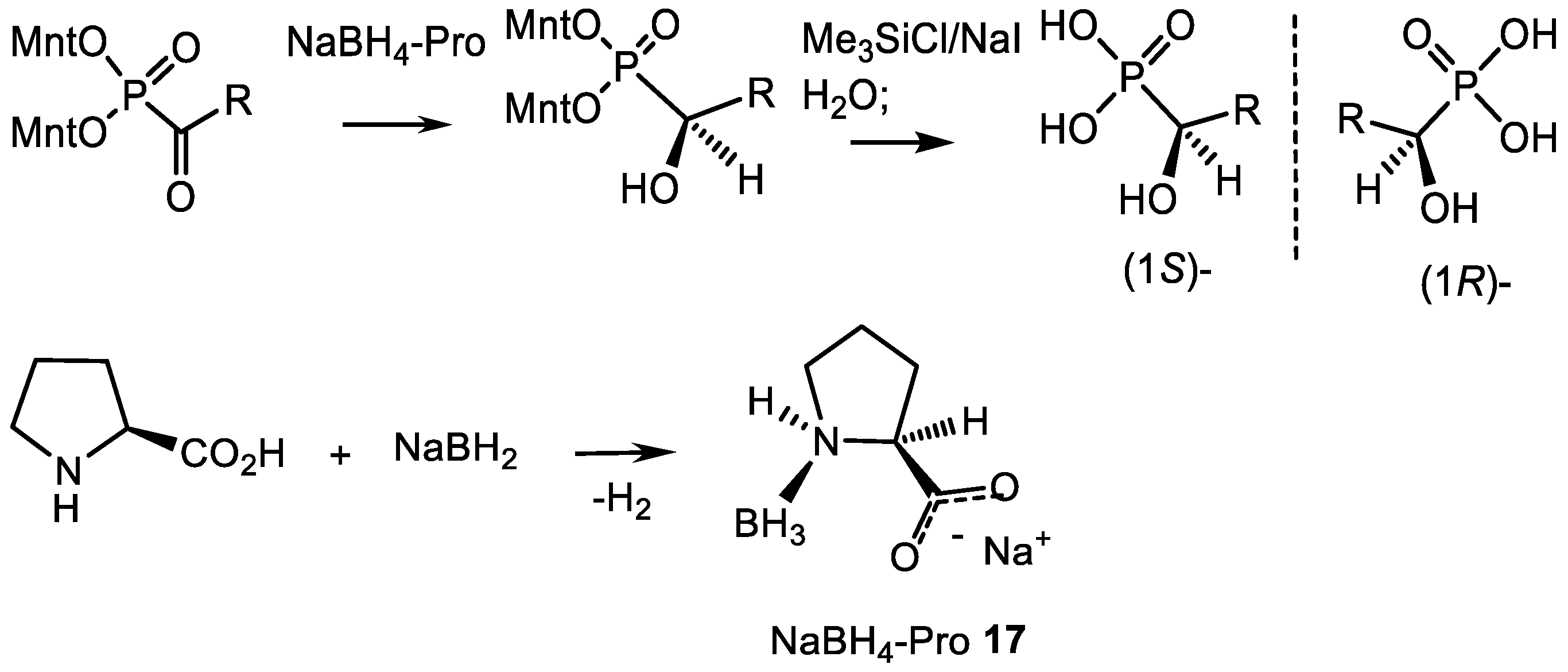
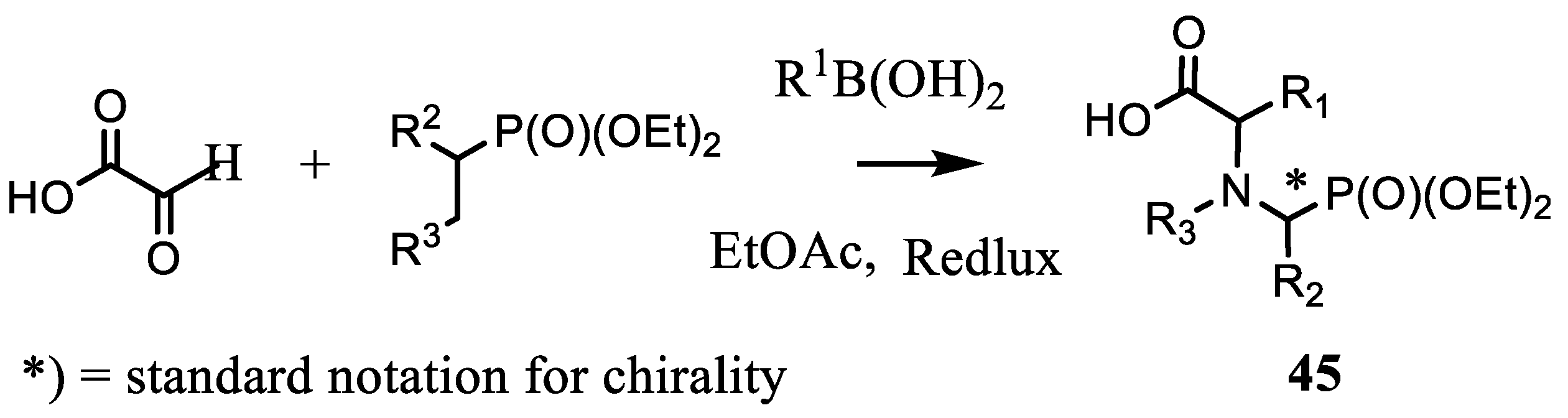
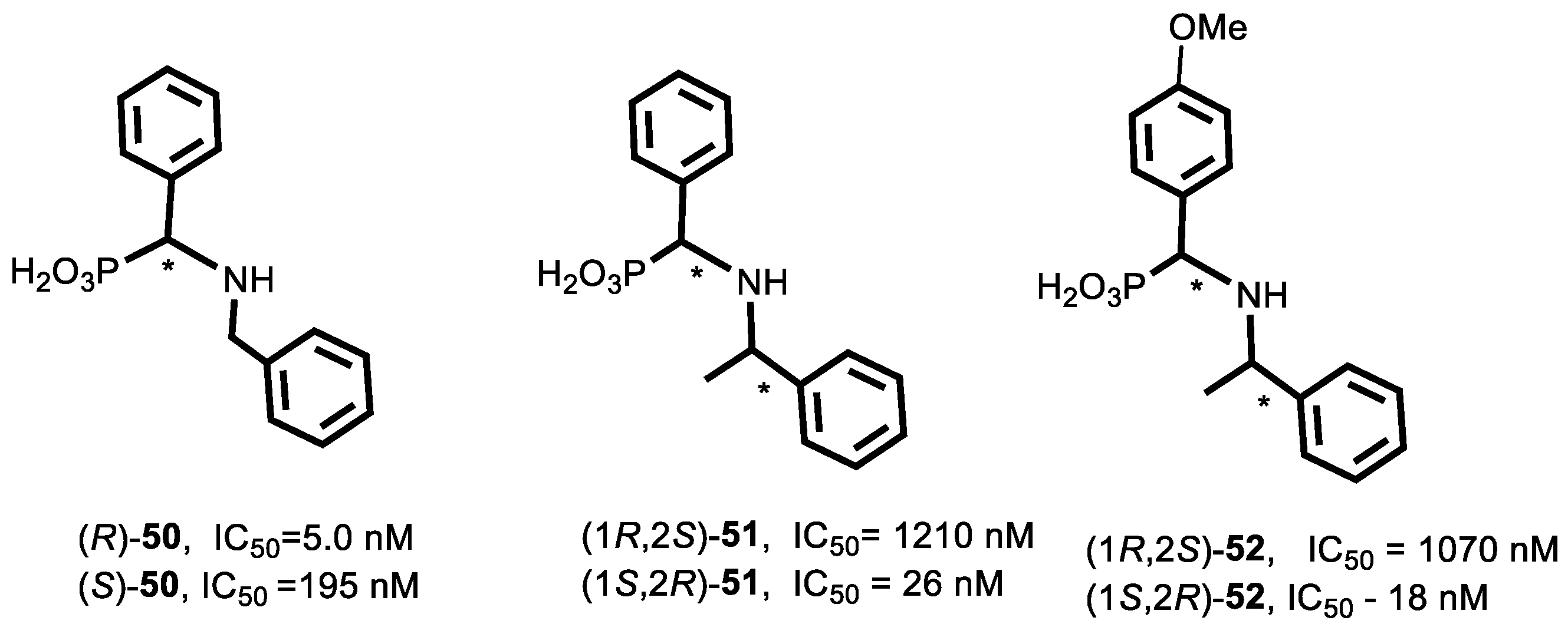
4.1. Synthesis of Phosphonic Acids Using Natural Amino Acids as Reagents
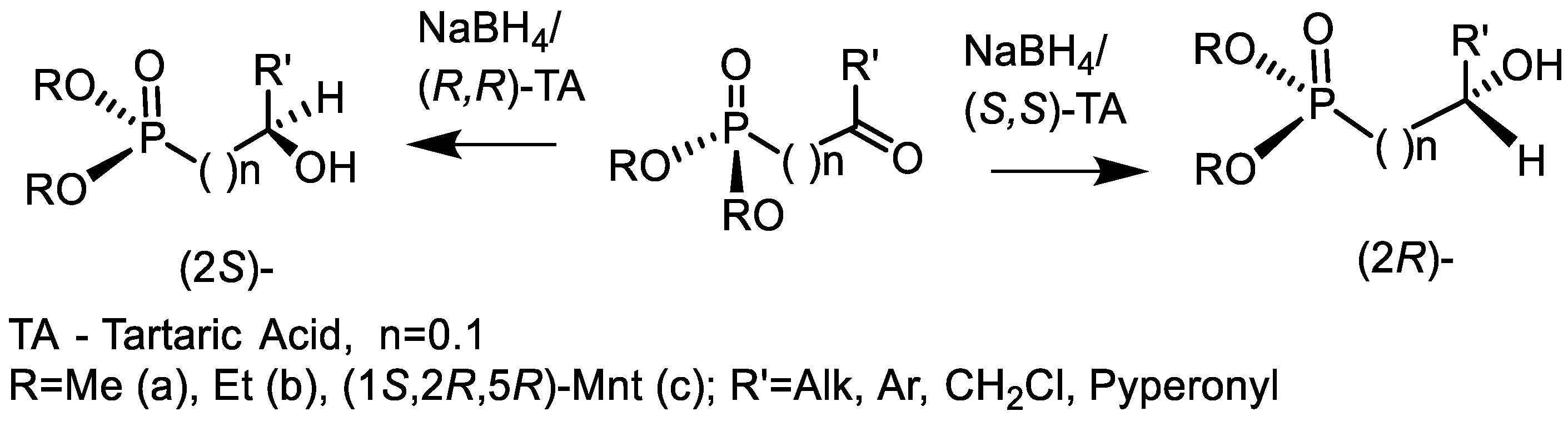
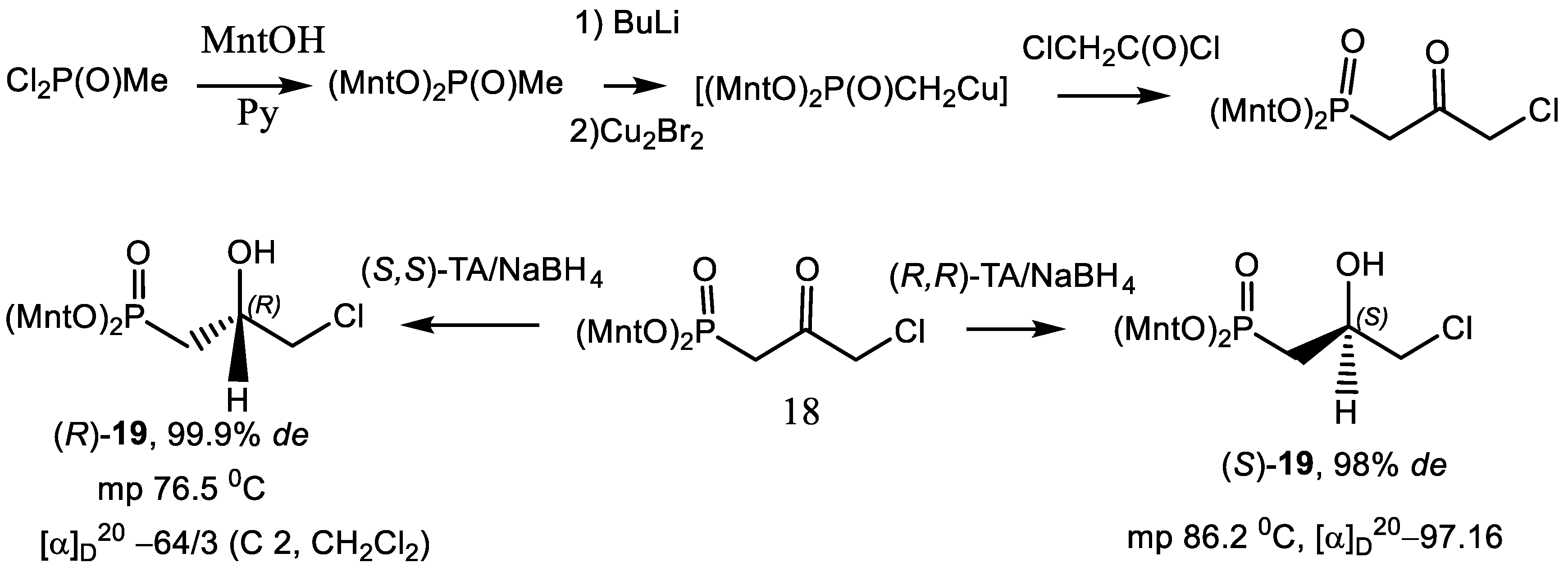
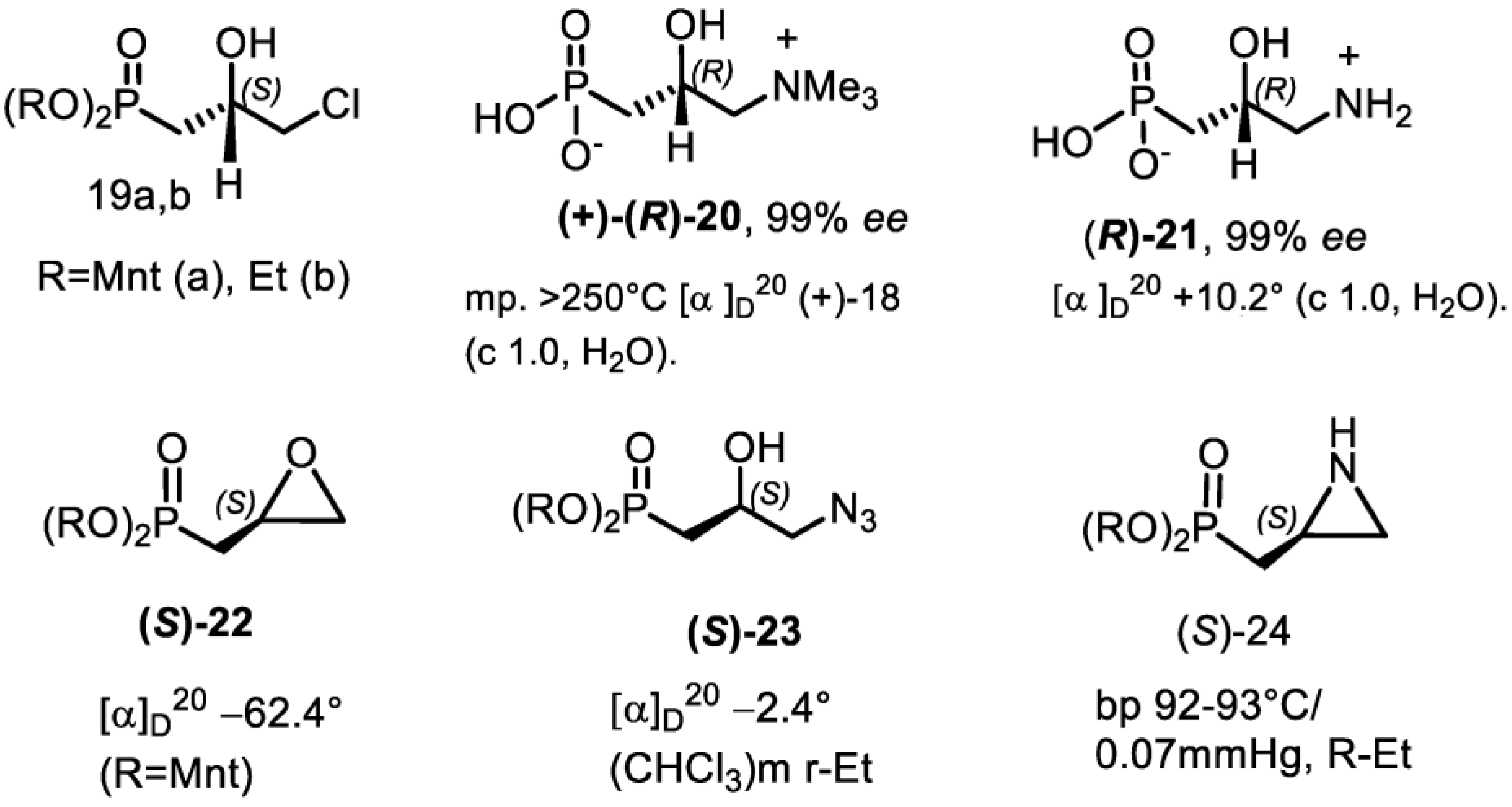
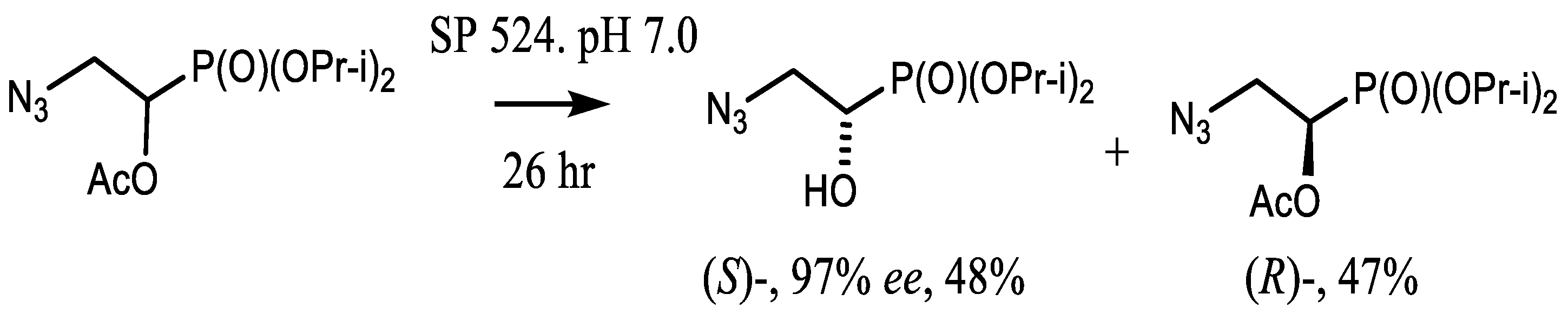
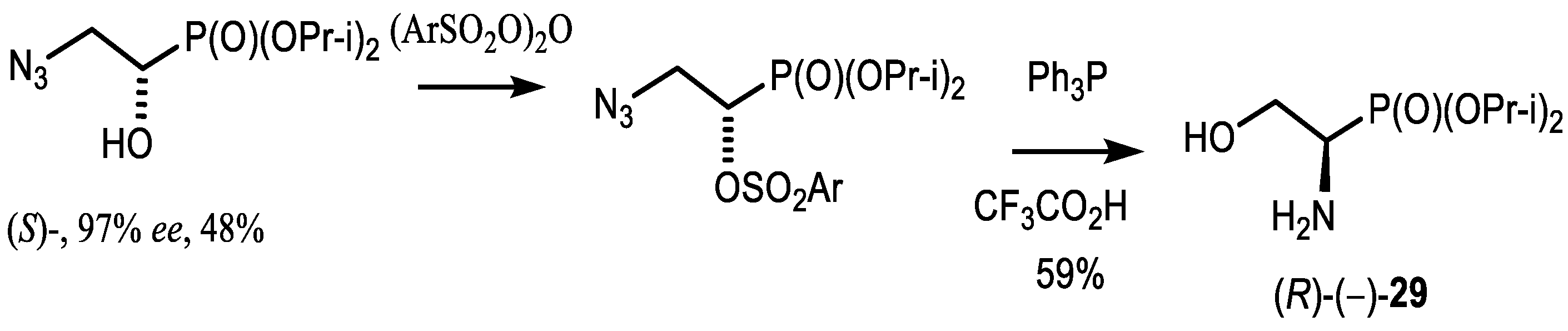
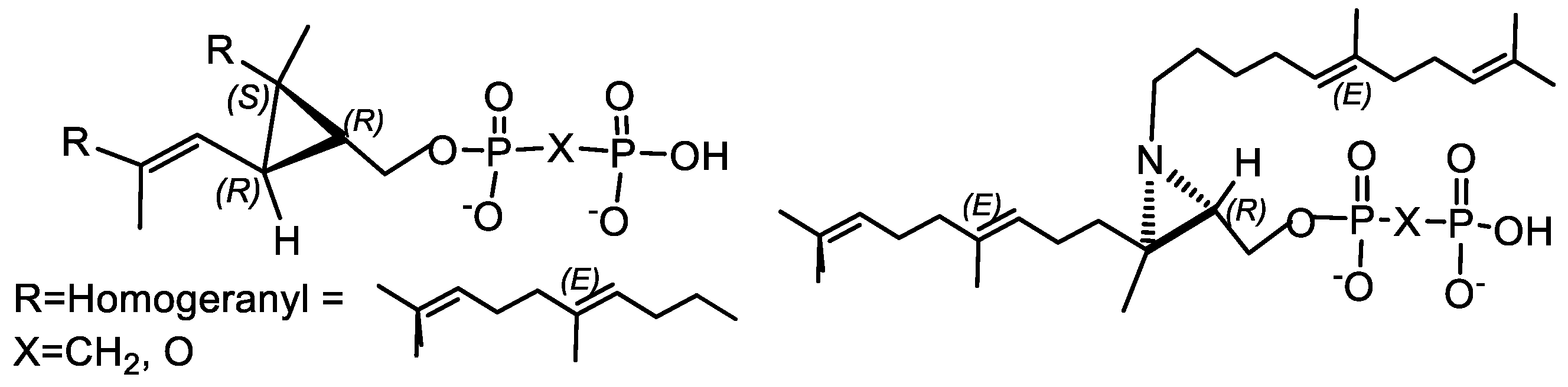
4.2. Bisphosphonates with an Asymmetric Center in the Side Chain
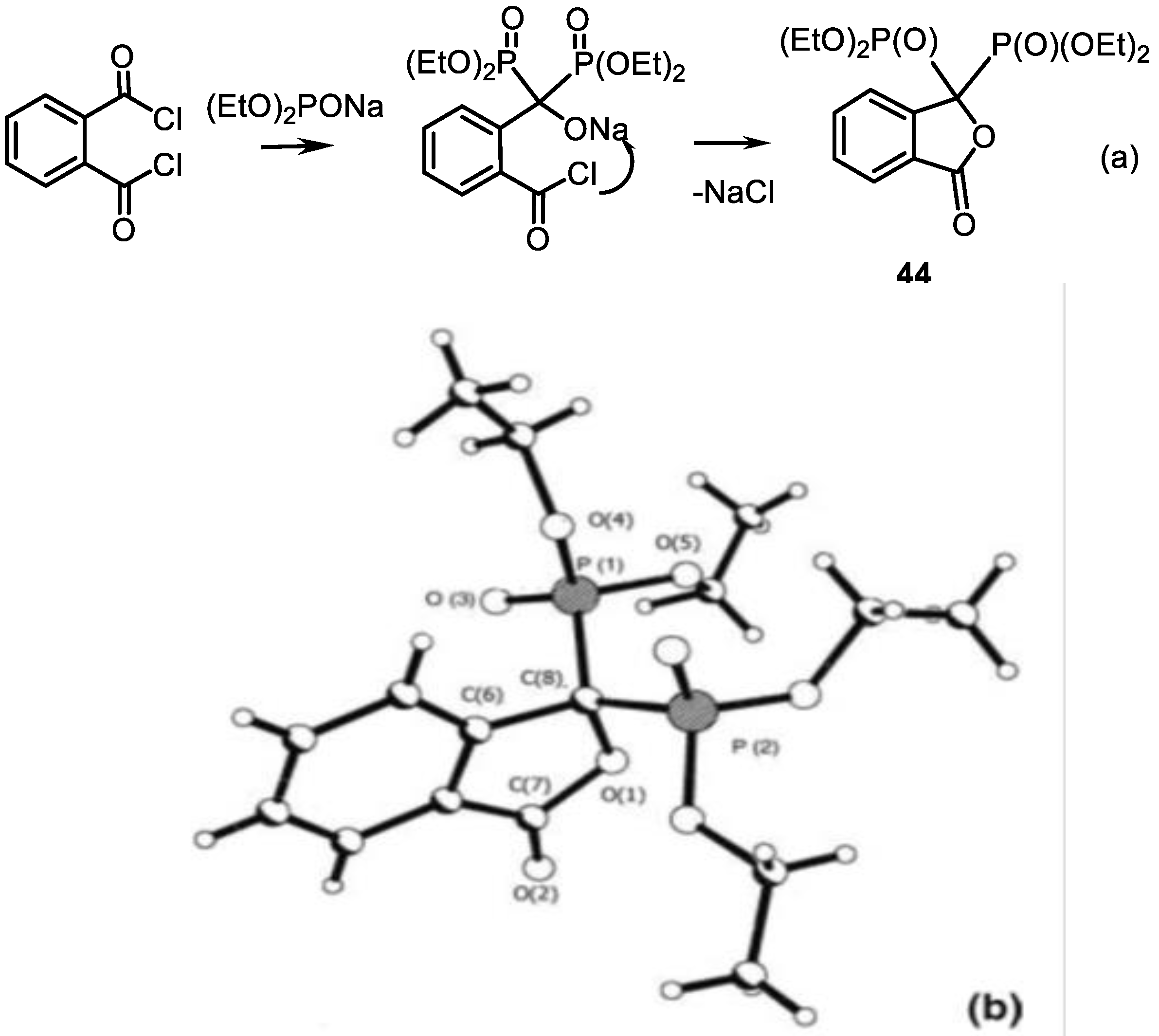
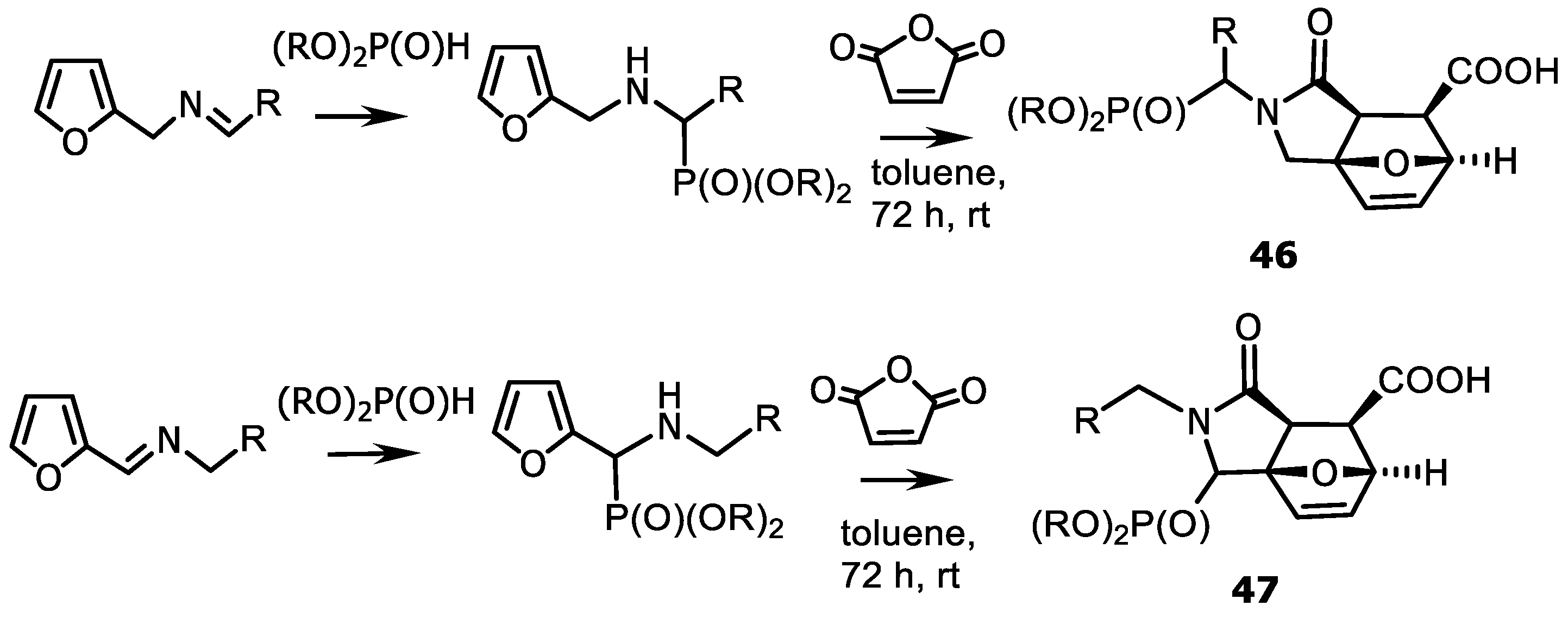
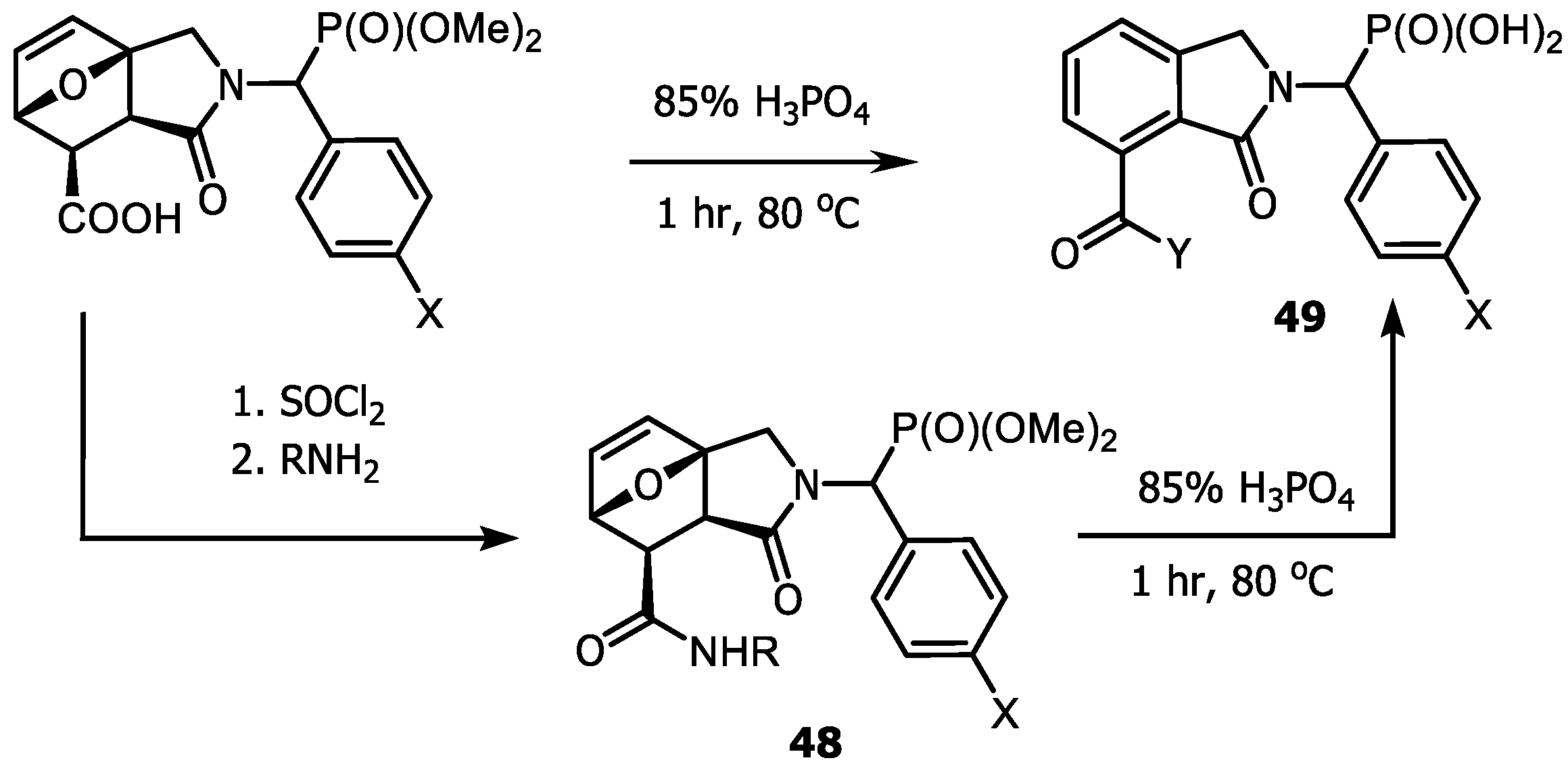
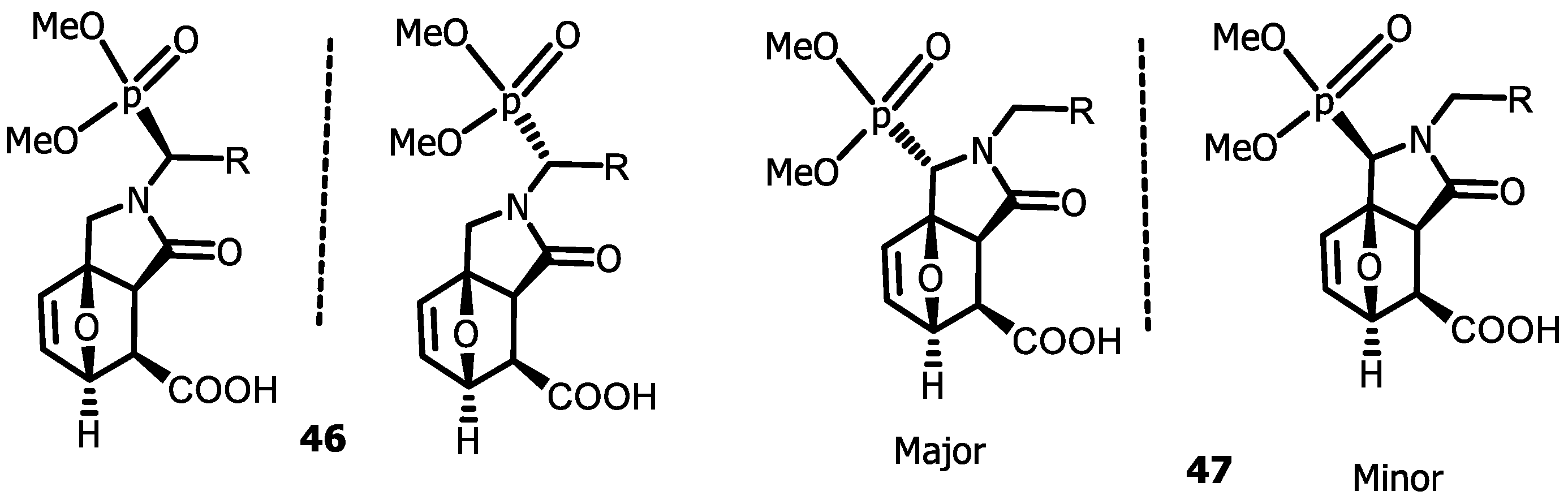
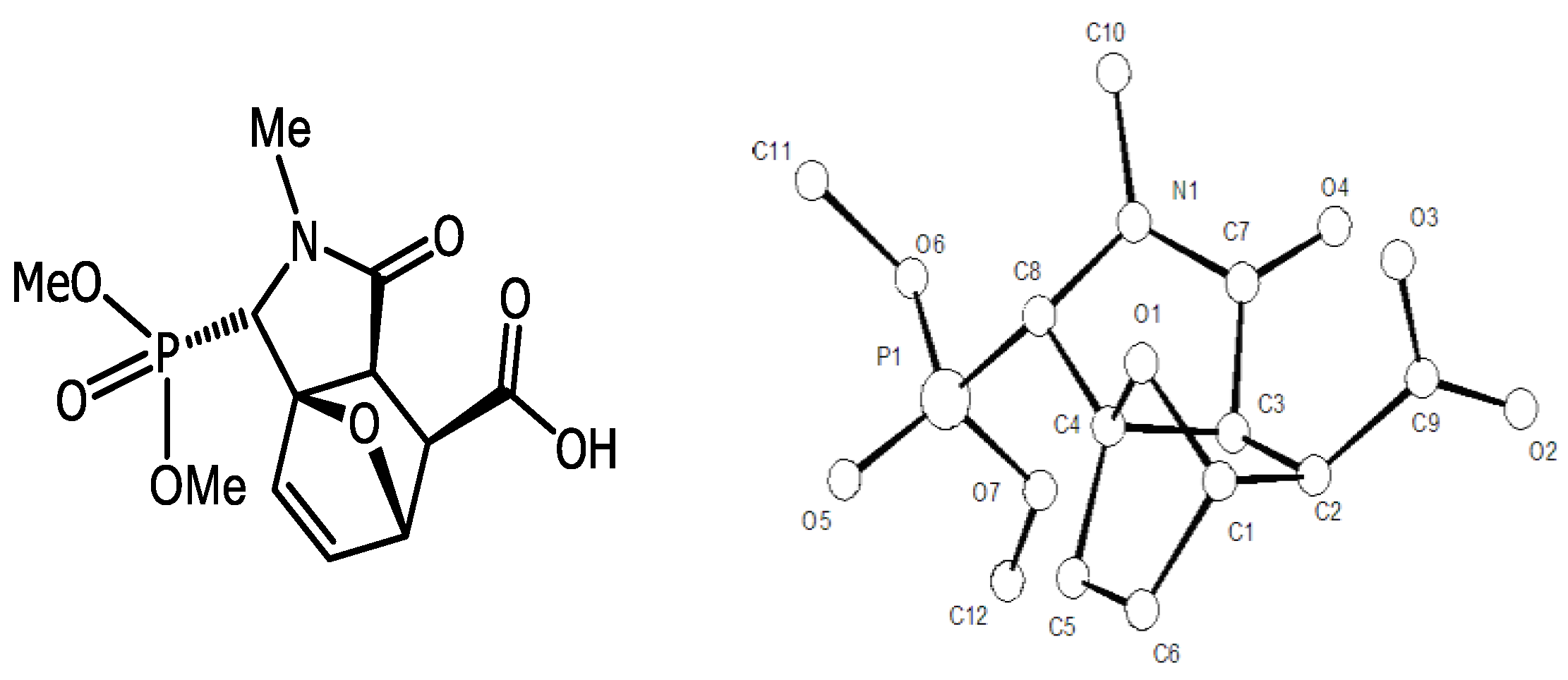
4.3. Synthesis of Isoindolinones
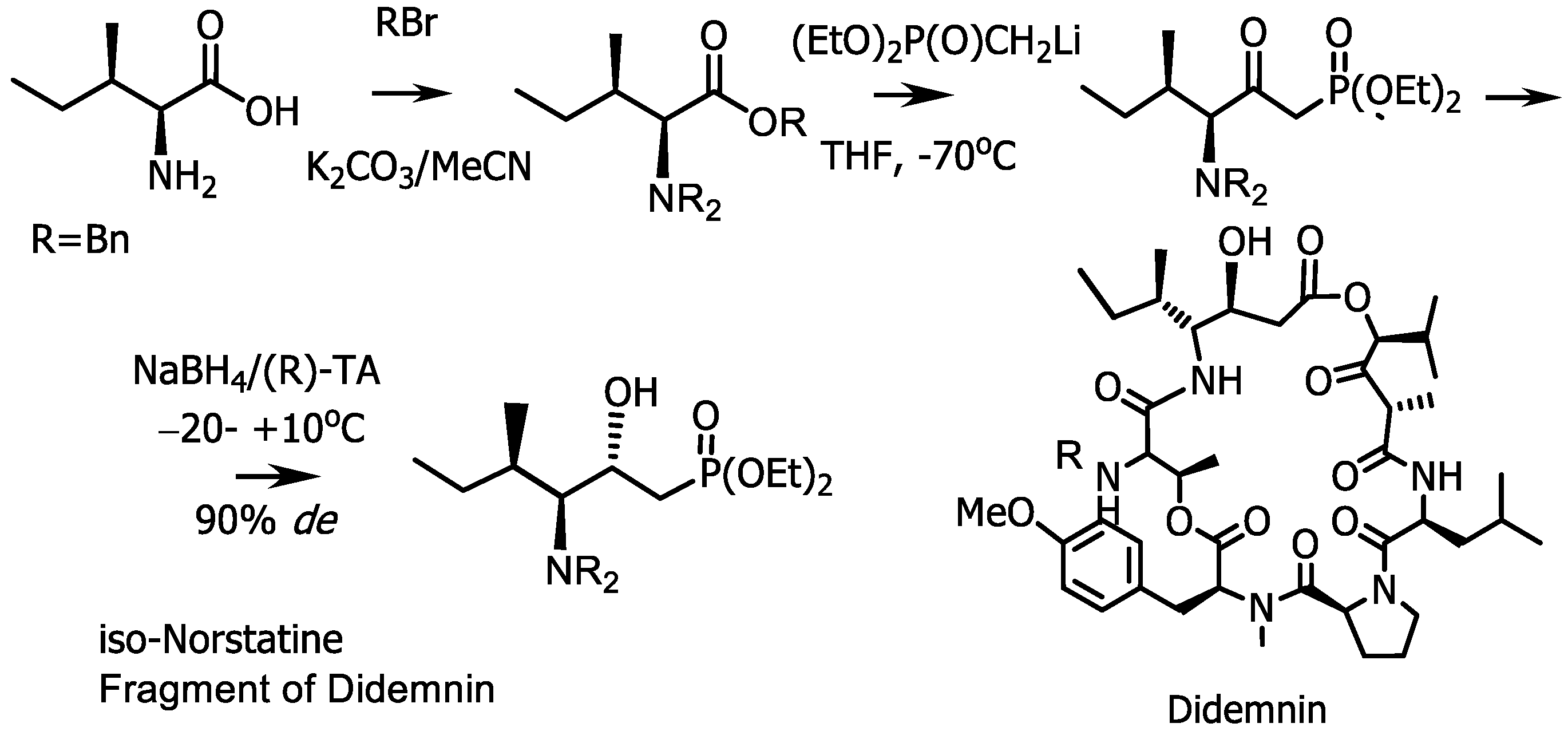
4.4. Synthesis of Phosphonic Analogue of Iso-Norstatine
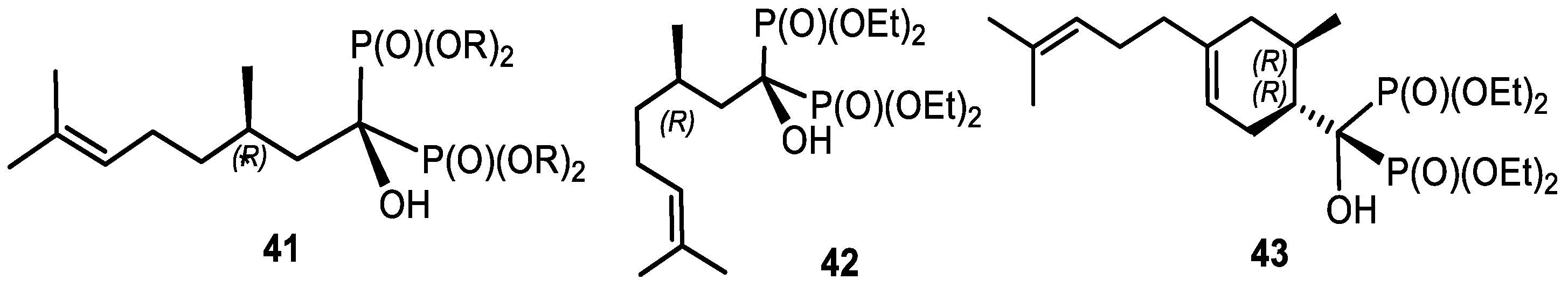
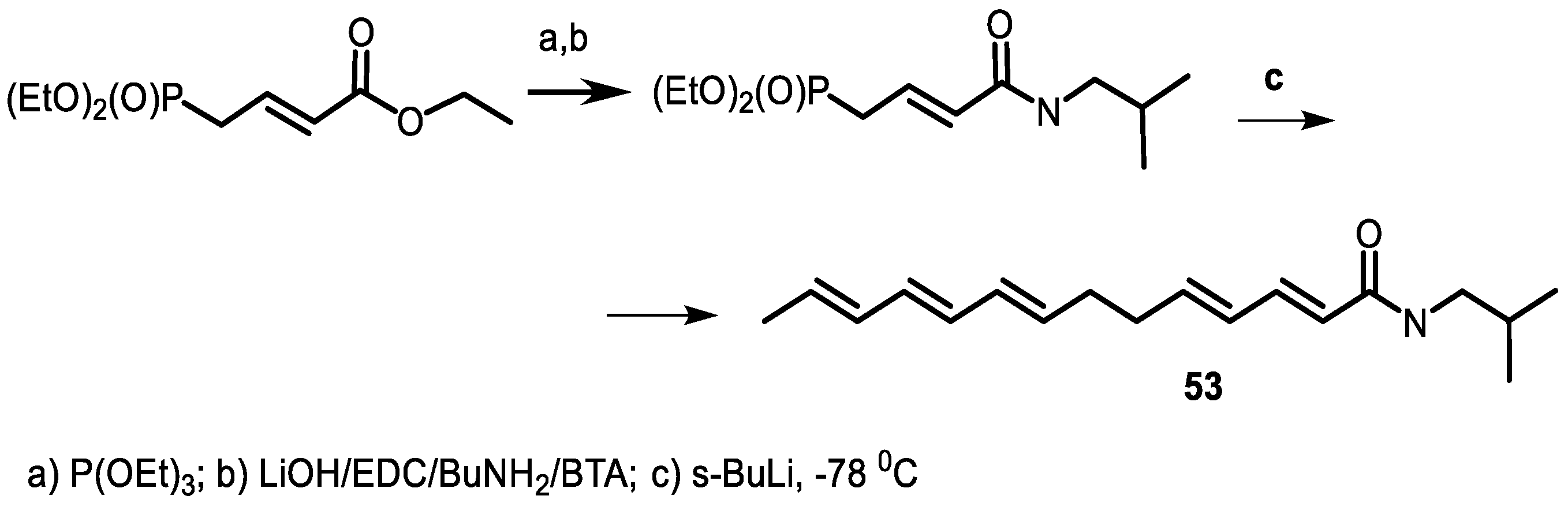
4.5. Tetradecapentaenoic Acid Derivatives
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Westheimer, F.H. Why Nature Chose Phosphates. Science 1987, 235, 1173–1178. [Google Scholar] [CrossRef]
- Hein, J.E.; Blackmond, D.G. On the origin of single chirality of amino acids and sugars in biogenesis. Acc. Chem. Res. 2012, 45, 2045–2054. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Stereochemistry of electrophilic and nucleophilic substitutions at phosphorus. Pure Appl. Chem. 2019, 91, 43–57. [Google Scholar] [CrossRef]
- Bernstein, M. Prebiotic materials from on and off the early Earth. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1689–1702. [Google Scholar] [CrossRef]
- Yamagata, Y.; Watanabe, H.; Saitoh, M. Volcanic production of polyphosphates and its relevance to prebiotic evolution. Nature 1991, 352, 516–519. [Google Scholar] [CrossRef]
- Krishnamurthy, R. Introduction: Chemical Evolution and the Origins of Life. Chem. Rev. 2020, 120, 4613–4615. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.W. Phosphorus in prebiotic chemistry. Philos. Trans. R. Soc. Lond B Biol. Sci. 2006, 361, 1743–1749. [Google Scholar] [CrossRef]
- Pasek, M.A. Schreibersite on the early earth: Scenarios for prebiotic Phosphorylation. Geosci. Front. 2017, 8, 329–335. [Google Scholar] [CrossRef]
- Pasek, M.A.; Kee, T.P.; Bryant, D.E.; Pavlov, A.A.; Lunine, J.I. Production of potentially prebiotic condensed phosphates by phosphorus redox chemistry. Angew. Chem. Int. Ed. 2008, 47, 7918–7920. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, Y.; Gao, X.; Xu, P. The international background of the origin of life. In Phosphorus Chemistry; Walter de Gruyter GmbH: Berlin, Germany, 2019; pp. 1–18. [Google Scholar]
- England, J. A New Physics Theory of Life. Available online: https://www.quantamagazine.org/a-new-thermodynamics-theory-of-the-origin-of-life-20140122/2013 (accessed on 28 January 2024).
- Kamerlin, S.C.L.; Sharma, P.K.; Prasad, R.B.; Warshel, A. Why nature really chose phosphate. Quart. Rev. Biophys. 2013, 46, 1–132. [Google Scholar] [CrossRef]
- Girard, C.; Kagan, H.B. Nonlinear Effects in Asymmetric Synthesis and Stereoselective Reactions: Ten Years of Investigation. Angew. Chem. Int. Ed. 1998, 37, 2922–2959. [Google Scholar] [CrossRef]
- Soai, K.; Kawasaki, T.; Matsumoto, A. Role of Asymmetric Autocatalysis in the Elucidation of Origins of Homochirality of Organic Compounds. Symmetry 2019, 11, 694. [Google Scholar] [CrossRef]
- Cintas, P.; Viedma, C. On the Physical Basis of Asymmetry and Homochirality. Chirality 2012, 24, 894–908. [Google Scholar] [CrossRef] [PubMed]
- Heck, P.R.; Greer, J.; Kööp, L.; Trappitsch, R.; Gyngard; Busemann, H.; Maden, C.; Ávila, J.N.; Davis, A.M.; Wieler, R. Lifetimes of interstellar dust from cosmic ray exposure ages of presolar silicon carbide. Proc. Nat. Acad. Sci. USA 2020, 117, 1884–1889. [Google Scholar] [CrossRef] [PubMed]
- Saks, V. (Ed.) Molecular System Bioenergetics: Energy for Life; Wiley-VCH: Weinheim, Germany, 2007; p. 633. ISBN 978-3-527-31787-5. [Google Scholar]
- Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Asymmetric Electrophilic Reactions in Phosphorus Chemistry. Symmetry 2020, 12, 108. [Google Scholar] [CrossRef]
- Yamamoto, T. Preferred dissociative mechanism of phosphate monoester hydrolysis in low dielectric environments. Chem. Phys. Lett. 2010, 500, 263–266. [Google Scholar] [CrossRef]
- Keglevich, G.; Kovács, T.; Csatlós, F. The Deoxygenation of Phosphine Oxides under Green Chemical Conditions. Heteroatom. Chem. 2015, 26, 199–205. [Google Scholar] [CrossRef]
- Muldoon, J.A.; Varga, B.R.; Deegan, M.M.; Chapp, T.W.; Eordogh, A.; Hughes, R.P.; Glueck, D.S.; Moore, C.E.; Rheingold, A.L. Inversion of Configuration at the Phosphorus Nucleophile in the Diastereoselective and Enantioselective Synthesis of P-Stereogenic syn-Phosphiranes from Chiral Epoxides. Angew. Chem. Int. Ed. 2018, 57, 5047–5051. [Google Scholar] [CrossRef]
- Kolodyazhnyi, O.I.; Grishkun, E.V.; Kolodyazhnaya, A.O.; Kolodyazhna, O.O.; Sheiko, S.Y. Generation of tert-Butyl-λ5-phosphanedione and Its Chemical Properties. Russ. J. Gen. Chem. 2018, 88, 919–924. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Recent Advances in Asymmetric Synthesis of P-Stereogenic Phosphorus Compounds. In Topics in Current Chemistry; Montchamp, J.-L., Ed.; Springer International Publication: Cham, Switzerland, 2015; Volume 361, pp. 161–237. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I. Asymmetric synthesis in organophosphorus chemistry. In Synthetic Methods, Catalysis and Application; Wiley-VCH: Weinheim, Germany, 2016; 360p. [Google Scholar]
- Kolodiazhnyi, O.I. Asymmetric catalysis as a method for the synthesis of chiral organophosphorus compounds. Tetrahedron Asymmetry 2014, 25, 865–922. [Google Scholar] [CrossRef]
- Trost, B.M. Asymmetric catalysis: An enabling science. Proc. Natl. Acad. Sci. USA 2004, 101, 5348–5355. [Google Scholar] [CrossRef]
- Oliana, M.; King, F.; Horton, P.N. Practical synthesis of chiral vinylphosphine oxides by direct nucleophilic substitution stereodivergent synthesis of aminophosphine ligands. J. Org. Chem. 2006, 71, 2472–2479. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Hatzakis, E.; McCarthy, S.M.; Reichl, K.D.; Lai, T.-Y.; Yennawar, H.P.; Radosevich, A.T. P–N Cooperative borane activation and catalytic hydroboration by a distorted phosphorous triamide platform. J. Am. Chem. Soc. 2017, 139, 6008–6016. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I.; Kolodiazhna, A.O. Multiple stereoselectivity in organophosphorus chemistry. Phosph. Sulf. Silicon. 2016, 191, 444–458. [Google Scholar] [CrossRef]
- Smaardijk, A.A.; Noorda, S.; van Bolhuis, F.; Wynberg, H. The absolute configuration of α-hydroxyphosphonates. Tetrahedron Lett. 1985, 26, 493. [Google Scholar] [CrossRef]
- Kolodyazhnaya, A.O.; Kukhar, V.P.; Kolodyazhnyi, O.I. Organic Catalysis of Phospha-Aldol Condensation. Russ. J. Gen. Chem. 2008, 78, 2043–2051. [Google Scholar] [CrossRef]
- Guliaiko, I.; Nesterov, V.; Sheiko, S.; Kolodiazhnyi, O.I.; Freytag, M.; Jones, P.G.; Schmutzler, R. Synthesis of Optically Active Hydroxyphosphonates. Heteroat. Chem. 2008, 19, 133–139. [Google Scholar] [CrossRef]
- Gryshkun, E.V.; Nesterov, V.; Kolodyazhnyi, O.I. Enantioselective reduction of ketophosphonates using adducts of chiral natural acids with sodium borohydride. Arkivoc 2012, 2012, 100–117. [Google Scholar] [CrossRef]
- Ordóñez, M.; Viveros-Ceballos, J.L.; Cativiela, C.; Sayago, F.J. An update on the stereoselective synthesis of α-aminophosphonic acids and derivatives. Tetrahedron 2015, 71, 1745–1784. [Google Scholar] [CrossRef]
- Nesterov, V.V.; Kolodiazhnyi, O.I. Efficient method for the asymmetric reduction of a- and b-ketophosphonates. Tetrahedron 2007, 63, 6720–6731. [Google Scholar] [CrossRef]
- Nesterov, V.V.; Kolodiazhnyi, O.I. Di(1R,2S,5R)-menthyl 2-Hydroxy-3-chloropropylphosphonate as a Useful Chiral Synthon for the Preparation of Enantiomerically Pure Phosphonic Acids. Synlett 2007, 2007, 2400–2404. [Google Scholar] [CrossRef]
- Nesterov, V.V. Asymmetric Reduction of Ketophosphonates. Ph.D. Thesis, IBONCH NAS of Ukraine, Kyiv, Ukraine, 2007; p. 0407U003652. Available online: https://dissertation.com.ua/node/671422 (accessed on 28 January 2024).
- Ordóñez, M.; González-Morales, A.; Ruíz, C.; De la Cruz-Cordero, R.; Fernández-Zertuche, M. Preparation of (R)- and (S)-γ-amino-β-hydroxypropylphosphonic acid from glycine. Tetrahedron Asymm. 2003, 14, 1775–1779. [Google Scholar] [CrossRef]
- Kolodiazhna, A.O. Asymmetric Synthesis of Modified Analogs of Natrural Compounds. Ph.D. Thesis, Institute of Bioorganic Chemistry and Petrol Chemistry, NAS of Ukraine, Kyiv, Ukraine, 2017. Available online: http://www.bpci.kiev.ua/rada/aref/autoref_Kolodyazhna_2017.pdf (accessed on 28 January 2024).
- Barfoot, C.W.; Harvey, J.E.; Kenworthy, M.N.; Kilburn, J.P.; Ahmed, M.; Taylor, R.J.K. Highly functionalised organolithium and organoboron reagents for the preparation of enantiomerically pure a-amino acids. Tetrahedron 2005, 61, 3403–3417. [Google Scholar] [CrossRef]
- Kolodiazhna, A.O.; Kolodiazhnyi, O. Catalytic Asymmetric Synthesis of C-Chiral Phosphonates. Symmetry 2022, 14, 1758. [Google Scholar] [CrossRef]
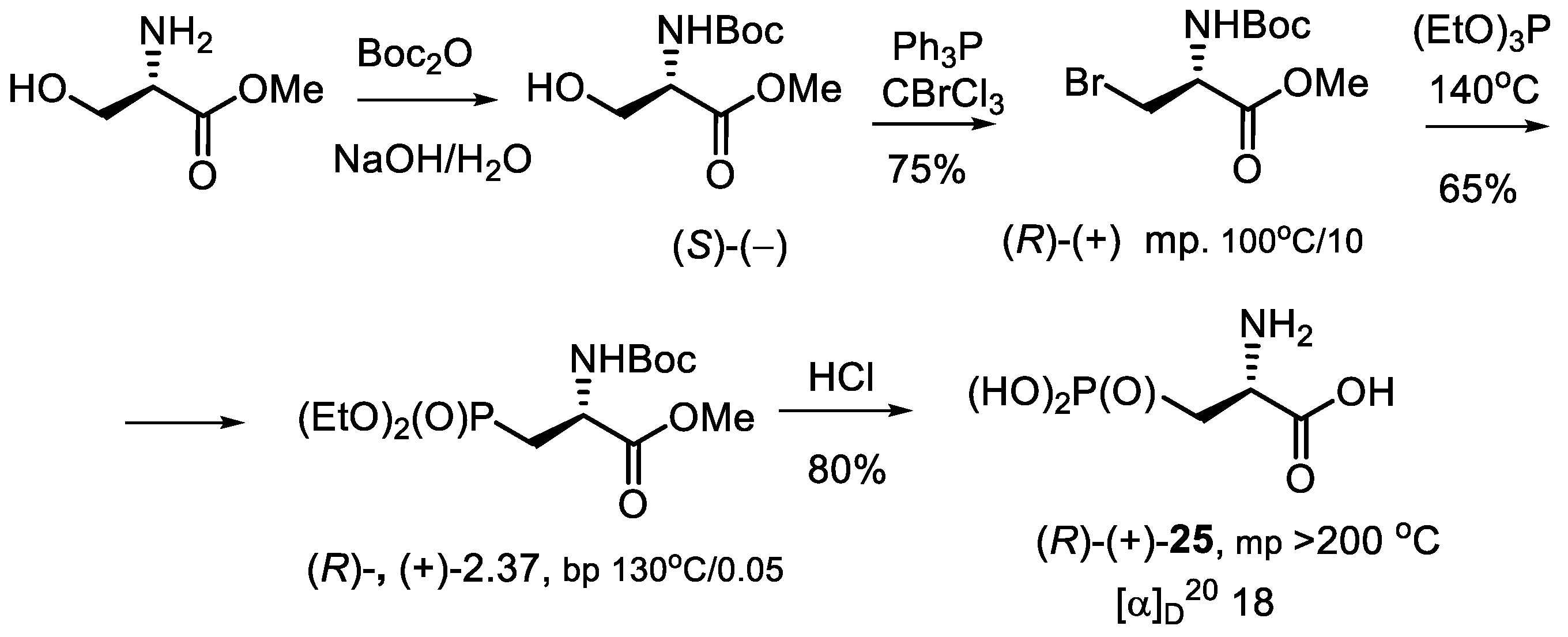
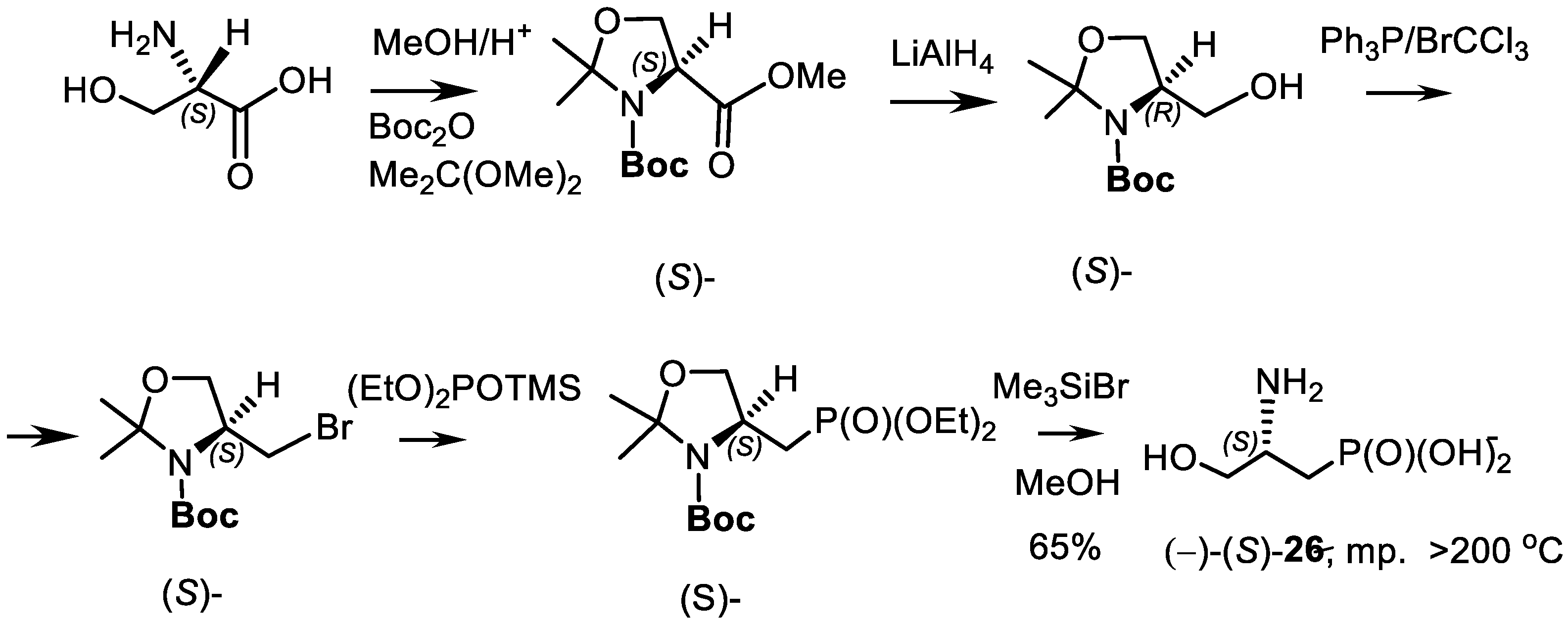
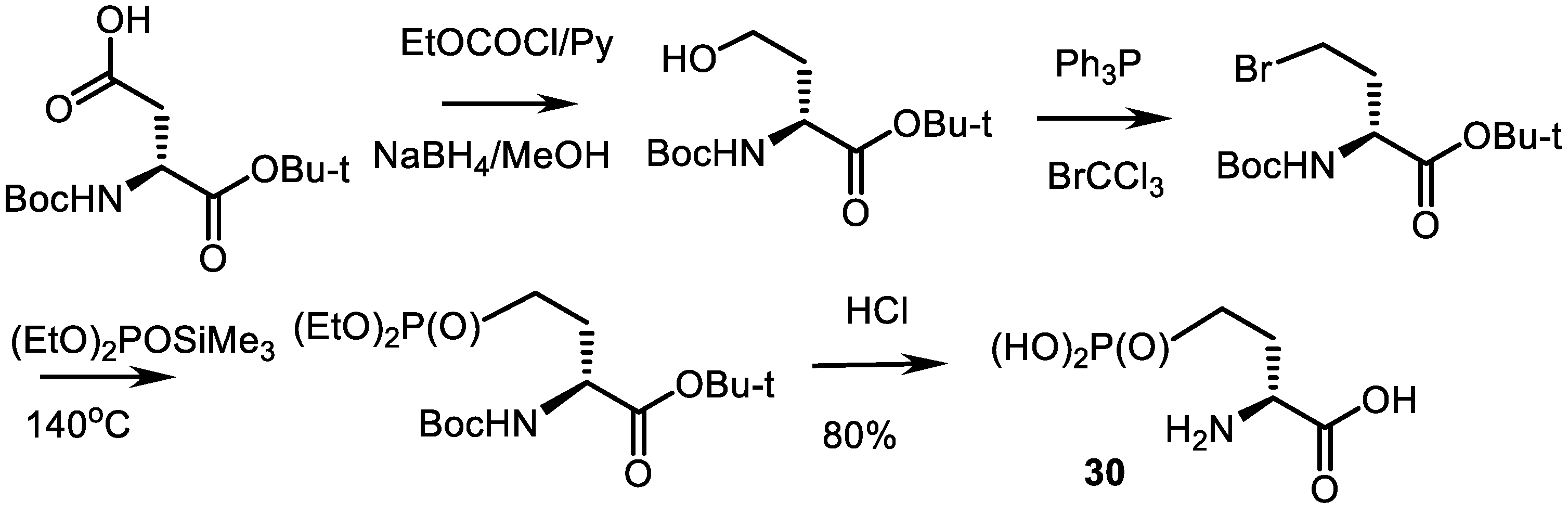
- Kolodyazhnyi, O.I.; Kolodyazhna, O.O. Synthesis of Enantiomerically Pure Phosphonic Analog of Natural Aspartic Acid. Russ. J. Gen. Chem. 2010, 80, 2519–2520. [Google Scholar] [CrossRef]
- Kolodyazhna, O.O.; Kolodyazhna, A.O.; Kolodyazhnyi, O.I. Synthesis of Phosphonic Analog of Glutamic Acid. Russ. J. Gen. Chem. 2013, 83, 777–778. [Google Scholar] [CrossRef]
- Hammerschmidt, F.; Lindner, W.; Wuggeniga, F.; Zarbl, E. Enzymes in organic chemistry. Part 10:1. Tetrahedron Asymm. 2000, 11, 2955–2964. [Google Scholar] [CrossRef]
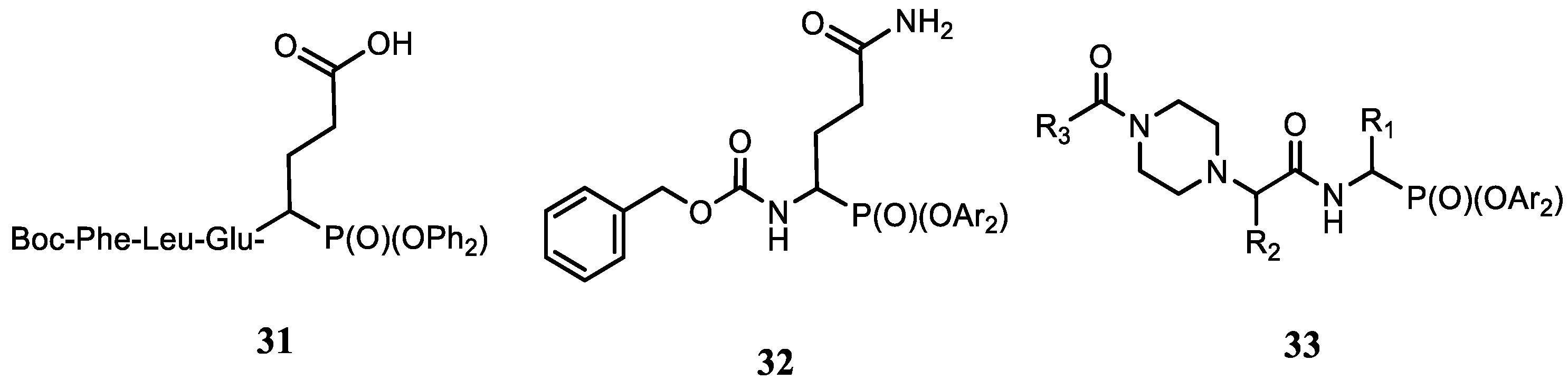
- Burchacka, E.; Skoreński, M.; Sieńczyk, M.; Oleksyszyn, J. Phosphonic analogues of glutamic acid as irreversible inhibitors of Staphylococcus aureus endoproteinase GluC: An efficient synthesis and inhibition of the human IgG degradation. Bioorg. Med. Chem. Lett. 2013, 23, 1412–1415. [Google Scholar] [CrossRef]
- Ewa, B.; Maciej, W.; Marcin, S.; Grzegorz, D.; Michał, Z.; Jan, P.; Józef, O. The development of first Staphylococcus aureus SplB protease inhibitors: Phosphonic analogues of glutamine. Bioorg. Med. Chem. Lett. 2012, 22, 5574–5578. [Google Scholar] [CrossRef]
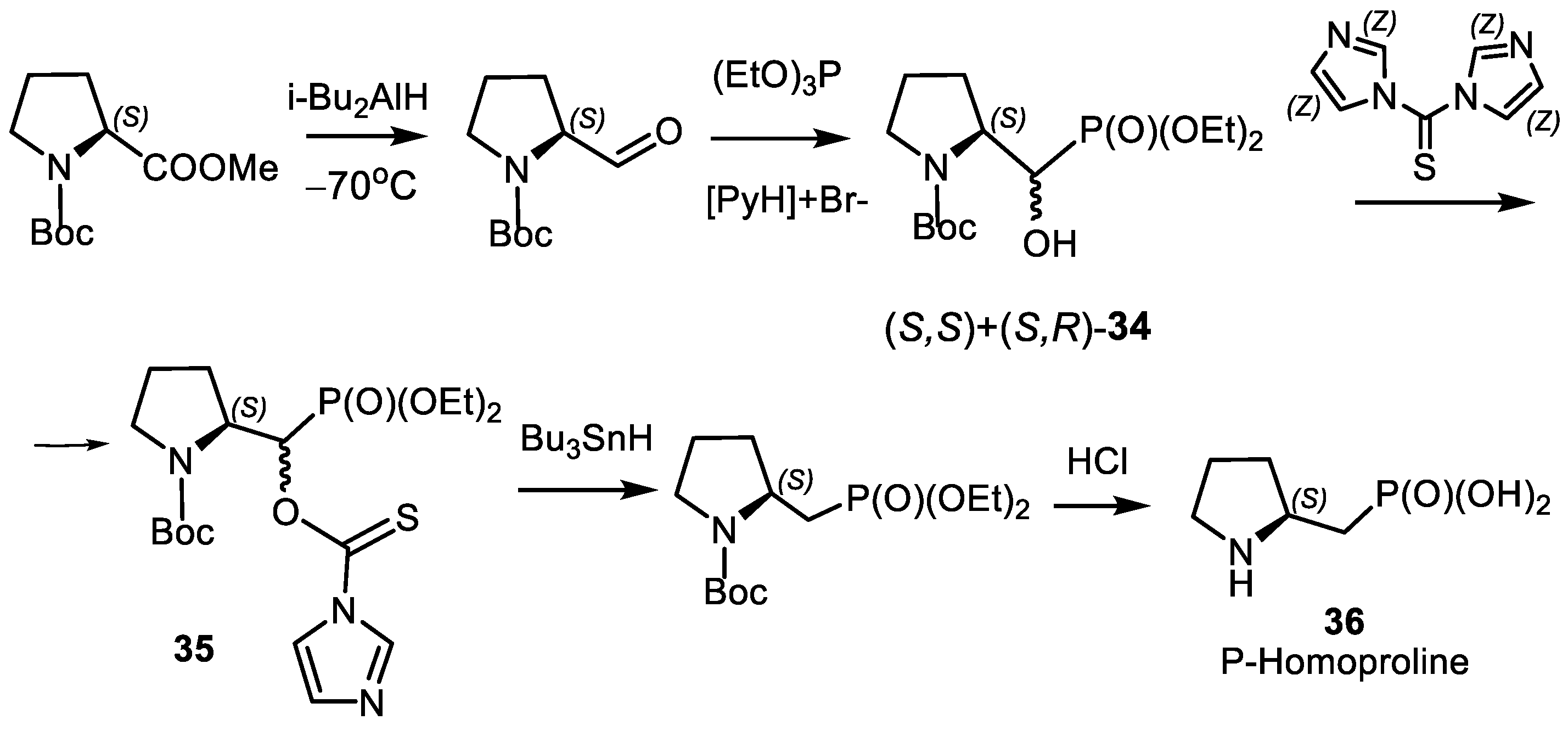
- Kolodyazhna, O.O.; Kolodyazhna, A.O.; Kolodyazhnyi, O.I. Synthesis of Phosphonic Analog of (S)-Homoproline. Russ. J. Gen. Chem. 2014, 84, 169–170. [Google Scholar] [CrossRef]
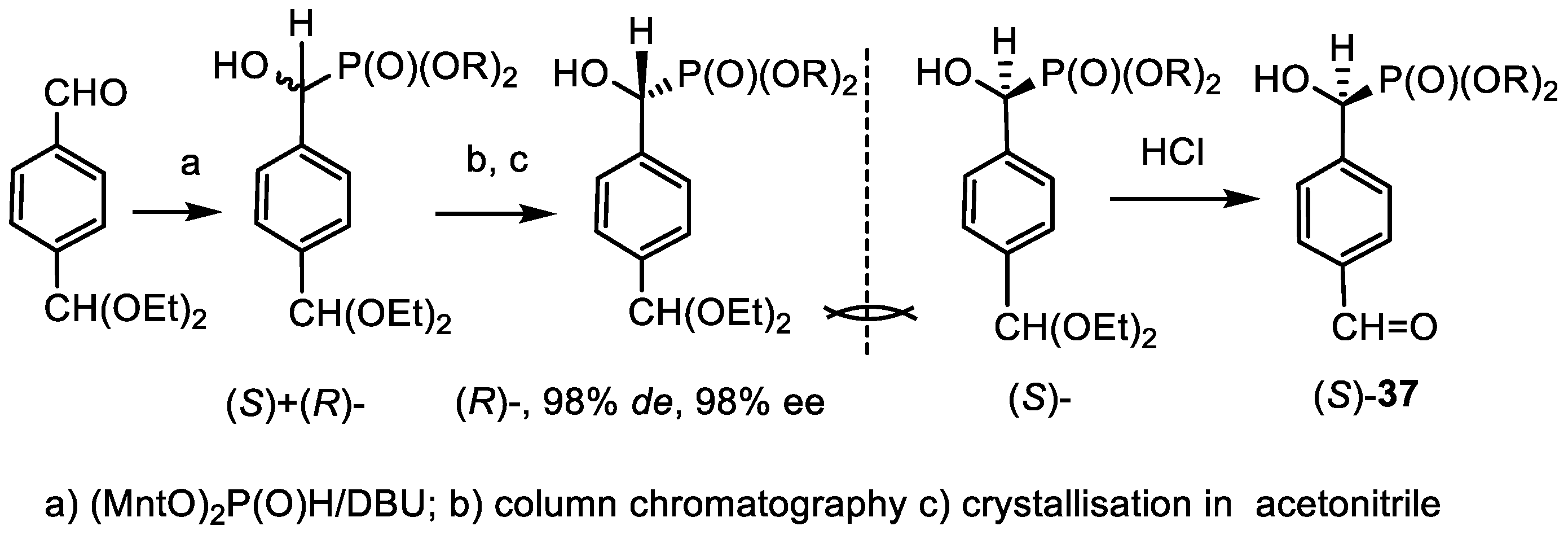
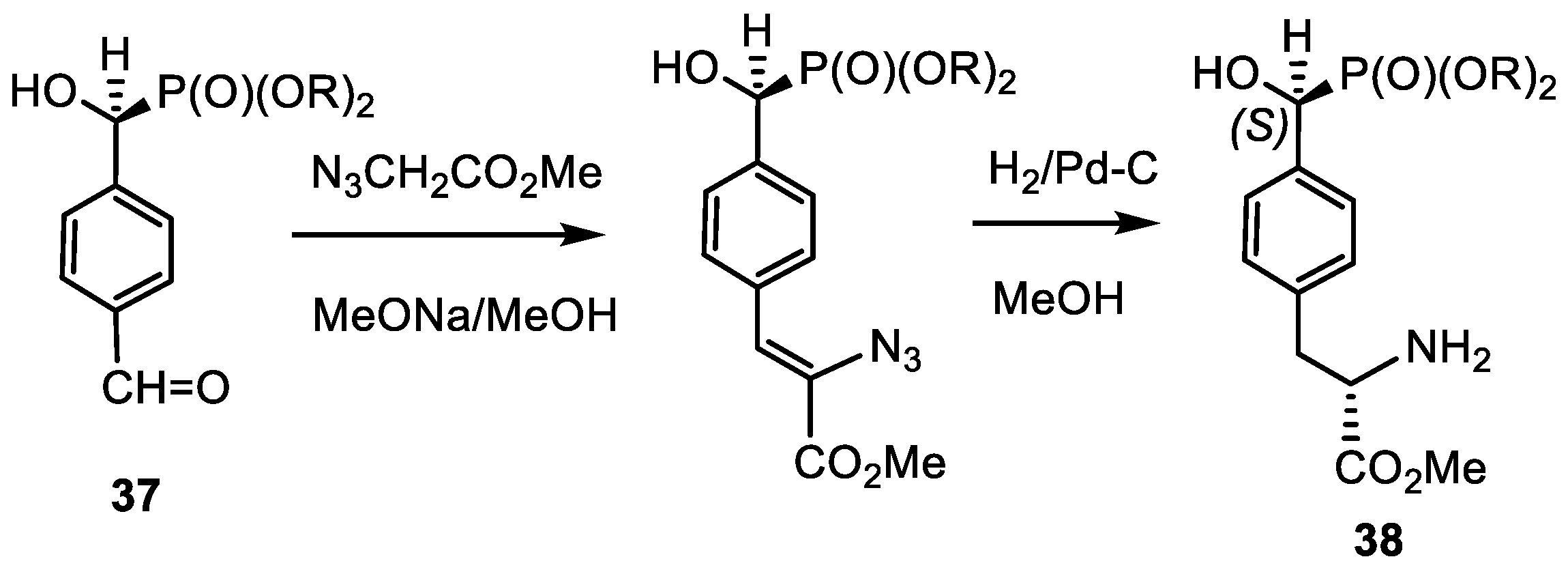
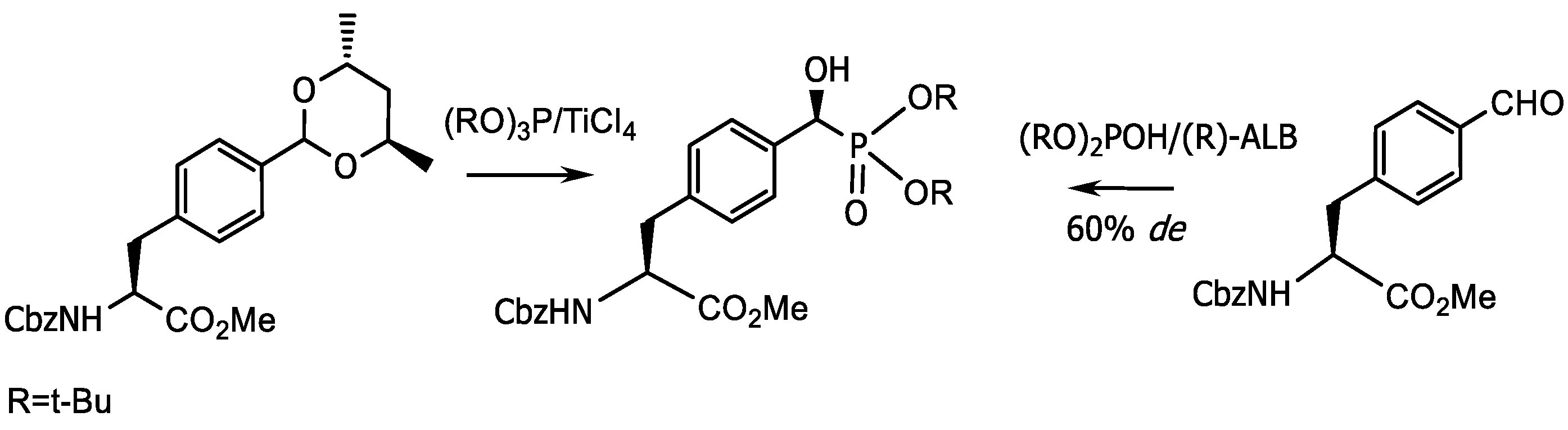
- Kolodyazhna, A.O.; Grishkun, E.V.; Kolodyazhnyi, O.I. Synthesis of Chiral Phosphonobenzaldehydes and Phosphonotyrosine. Russ. J. Gen. Chem. 2017, 87, 991–996. [Google Scholar] [CrossRef]
- Burke, T.R.; Smyth, M.S.; Nomizu, M.; Otaka, A.; Roller, P.P. Preparation of fluoro- and hydroxy-4-(phosphonomethyl)-D,L-phenylalanine suitably protected for solid-phase synthesis of peptides containing hydrolytically stable analogs of O-phosphotyrosine. J. Org. Chem. 1993, 58, 1336. [Google Scholar] [CrossRef]
- Fleisch, H. The role of bisphosphonates in breast cancer: Development of bisphosphonates. Breast Cancer Res. 2002, 4, 30–34. [Google Scholar] [CrossRef]
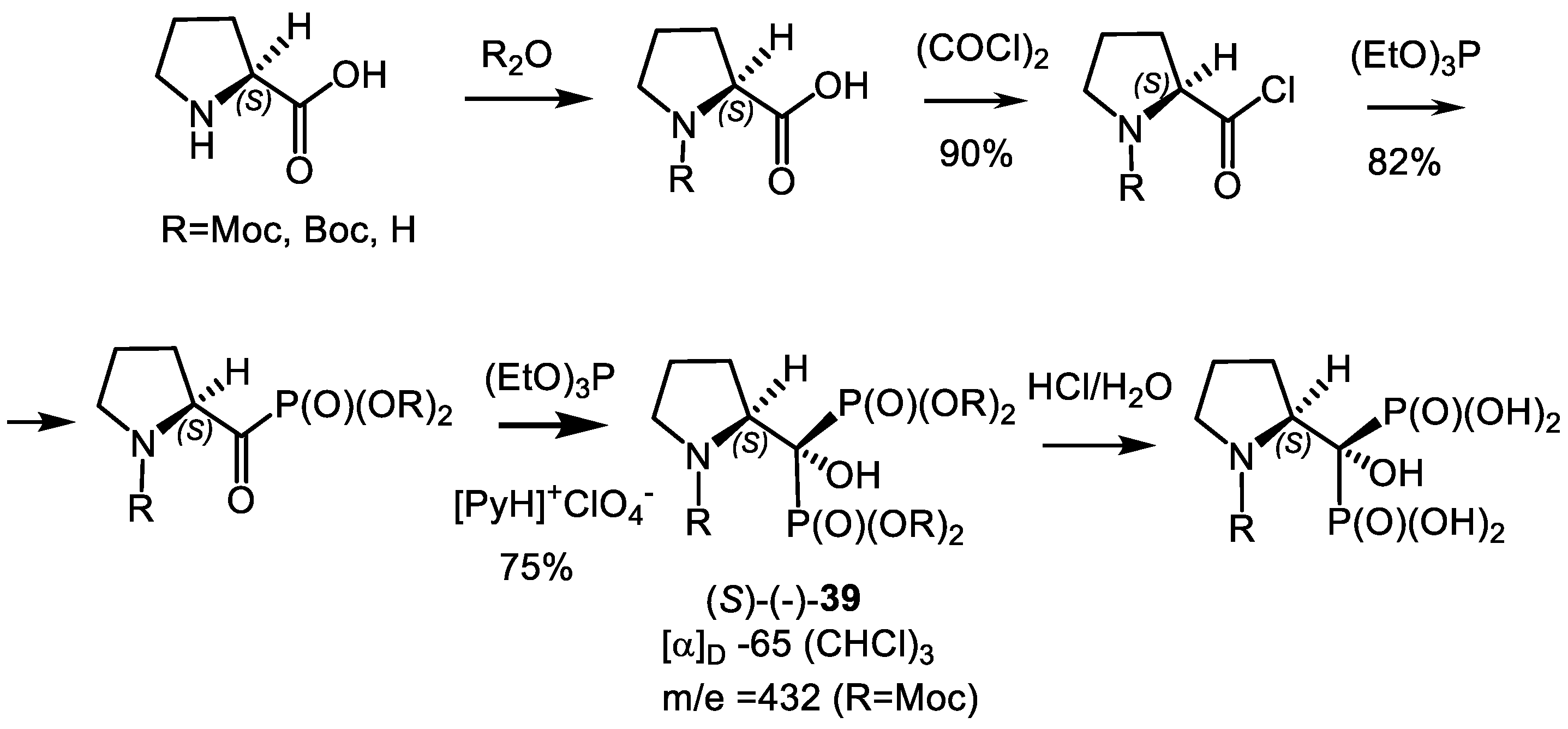
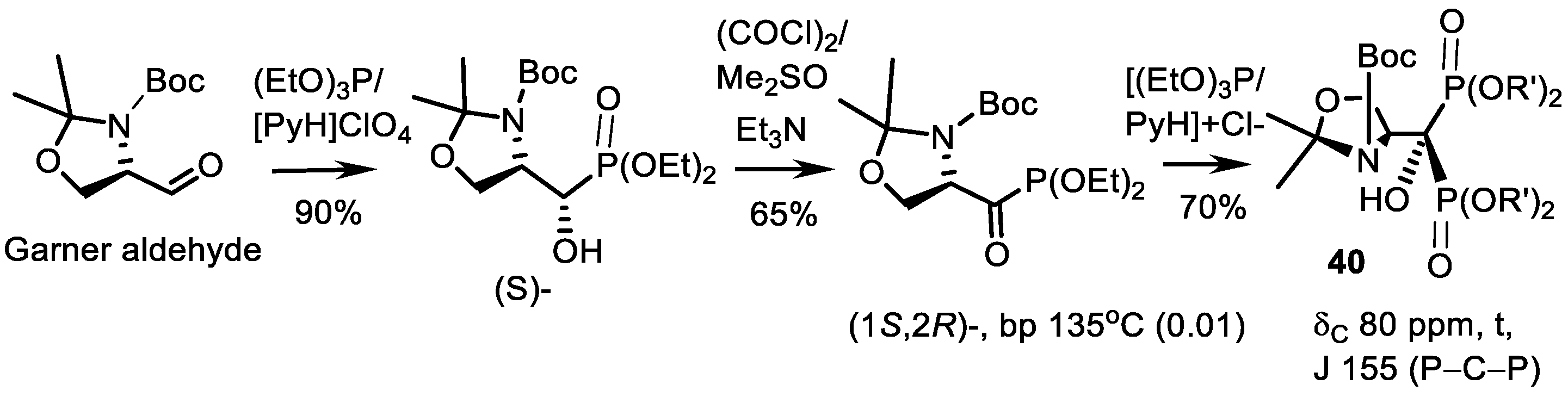
- Kolodyazhna, O.O.; Kolodyazhnyi, O.I. Chiral N-Moc-Pyrrolidine Bisphosphonate. Russ. J. Gen. Chem. 2011, 81, 145–146. [Google Scholar] [CrossRef]
- Zemlianoy, V.N.; Chernega, A.N.; Gutovm, A.V.; Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Synthesis of 3,3-Bis(diethylphosphono)-1-(3H)-isobenzofuranone and Its Chemical Properties. Phosphorus Sulfur Silicon Relat. Elem. 2011, 186, 481–488. [Google Scholar] [CrossRef]
- Magnin, D.R.; Biller, S.A.; Dickson, J.K., Jr.; Logan, J.V.; Lawrence, R.M.; Chen, Y.; Sulsky, R.B.; Ciosek, C.P., Jr.; Harrity, T.W.; Jolibois, K.G.; et al. 1,l-Bisphosphonate Squalene Synthase 925 Inhibitors: Interplay between the Isoprenoid Subunit and the Diphosphate Surrogate. J. Med. Chem. 1995, 38, 2596–2605. [Google Scholar] [CrossRef]
- Minard, C.; Nakhjiri, M.; Negahbani, A.; Kashemirov, B.A.; McKenna, C.E. New Chirally Modified Bisphosphonates for Synthesis of Individual Beta,Gamma-CHX-Deoxynucleotide Diastereomers Pouya Haratipour. Phosph. Sulpf. Silicon 2019, 194, 329–330. [Google Scholar] [CrossRef]
- Zuo, Z.; Kim, R.; Watson, D. Synthesis of Axially Chiral 2,2′-Bisphosphobiarenes via a Nickel-Catalyzed Asymmetric Ullmann Coupling: General Access to Privileged Chiral Ligands without Optical Resolution. ChemRxiv, 2020; preprint. [Google Scholar] [CrossRef]
- Kolodiazhna, A.O.; Kolodiazhnyi, O.I. Chiral organophosphorus pharmaceuticals: Properties and 2 Application. Symmetry 2023, 15, 1550. [Google Scholar] [CrossRef]
- Koohang, A.; Bailey, J.L.; Coates, R.M.; Erickson, H.K.; Owen, D.; Poulter, C.D. Enantioselective Inhibition of Squalene 901 Synthase by Aziridine Analogues of Presqualene Diphosphate. J. Org. Chem. 2010, 75, 4769–4777. [Google Scholar] [CrossRef]
- Wasko, B.M.; Smits, J.P.; Shull, L.W.; Wiemer, D.F.; Hohl, R.J. A novel bisphosphonate inhibitor of squalene synthase 904 combined with a statin or a nitrogenous bisphosphonate in vitro. J. Lipid Res. 2011, 52, 1957–1964. [Google Scholar] [CrossRef]
- Schevchuk, M.V.; Metelitsa, L.A.; Charochkina, L.L.; Mogilevich, S.E.; Rusanov, E.B.; Sorochinsky, A.E.; Khilya, V.P.; Romanenko, V.D.; Kukhar, V.P. Synthesis of N-(phosphonomethyl)glycine derivatives and studies of their immunotropic activity. Russ. Chem. Bull. Int. Ed. 2011, 60, 712–718. [Google Scholar] [CrossRef]
- Shevchuk, M.V.; Sorochinsky, A.E.; Khilya, V.P.; Romanenko, V.; Kukhar, V.P. Utilization of Aminophosphonates in the Petasis Boronic Acid Mannich V. D. Reaction Petasis Boronic Acid Manicch Reaction. Synlett 2010, 2010, 73–76. [Google Scholar] [CrossRef]
- Marrs, E.C.L.; Varadi, L.; Bedernjak, A.F.; Day, K.M.; Gray, M.; Jones, A.L.; Cummings; Anderson, S.P.; Anderson, R.J.; Perry, J.D. Phosphonopeptides Revisited, in an Era of Increasing Antimicrobial Resistance. Molecules 2020, 25, 1445. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, K.K.A.; Van Der Donk, W.A. New insights into the biosynthesis of fosfazinomycin. Chem. Sci. 2016, 7, 5219–5223. [Google Scholar] [CrossRef]
- Kachkovskyi, G.O.; Kolodiazhnyi, O.I. α-Acylaminophosphonates possessing epoxyisoindolone moiety. Tetrahedron 2007, 63, 12576–12582. [Google Scholar] [CrossRef]
- Vovk, A.I.; Mischenko, I.M.; Tanchuk, V.Y.; Kachkovskii, G.A.; Sheiko, S.Y.; Kolodyazhnyi, O.I.; Kukhar, V.P. Stereoselectivity of binding of a-(N-benzylamino)benzylphosphonic acids to prostatic acid phosphatase. Bioorg. Med. Chem. Lett. 2008, 18, 4620–4623. [Google Scholar] [CrossRef]
- Kachkovskyi, G.O.; Kolodiazhnyi, O.I. Synthesis of the Phosphonoanalogue of Benzoc.pyroglutamic Acid. Phosph. Sulf. Silicon 2010, 185, 2441–2448. [Google Scholar] [CrossRef]
- Kolodiazhnyi, O.I.; Guliaiko, I. Asymmetric Syntheses of New Phosphonotaxoids. Phosph. Sulfur. Silicon 2008, 183, 677. [Google Scholar] [CrossRef]
- Gulyaiko, I.V.; Kolodyazhnyi, O.I. Synthesis of a Phosphorus Analog of iso-Statine. Russ. J. Gen. Chem. 2008, 78, 1626–1627. [Google Scholar] [CrossRef]
- Chruma, J.J.; Cullen, D.J.; Bowman, L.; Toy, P.H. Polyunsaturated fatty acid amides from the Zanthoxylum genus—From culinary curiosities to probes for chemical biology. Nat. Prod. Rep. 2018, 35, 54–74. [Google Scholar] [CrossRef]
- Mugnaini, C.; Corellim, F. Total Synthesis of δ-Sanshool and Analogues Thereof. Synthesis 2016, 48, 2085–2092. [Google Scholar] [CrossRef]
- Aoki, K.; Igarashi, Y.; Nishimura, H. I Morishita, K Usui Application of iron carbonyl complexation to the selective total synthesis of sanshools. Tetrahedron Lett. 2012, 53, 6000–6003. [Google Scholar] [CrossRef]
- Corey, E.J.; Fuchs, P.L. A synthetic method for formylethynyl conversion (RCHO→RC≡CH or RC≡CR′). Tetrahedron Lett. 1972, 13, 3769–3772. [Google Scholar] [CrossRef]
- Trost, B.M.; Kazmaier, U. Internal Redox Catalyzed by Triphenylphosphine. J. Amer. Chem. Soc. 1992, 114, 7933–7935. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Preti, D.; Materazzi, S.; Geppetti, P. Transient Receptor Potential Ankyrin 1 (TRPA1) Channel as Emerging Target for Novel Analgesics and Anti-Inflammatory Agents. J. Med. Chem. 2010, 53, 5085–5107. [Google Scholar] [CrossRef] [PubMed]
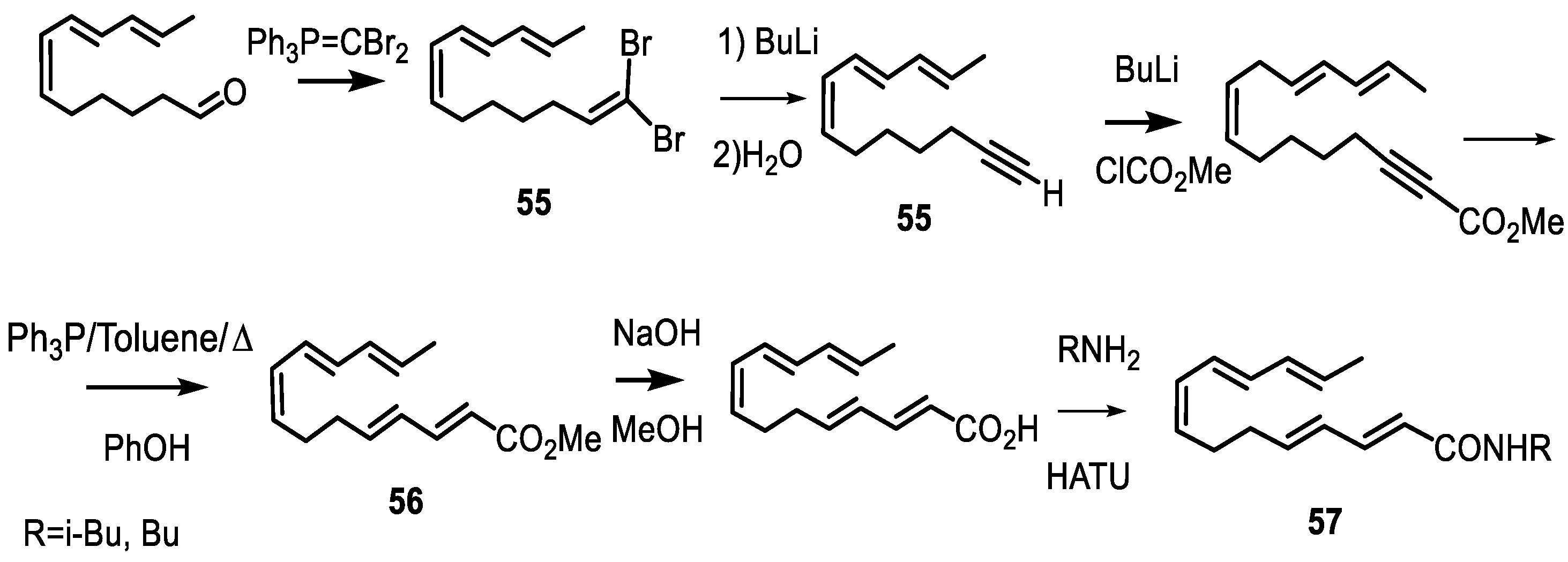
- Kolodyazhna, A.O.; Kolodyazhnyi, O.I. Synthesis of Tetradecapentaenoic Acid Derivatives. Russ. J. Gen. Chem. 2019, 89, 1998–2004. [Google Scholar] [CrossRef]
- Kolodiazhna, A.; Kolodiazhnyi, O. Stereoselective syntheses of sanshool derivatives. Phosph. Sulf. Silicon 2019, 194, 275–276. [Google Scholar] [CrossRef]
- Rádai, Z.; Keglevich, G. Synthesis and Reactions of Hydroxyphosphonates. Molecules 2018, 23, 1493. [Google Scholar] [CrossRef]











































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
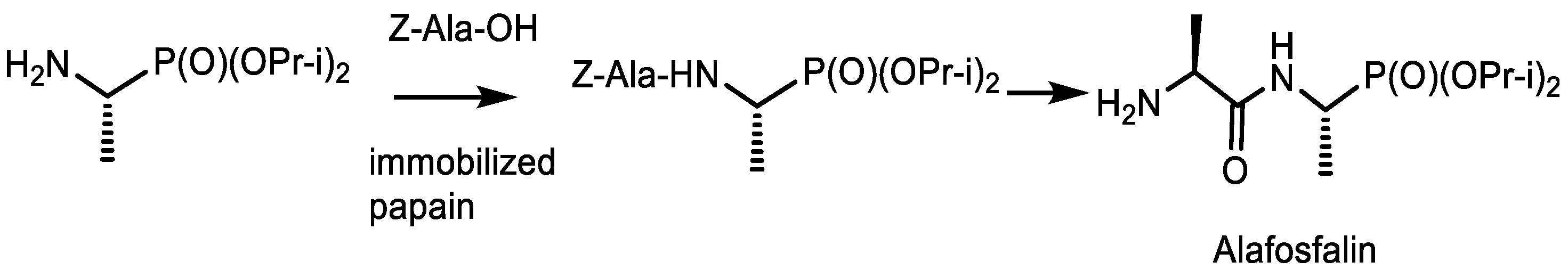{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | R | R’ | R*OH | B | Ratio | Ref. |
|---|---|---|---|---|---|---|
| 1 | Ph | Me | GF | Et3N | 90:10 | [23] |
| 2 | Ph | Et | GF | Et3N | 96:4 | [23] |
| 3 | Ph | i-Bu | GF | Et3N | 95:5 | [23] |
| 4 | Ph | PhCH2 | GF | Et3N | ~100:0 | [23] |
| 5 | Ph | PhCH2 | GF | Py | 25:75 | [24] |
| 6 | Ph | Me | (1S)-Borneol | DMAP | 4:1 | [24] |
| 7 | Ph | Me | L-Menthol | DMAP | 1:1 2:1 | [24] |
| 8 | Ph | Me | (−)-Isopinocampheol | DMAP | 1:1 | [23] |
| 9 | Ph | Me | (+)-isoborneol | DMAP | 74:26 | [23] |
| 10 | 4-An | Ph | (−)-Menthol | Et3N | 4:1 | [23] |
| Entry | R1 | CR32 | B | Solvent | Yields (%) | (SP):(RP) | Ref |
|---|---|---|---|---|---|---|---|
| 1 | Me | CMe2 | Et3N | Toluene | 70 | 90:10 | [23,24] |
| 2 | Et | CMe2 | Et3N | Toluene | 70 | 96:4 | [23] |
| 3 | i-Bu | CMe2 | Et3N | Toluene | 70 | 95:5 | [23] |
| 5 | Bn | CMe2 | Py | THF | 70 | 25:75 | [23] |
| 6 | Me | c-C5H10 | Et3N | Toluene | 75 | 95:5 | [23,27] |
| 7 | Me | c-C5H10 | Et3N | THF | 70 | 95:5 | [23,27] |
| 8 | Me | c-C5H10 | Et3N | CH2Cl2 | 70 | 87:13 | [23] |
| 9 | Et | c-C5H10 | Et3N | Toluene | 93 | 93:7 | [23] |
| 10 | Et | c-C5H10 | Py | THF | 94 | 30:70 | [23] |
| 11 | i-Pr | c-C5H10 | Et3N | Toluene | 92 | 86:14 | [23] |
| 12 | Bn | c-C5H10 | Et3N | Toluene | 95 | 90:10 | [23,24] |
| 13 | o-An | c-C5H10 | Py | THF | 94 | 55:45 | [23] |
| 14 | 1-Nphth | c-C5H10 | Et3N | Toluene | 87 | 40:60 | [20] |
| пп | Solvent | B | Temp. °C | 1:2:B | dr a |
|---|---|---|---|---|---|
| 1 | Benzene | Et3N | 20 | 1:1:1 | 8:92 |
| 2 | Toluene | Et3N | 70 | 1:1:1 | 16:84 |
| 3 | Toluene | DABCO | 20 | 1:1:1 | 25:75 |
| 4 | Toluene | PEA | 20 | 1:1:1 | 17.5:82.5 |
| 5 | Toluene | DBU | 20 | 1:1:1 | 42:58 |
| 6 | Ether | Et3N | 20 | 1:1:1 | 12:88 |
| 7 | Hexane | Et3N | 20 | 1:1:1 | 20:80 |
| 8 | THF | Et3N | 20 | 1:1:1 | 38:62 |

| Entry | R | R’ | n | Yields (%) | Auxiliary | Configuration | ee (%) |
|---|---|---|---|---|---|---|---|
| 1 | Ph | Mnt | 0 | 90 | L-Pro | S | 52.6 |
| 2 | 2-F-C6H4 | Mnt | 0 | 90 | L-Pro | S | 79.2 |
| 3 | 2-An | Mnt | 0 | 90 | L-Pro | S | 60.6 |
| 3 | Ph | Mnt | 0 | 95 | L-TA | R | 92.4 |
| 4 | Ph | Mnt | 0 | 98 | D-TA | S | 46 |
| 5 | 2-F-C6H4 | Mnt | 0 | 97 | L-TA | S | 80.5 |
| 6 | 2-An | Mnt | 0 | 96 | L-TA | S | 74 |
| 7 | Pyperonyl | Mnt | 0 | 97 | L-TA | S | 96 |
| 8 | i-Pr | Mnt | 0 | 97.6 | L-TA | S | 68 |
| 9 | Ph | Et | 0 | 95 | L-TA | S | 60 |
| 10 | Ph | Et | 0 | 94 | D-TA | R | 60 |
| 11 | CH2Cl | Et | 1 | 86 | L-TA | S | 80 |
| 12 | CH2Cl | Et | 1 | 82 | D-TA | R | 80 |
| 13 | CH2Cl | Mnt | 1 | 94 | L-TA | S | 96 |
| 14 | CH2Cl | Mnt | 1 | 80 | D-TA | R | 82 |
| 15 | Ph | Et | 1 | 95 | D-TA | S | 44 |
| Comp-d | R1 | R2 | R3 | dr | Yields (%) |
|---|---|---|---|---|---|
| 45a | PhCH=CH- | 4-An | H | 90:10 | 75 |
| 45b | 4-An | 4-An | H | 90:10 | 76 |
| 45c | 5-Benzo[1,3]dioxole | 4-An | H | 90:10 | 53 |
| 45d | 1-Thienyl | 4-An | H | 90:10 | 45 |
| 45e | PhCH-CH- | Ph | Bn | 95:5 | 80 |
| 45f | 4-An | Ph | Bn | 95:5 | 95 |
| 45g | 5-Benzo[1,3]dioxole | Ph | Bn | 95:5 | 88 |
| 45h | 1-Thienyl | Ph | Bn | 95:5 | 69 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolodiazhnyi, O.I.; Kolodiazhna, A.O. Stereoselective Syntheses of Organophosphorus Compounds. Symmetry 2024, 16, 342. https://doi.org/10.3390/sym16030342
Kolodiazhnyi OI, Kolodiazhna AO. Stereoselective Syntheses of Organophosphorus Compounds. Symmetry. 2024; 16(3):342. https://doi.org/10.3390/sym16030342
Chicago/Turabian StyleKolodiazhnyi, Oleg I., and Anastasy O. Kolodiazhna. 2024. "Stereoselective Syntheses of Organophosphorus Compounds" Symmetry 16, no. 3: 342. https://doi.org/10.3390/sym16030342