In recent decades, azides have received a lot of interest as useful and versatile intermediates in synthetic organic chemistry, serving as precursors among others of amines, amides, or heterocycles like pyrroles, pyridines, and 1,2,3-triazoles [
6,
7,
8,
9]. Additionally, they are commonly employed in chemical biology and pharmaceutical chemistry [
10].
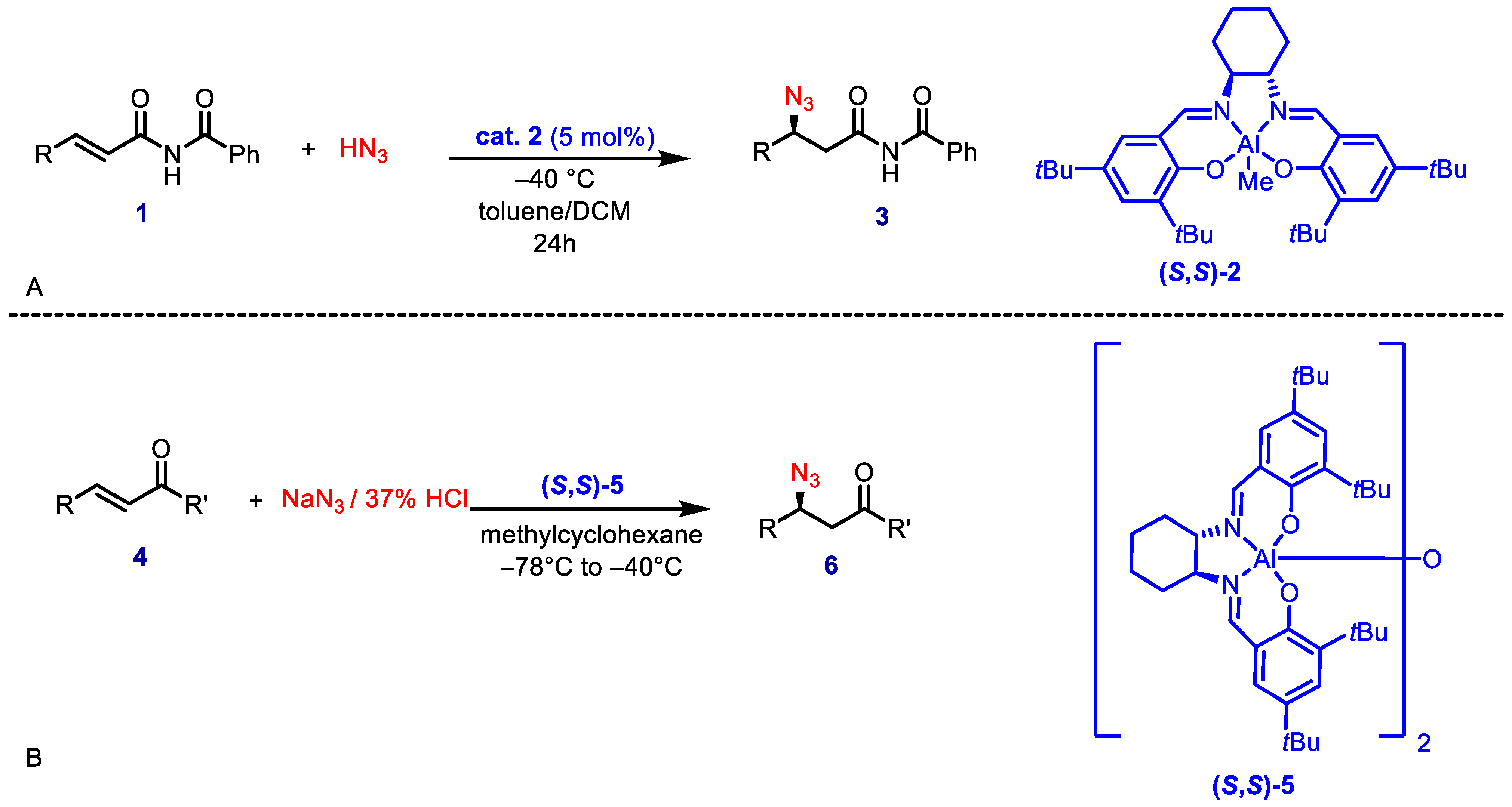
In 1999, Jacobsen and coworkers described the asymmetric synthesis of β-amino acid derivatives via conjugate addition of hydrazoic acid to unsaturated imides (
Scheme 1A) in the presence of a chiral (salen)Al(III) complex [
12]. Subsequently, the same research group reported the asymmetric hydroazidation of α,β-unsaturated ketones (
Scheme 1B) using a similar catalytic system [
13]. The major drawback of this highly enantioselective methodology is the high toxicity and explosivity of hydrazoic acid.
2.1. Organocatalyzed Enantioselective Hydroazidation
Various research groups, with the purpose of meeting green chemistry principles, explored enantioselective metal-free catalyzed azidation reactions under mild conditions, avoiding the direct use of hydrazoic acid as a nucleophilic azide source.
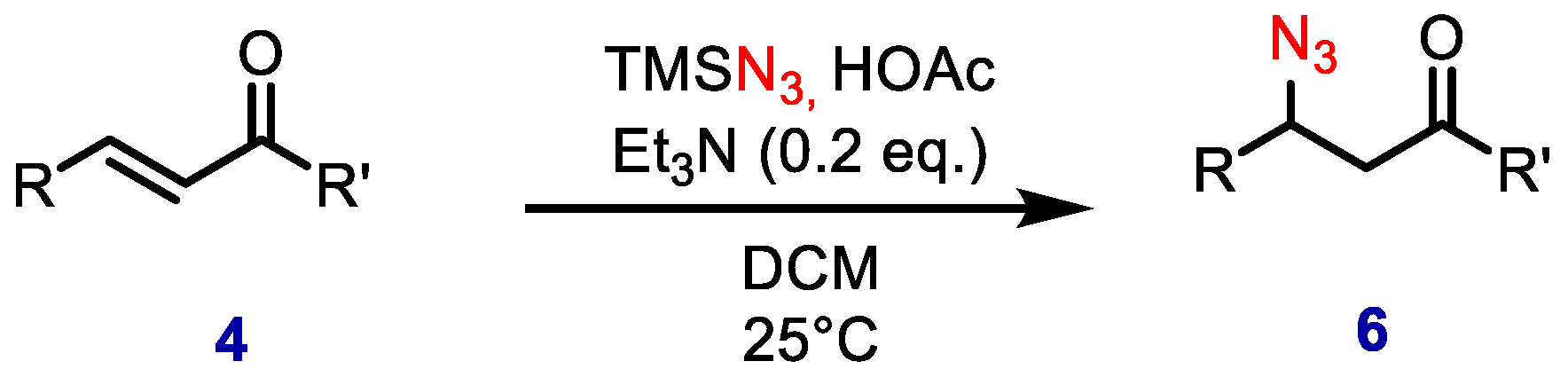
The first organocatalytic hydroazidation was reported by Miller and collaborators in 1999 [
14]. Tertiary amines were employed as catalysts for the β-azidation of α,β-unsaturated carbonyl compounds. The azide source was generated by mixing TMSN
3 and AcOH (
Scheme 2).
In 2000, the same research group reported the asymmetric organocatalytic hydroazidation promoted by the small β-turn peptide derivative
7, armed with a τ-(benzyl)-His residue (
Table 1) [
15].
Based on these results, Miller and coworkers in 2002 elaborated an enantioselective azidation/cycloaddition sequence achieving optically enriched triazoles and triazolines [
16].
Various organocatalytic systems have been developed to carry out the enantioselective conjugate addition of the azide group to unsaturated nitroalkenes.
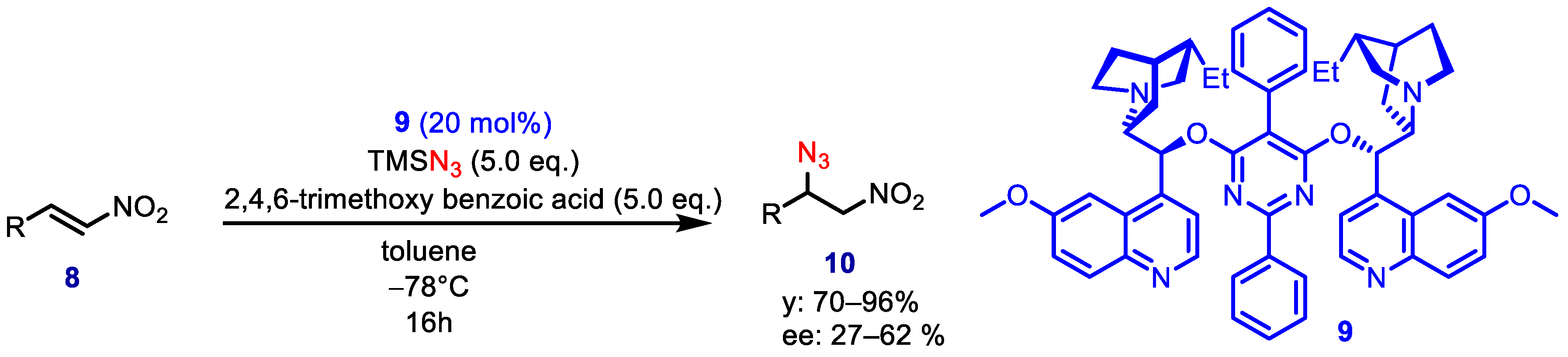
In 2007, Jørgensen and coworkers described the first asymmetric conjugate addition of azide to α,β-unsaturated nitroalkenes catalyzed by
Cinchona alkaloids derivatives [
17]. In this methodology, the simultaneous presence of TMSN
3 and a carboxylic acid provided hydrazoic acid in
situ. The best
Cinchona alkaloid-derived catalyst
9 led to adducts in high conversions but moderate enantioselectivities (27–62% ee) (
Scheme 3).
This process turned out to be strongly dependent on the steric and electronic nature of the acid additive. As a matter of example, the reaction of 1-nitro-hept-1-ene performed in the presence of benzoic acid led to 50% ee, whereas AcOH and 2,4,6-trimethoxy benzoic acid furnished 57% ee and 62% ee, respectively.
In 2015, Della Sala and collaborators reported the asymmetric hydroazidation of nitroalkenes promoted by the secondary amine-thiourea catalyst (
11) [
18]. After a thorough screening of bifunctional catalysts, the asymmetric hydroazidation of various nitroalkenes in the presence of TMSN
3 and AcOH was achieved with a good level of enantioselectivity (71–82% ee), as reported in
Table 2. The only exception, in terms of enantioselectivity (39% ee), is represented by nitrostyrene (
Table 2, entry 7). However, it would be stressed that this is the first example of asymmetric hydroazidation of nitrostyrenes.
A tandem hydroazidation–hydroxylation reaction of α,β-unsaturated aldehydes was realized by Jang in 2014 by using TMSN
3, TEMPO, FeCl
3·6H
2O, and the Jørgensen–Hayashi catalyst
13 [
19]. This methodology afforded optically active α,β-disubstituted aldehydes, which are key intermediates of biologically interesting β-amino α-hydroxy esters [
20,
21,
22]. Under the optimized reaction conditions, diverse α,β-unsaturated aldehydes were used for the tandem azido/TEMPO addition, achieving moderate yields (42–71%) and good enantioselectivities (71–90% ee) (
Table 3).
Luo and coworkers reported, in 2017, the first example of asymmetric hydroazidation of α-substituted vinyl ketones carried out with TMSN
3 and a chiral primary tertiary diamine catalyst (
17) [
23]. This transformation was performed under mild reaction conditions, achieving good yields (56–91%) and enantioselectivities, as reported in
Table 4.
With the aim of exploring the ability of hydrogen bonding amine bifunctional organocatalysts to activate TMSN
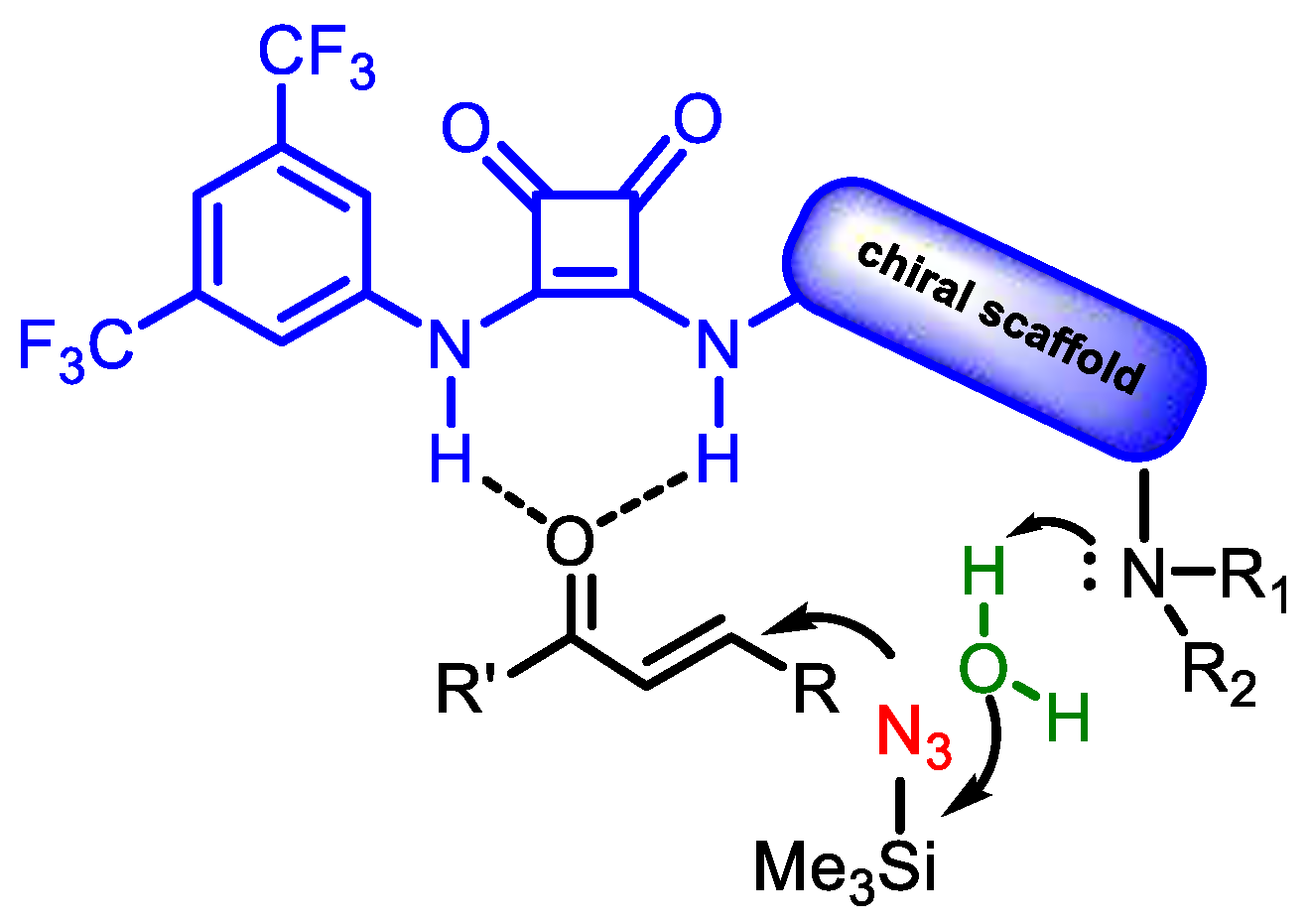
3 and direct the enantioselective addition to Michael acceptors, Aleman and coworkers, in 2019, reported the asymmetric hydroazidation of α,β-unsaturated ketones using the bifunctional squaramide
19. This catalyst proved capable of simultaneously activating the enone and the TMSN
3 without using any carboxylic acid additive [
24]. The presence of trace amounts of water was found to be essential to activate TMSN
3 and promote the conjugate addition without generating free hydrazoic acid. DFT calculations demonstrated that the desilylation of TMSN
3 and generation of azide anion is carried out by an H
2O molecule pre-coordinated to the tertiary nitrogen atom of the catalyst (
Figure 1).
Using the optimized reaction condition, various differently substituted α,β-unsaturated ketones were tested, resulting in good yields and enantioselectivities as described in
Table 5.
2.2. Organocatalyzed Enantioselective Hydrocyanation
The asymmetric conjugate addition of cyanide to α,β-unsaturated carbonyl derivatives was first accomplished by Jacobsen [
25,
26,
27] using chiral aluminum salen complexes and by Shibasaki [
28] using chiral gadolinium catalyst, producing highly valuable chiral building blocks for pharmaceuticals.
Bifunctional compounds, such as β-amino acids, can be synthesized from β-nitro nitriles. The simple pathway to such molecules, according to an intuitive retrosynthesis study, involves a direct conjugate cyanide addition to nitroalkenes. The great tendency of nitroalkenes to polymerize under basic conditions, however, limits the development of this reaction.
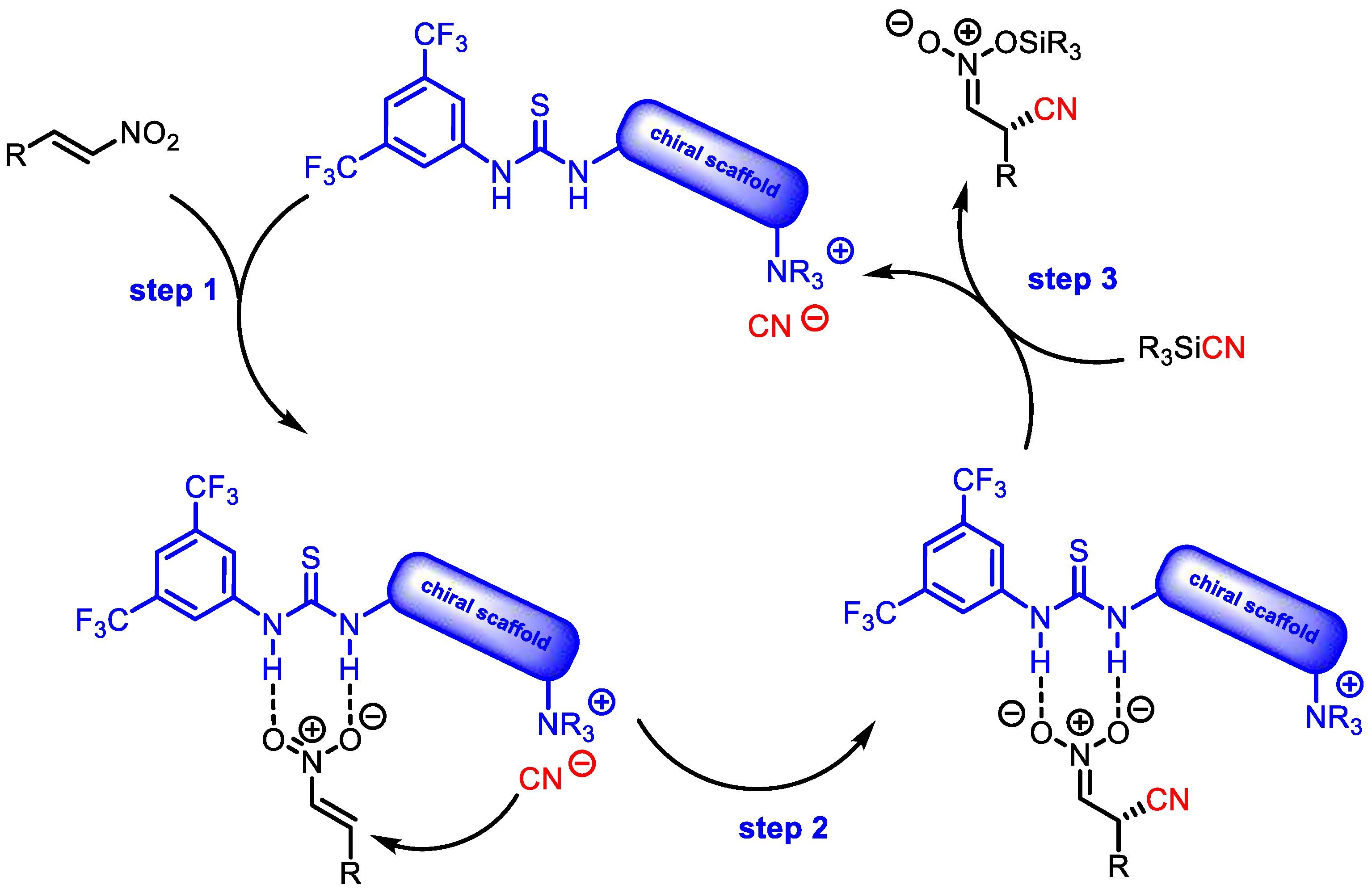
In 2010, Lassaletta and coworkers decided to explore the asymmetric unprecedented cyanosilylation of nitroalkenes [
29]. The employment of hydrogen bonding bifunctional tertiary amine organocatalysts resulted in disappointing conversions, whereas much better performances were achieved by using bifunctional
Cinchona alkaloids derived from halide or cyanide ammonium salts. After an in-depth screening of
Cinchona alkaloid derivatives, the model reaction was efficiently catalyzed by
21 in TBME. The products
22 were always produced with excellent yields and good enantioselectivities when with a variety of aliphatic substrates (
Table 6). The authors proposed a mechanism involving the activation of TMSCN triggered by the nucleophilic attack of the halide or cyanide anion (
Figure 2).
In the key stereoselective cyanation step, the CN– counterion attacks the substrate bound to the thiourea moiety.
Both methods of Jacobsen and Lassaletta use trimethylsilyl cyanide (TMSCN), an expensive source of cyanide ions. In 2010, Ricci and collaborators [
30] started their investigation using acetone cyanohydrin as a cyanide donor under phase-transfer conditions for the addition to β,β-disubstituted nitroolefins promoted by
Cinchona alkaloids derived catalysts (
Table 7).
The organocatalytic ion pair is generated by the base-promoted decomposition of cyanohydrin liberating cyanide ion. The transfer of the C-nucleophile to the electrophilic nitroolefin’s conjugated site then occurs.
Some years later, Deng and coworkers [
31] employed cupreidinium salts for the asymmetric 1,4-addition of cyanide to enones with acetone cyanohydrin and Cs
2CO
3 in toluene/CHCl
3 (
Scheme 4).
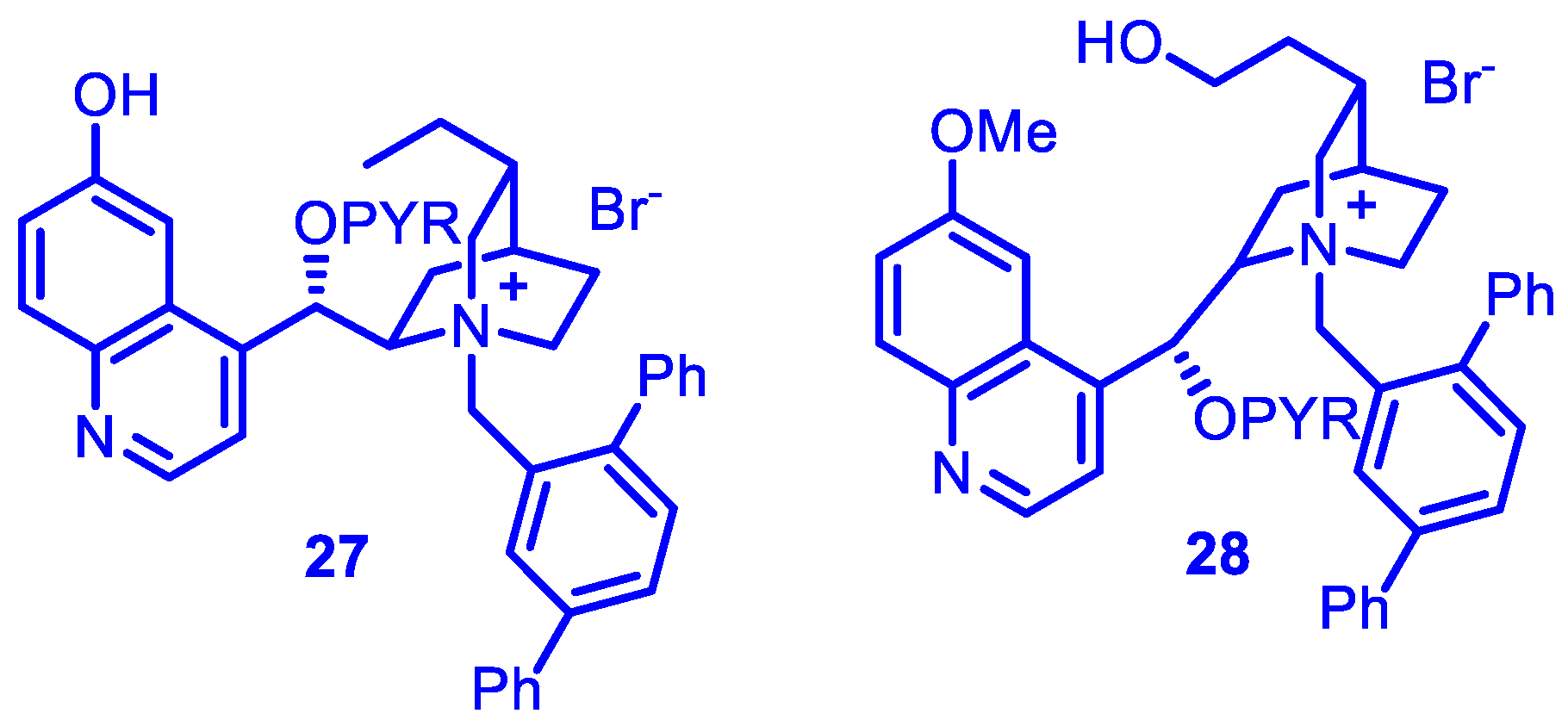
Using the best PTC catalysts (
27 and
28) (
Figure 3), a wide range of acyclic enones bearing linear and branched alkyl groups as the β substituents performed satisfactorily (
Table 8).
In 2010, Chen and coworkers described an enantioselective 1,4-addition of TMSCN to aromatic chalcones catalyzed by a chiral sodium phosphate [
32]. The catalytic sodium salt was generated in situ from the corresponding phosphoric acid and sodium hydroxide. After a screening of BINOL-derived phosphoric acid salts, the best catalysts was found to be a derivative bearing bulky adamantyl groups at 3,3′ positions with excellent yields (86–99%) and moderate enantioselectivities (53–72% ee).
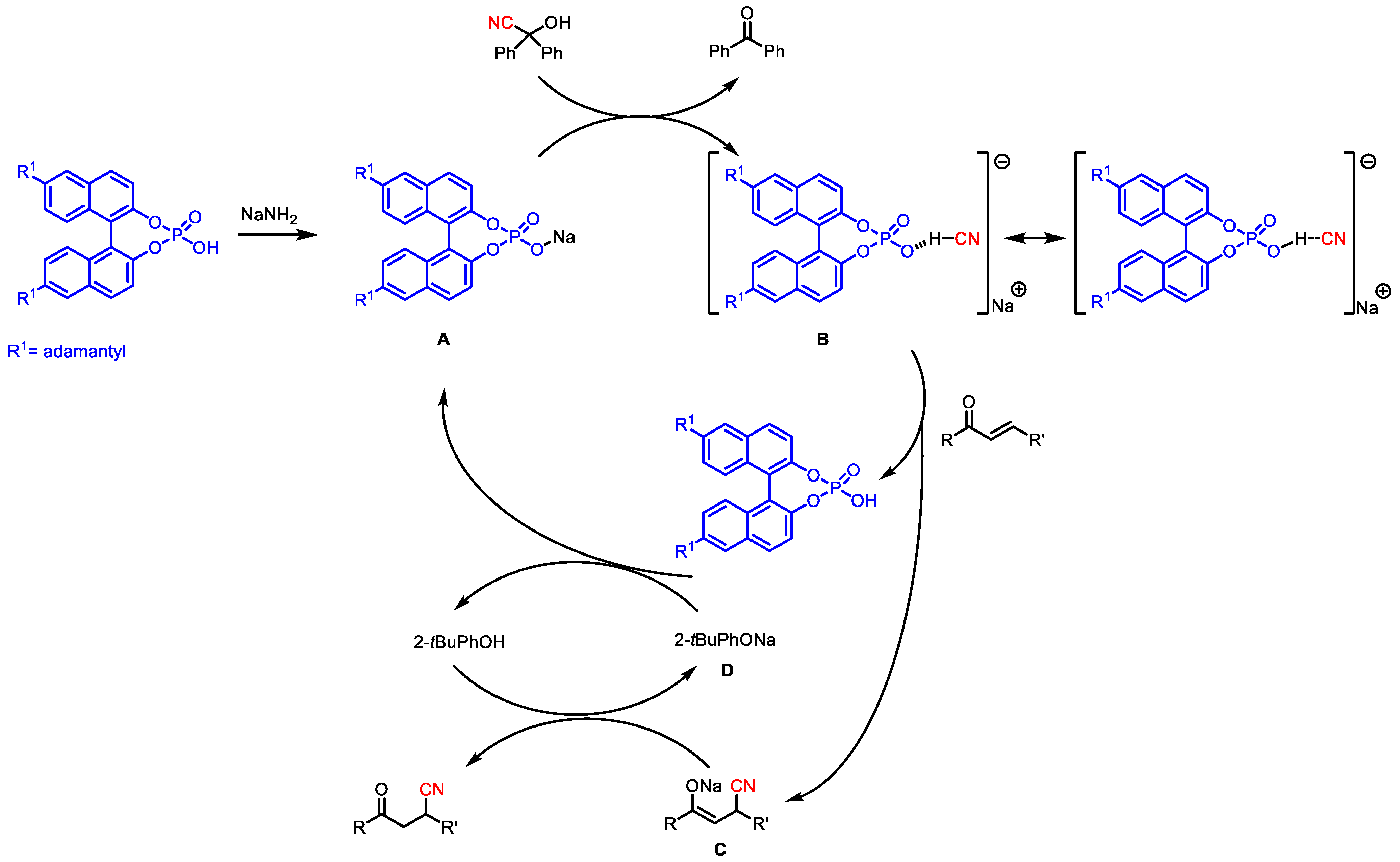
Later, in 2013, the same research group reported the asymmetric conjugate hydrocyanation of enones with benzophenone cyanohydrin catalyzed by an anionic chiral phosphate catalyst [
33]. The best catalyst was
31, bearing adamantyl substituents at 6,6′ positions. In the scope of reaction (
Table 9), all the chalcone analogs exhibited excellent enantioselectivities (92–98% ee) with the exclusive formation of 1,4-adducts up to 96% yields.
A possible mechanism is described in
Figure 4: firstly, the cyanohydrin decomposes into HCN, reacting with the in situ generated sodium phosphate A, the real catalyst, to form the negative-charged intermediate B. After being altered by the chiral anion via hydrogen bonding, the HCN nucleophile gave an asymmetric conjugate addition to the enone to produce a cyano-enolate C. This is then acidified by the phenol additive to produce sodium phenolate D and the hydrocyanation product. Finally, the phenolate D deprotonates the chiral phosphoric acid, regenerating A.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
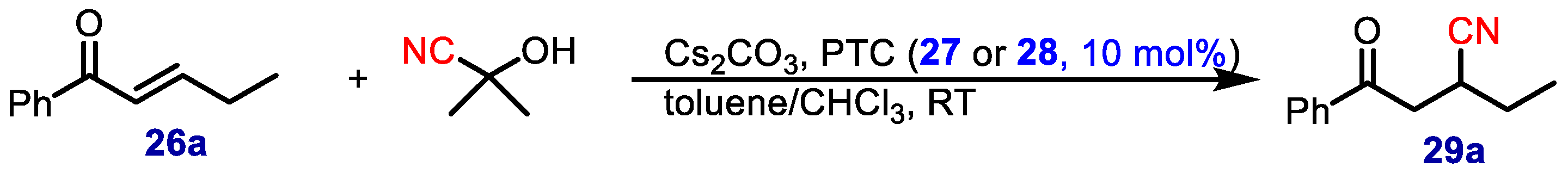{kind=link}









































