
A Mini-Review of the Kinetic Energy Partition Method in Quantum Mechanics

Abstract
:1. Introduction
2. Theory of One-Body Problem Interacting with N Force Centers
3. Theory of N-Body KEP
4. One-Dimensional Model Potentials
4.1. Double Zero-Range Potentials in One-Dimensional System
4.2. Triple Zero-Range Potentials in One-Dimensional System
4.3. N Harmonic Oscillator Interactions in One-Dimensional Schrödinger Equation
4.4. Three-Body Problem and Many Interactions of One-Dimensional Moshinsky Atoms
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arteca, G.A.; Fernández, F.M.; Castro, E.A.; Arteca, G.A.; Fernández, F.M.; Castro, E.A. Large Order Perturbation Theory and Summation Methods in Quantum Mechanics; Springer: Berlin/Heidelberg, Germany, 1990. [Google Scholar]
- Chen, Y.H.; Greene, C.H. P-wave Efimov physics implications at unitarity. Phys. Rev. A 2023, 107, 033329. [Google Scholar] [CrossRef]
- Esry, B.D.; Greene, C.H. Spontaneous spatial symmetry breaking in two-component Bose-Einstein condensates. Phys. Rev. A 1999, 59, 1457–1460. [Google Scholar] [CrossRef]
- Higgins, M.D.; Greene, C.H. Three and four identical fermions near the unitary limit. Phys. Rev. A 2022, 106, 023304. [Google Scholar] [CrossRef]
- Andrews, D.L. Symmetry and quantum features in optical vortices. Symmetry 2021, 13, 1368. [Google Scholar] [CrossRef]
- Fernandez, F.M. Introduction to Perturbation Theory in Quantum Mechanics; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Higgins, M.D.; Greene, C.H.; Kievsky, A.; Viviani, M. Nonresonant density of states enhancement at low energies for three or four neutrons. Phys. Rev. Lett. 2020, 125, 052501. [Google Scholar] [CrossRef]
- Znojil, M. Perturbation theory near degenerate exceptional points. Symmetry 2020, 12, 1309. [Google Scholar] [CrossRef]
- Mineo, H.; Chao, S.D. Split kinetic energy method for quantum systems with competing potentials. Ann. Phys. 2012, 327, 2061–2073. [Google Scholar] [CrossRef]
- Chen, Y.; Chao, S.D. Negative Mass Can Be Positively Useful in Quantum Mechanics. J. Chin. Chem. Soc. 2018, 65, 654–666. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Chao, S.D. The kinetic energy partition method applied to a confined quantum harmonic oscillator in a one-dimensional box. Chin. J. Phys. 2018, 56, 584–592. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Chao, S.D. Kinetic energy partition method applied to ground state helium-like atoms. J. Chem. Phys. 2017, 146, 124120. [Google Scholar] [CrossRef]
- Benavides-Riveros, C.L.; Toranzo, I.V.; Dehesa, J.S. Entanglement in N-harmonium: Bosons and fermions. J. Phys. B At. Mol. Opt. Phys. 2014, 47, 195503. [Google Scholar] [CrossRef]
- Fernández, F.M. Comment on: “The kinetic energy partition method applied to quantum eigenvalue problems with many harmonic-oscillator potentials” by Y.-H. Chen and SD Chao. J. Math. Chem. 2018, 56, 1511–1514. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Chao, S.D. Solving many-body Schrödinger equations with kinetic energy partition method. Ann. Phys. 2018, 388, 54–68. [Google Scholar] [CrossRef]
- Armstrong, J.R.; Volosniev, A.G.; Fedorov, D.V.; Jensen, A.S.; Zinner, N.T. Analytic solutions of topologically disjoint systems. J. Phys. A Math. Theor. 2015, 48, 085301. [Google Scholar] [CrossRef]
- Mehta, N.P. Born-Oppenheimer study of two-component few-particle systems under one-dimensional confinement. Phys. Rev. A 2014, 89, 052706. [Google Scholar] [CrossRef]
- Armstrong, J.R.; Zinner, N.T.; Fedorov, D.V.; Jensen, A.S. Analytic harmonic approach to the N-body problem. J. Phys. B At. Mol. Opt. Phys. 2011, 44, 055303. [Google Scholar] [CrossRef]
- Alon, O.E. Analysis of a trapped Bose–Einstein condensate in terms of position, momentum, and angular-momentum variance. Symmetry 2019, 11, 1344. [Google Scholar] [CrossRef]
- Vitória, R.L.L.; Belich, H. Harmonic oscillator in an environment with a pointlike defect. Phys. Scr. 2019, 94, 125301. [Google Scholar] [CrossRef]
- Pattanayak, A.; Dixit, G. Influence of vacancy defects in solid high-order harmonic generation. Phys. Rev. A 2020, 101, 013404. [Google Scholar] [CrossRef]
- Kościk, P.; Kuroś, A.; Pieprzycki, A.; Sowiński, T. Pair-correlation ansatz for the ground state of interacting bosons in an arbitrary one-dimensional potential. Sci. Rep. 2021, 11, 13168. [Google Scholar] [CrossRef]
- Wan, J.-Y.; Wang, X.; Zhang, X.; Meng, Y.-L.; Wang, W.-L.; Sun, Y.; Liu, L. Quasi-one-dimensional diffuse laser cooling of atoms. Phys. Rev. A 2022, 105, 033110. [Google Scholar] [CrossRef]
- Kościk, P. Quantum entanglement of two harmonically trapped dipolar particles. Few-Body Syst. 2015, 56, 107–114. [Google Scholar] [CrossRef]
- Mistakidis, S.; Volosniev, A.; Barfknecht, R.; Fogarty, T.; Busch, T.; Foerster, A.; Schmelcher, P.; Zinner, N. Few-body Bose gases in low dimensions—A laboratory for quantum dynamics. Phys. Rep. 2023, 1042, 1–108. [Google Scholar] [CrossRef]
- Greene, C.H.; Giannakeas, P.; Pérez-Ríos, J. Universal few-body physics and cluster formation. Rev. Mod. Phys. 2017, 89, 035006. [Google Scholar] [CrossRef]
- Cohen, L.; Lee, C. Exact reduced density matrices for a model problem. J. Math. Phys. 1985, 26, 3105–3108. [Google Scholar] [CrossRef]








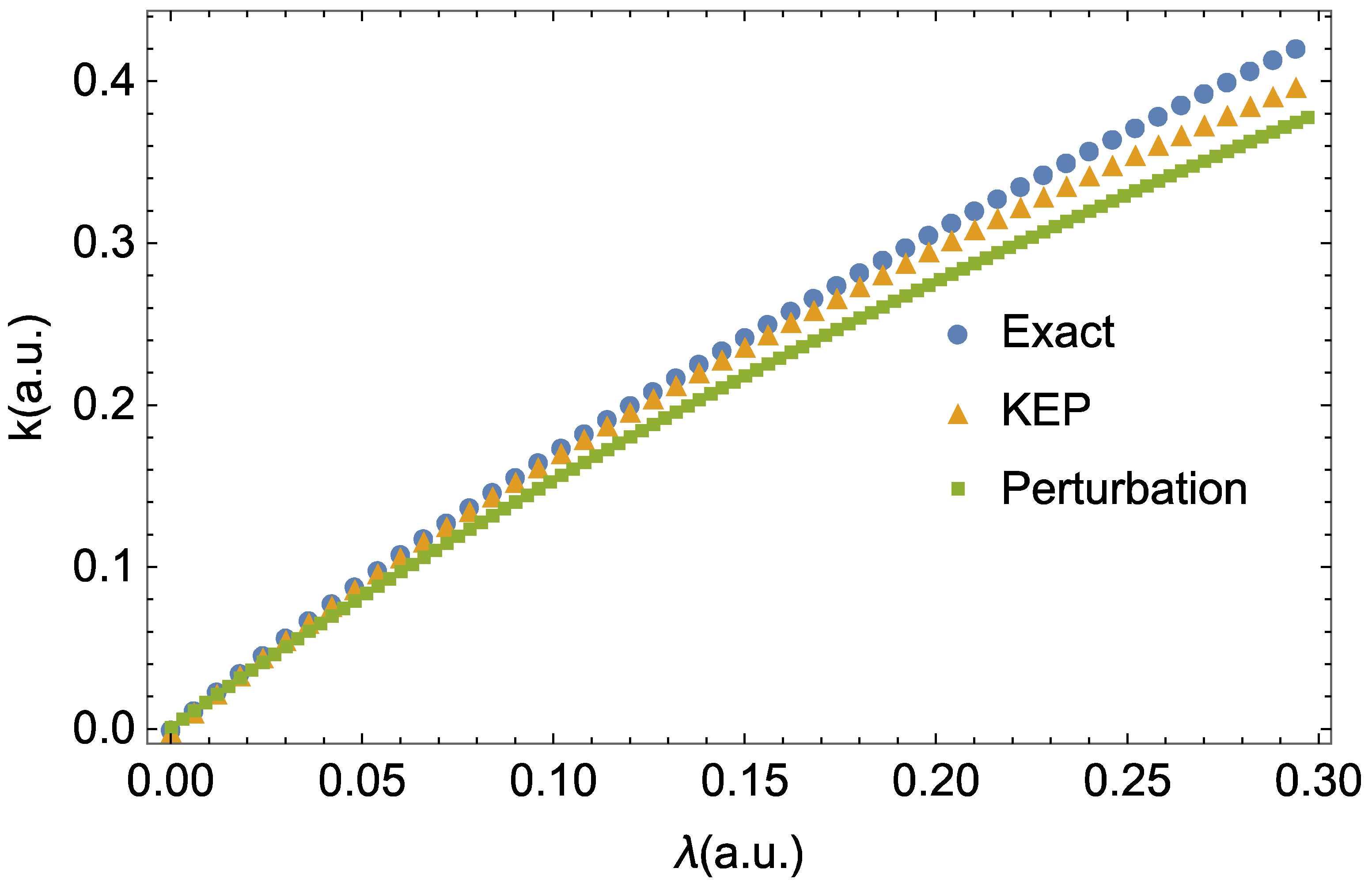{kind=link}
{kind=link}
{kind=link}
{kind=link}
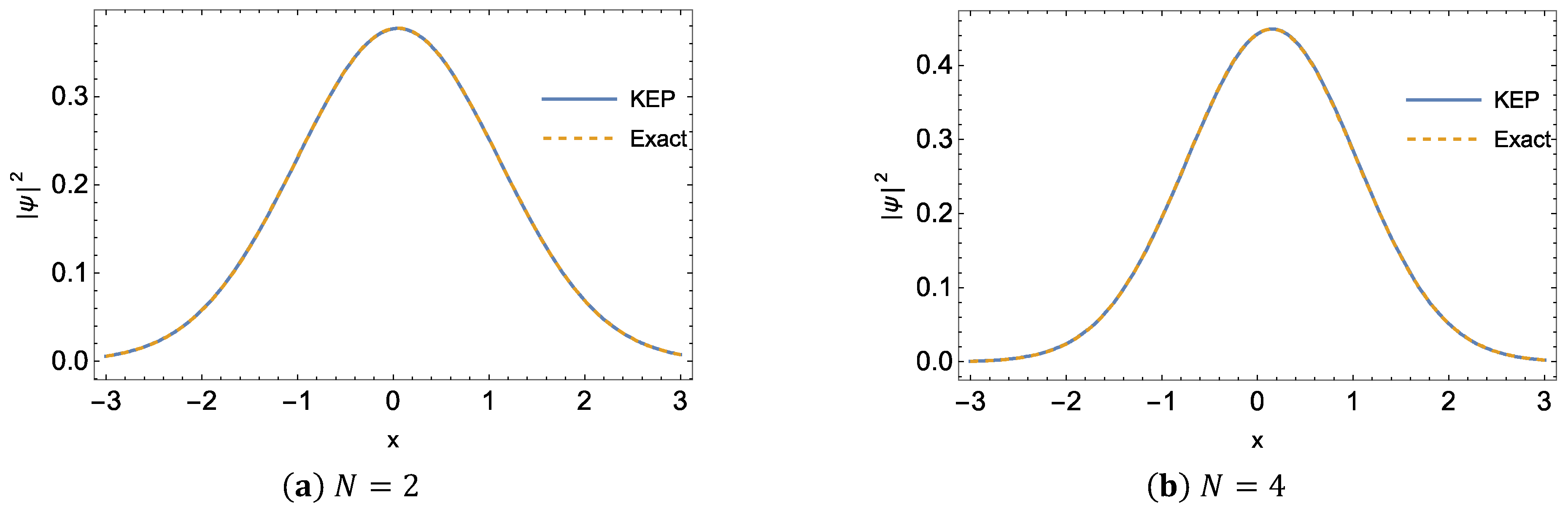{kind=link}
{kind=link}
{kind=link}
| N | KEP (a.u.) | Exact (a.u.) | Error (%) |
|---|---|---|---|
| 1 | 0.15811 | 0.15811 | 0 |
| 2 | 0.22386 | 0.22386 | 0 |
| 3 | 0.27486 | 0.27486 | 0 |
| 4 | 0.31873 | 0.31873 | 0 |
| 5 | 0.35848 | 0.35855 | 0.0195 |
| 6 | 0.39419 | 0.39605 | 0.4696 |
| 7 | 0.43236 | 0.43233 | 0.0069 |
| 8 | 0.46822 | 0.46821 | 0.0021 |
| 9 | 0.50433 | 0.50434 | 0 |
| 10 | 0.54154 | 0.54125 | 0.0535 |
| k/K | KEP | AKEP | Exact | Error for KEP (%) | Error for AKEP (%) |
|---|---|---|---|---|---|
| 2.0 | 0.101 | 0.098 | 0.071 | 42.5 | 39.13 |
| 2.1 | 0.106 | 0.103 | 0.088 | 19.6 | 17.1 |
| 2.5 | 0.123 | 0.121 | 0.114 | 7.6 | 6.1 |
| 3.0 | 0.142 | 0.141 | 0.137 | 4.2 | 3.2 |
| 3.5 | 0.160 | 0.158 | 0.155 | 3.1 | 2.3 |
| 4.0 | 0.175 | 0.174 | 0.171 | 2.6 | 2.0 |
| 4.5 | 0.190 | 0.189 | 0.185 | 2.4 | 1.9 |
| 5.0 | 0.203 | 0.202 | 0.198 | 2.4 | 1.9 |
| 5.5 | 0.216 | 0.215 | 0.211 | 2.4 | 2.0 |
| 6.0 | 0.228 | 0.227 | 0.223 | 2.4 | 2.1 |
| 6.5 | 0.239 | 0.239 | 0.234 | 2.5 | 2.2 |
| 8.0 | 0.271 | 0.270 | 0.264 | 2.7 | 2.4 |
| 10.0 | 0.308 | 0.308 | 0.300 | 2.9 | 2.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-H.; Wu, I.-H.; Chao, S.D. A Mini-Review of the Kinetic Energy Partition Method in Quantum Mechanics. Symmetry 2024, 16, 290. https://doi.org/10.3390/sym16030290
Chen Y-H, Wu I-H, Chao SD. A Mini-Review of the Kinetic Energy Partition Method in Quantum Mechanics. Symmetry. 2024; 16(3):290. https://doi.org/10.3390/sym16030290
Chicago/Turabian StyleChen, Yu-Hsin, I-Huan Wu, and Sheng D. Chao. 2024. "A Mini-Review of the Kinetic Energy Partition Method in Quantum Mechanics" Symmetry 16, no. 3: 290. https://doi.org/10.3390/sym16030290