
M6A-BERT-Stacking: A Tissue-Specific Predictor for Identifying RNA N6-Methyladenosine Sites Based on BERT and Stacking Strategy

Abstract
:1. Introduction
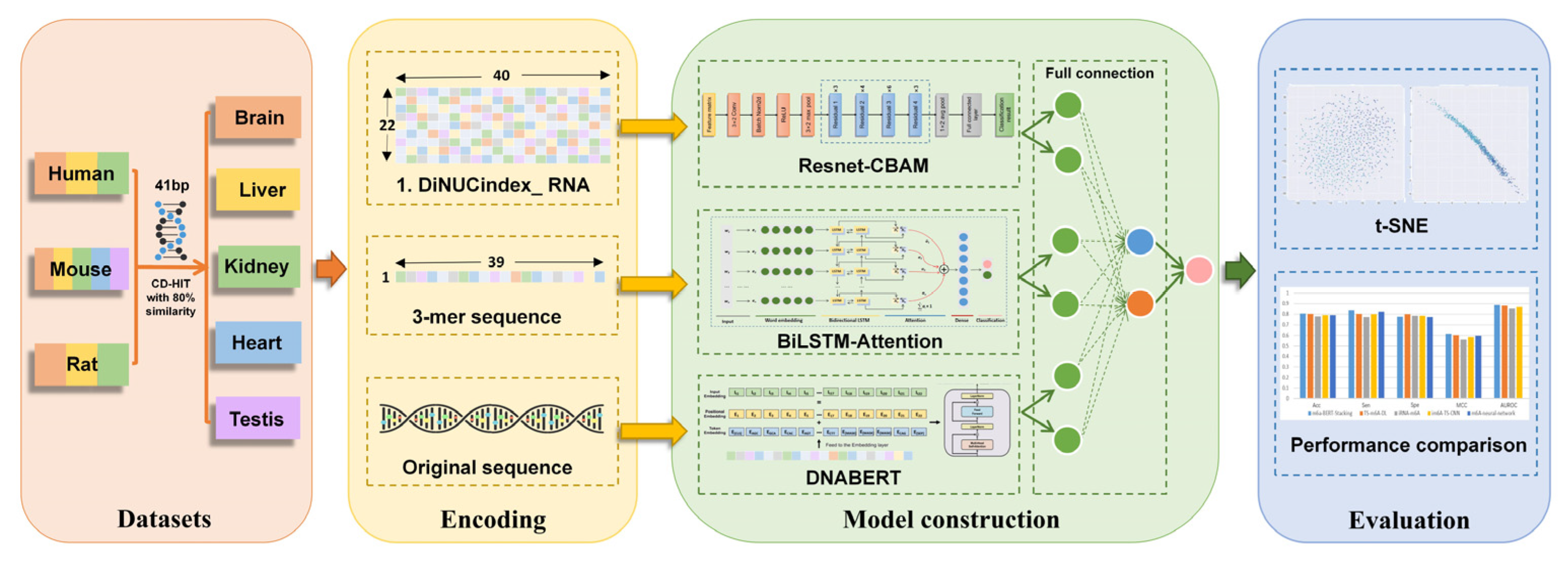
2. Materials and Methods
2.1. Benchmark Datasets
2.2. Feature Encoding Algorithms
2.2.1. DiNUCindex_RNA
2.2.2. K-mer Word Segmentation
2.3. Deep Learning Model Architecture
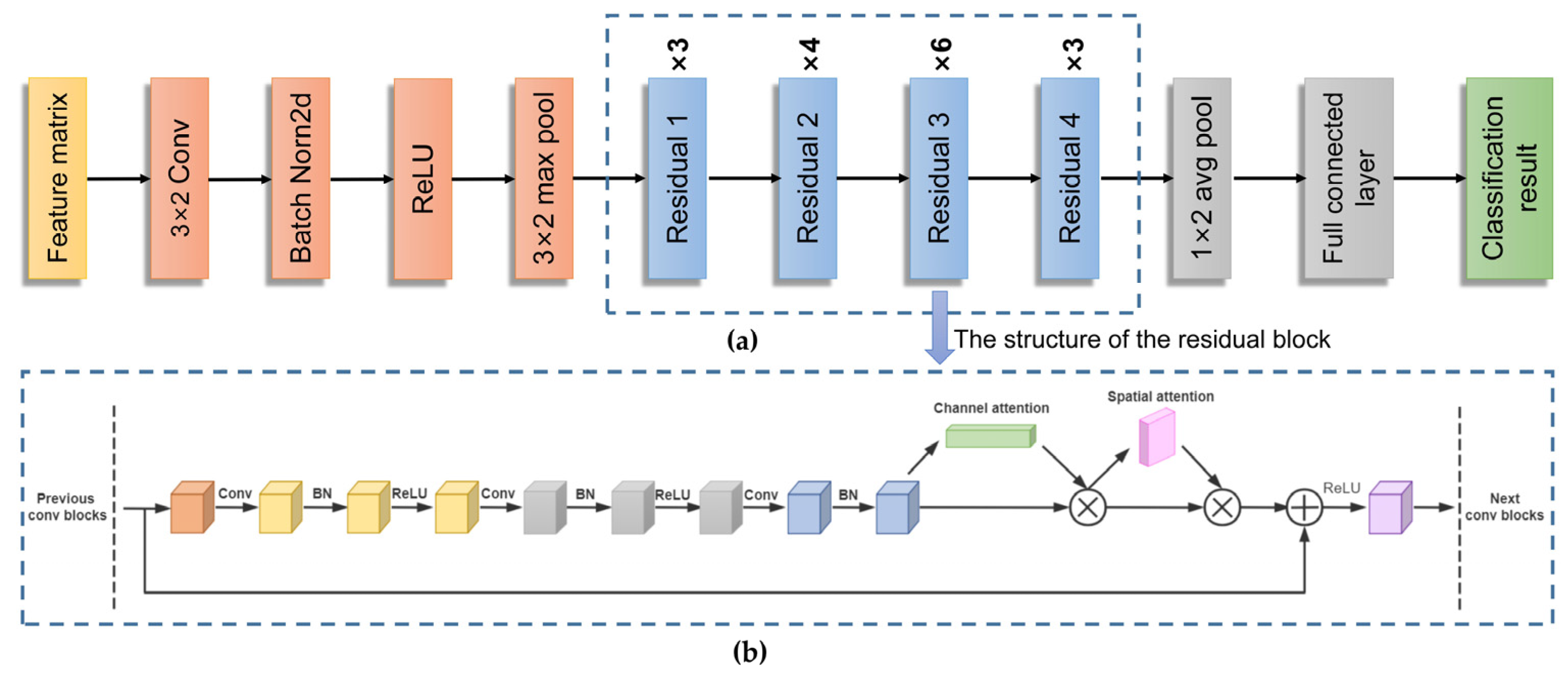
2.3.1. Resnet-CBAM
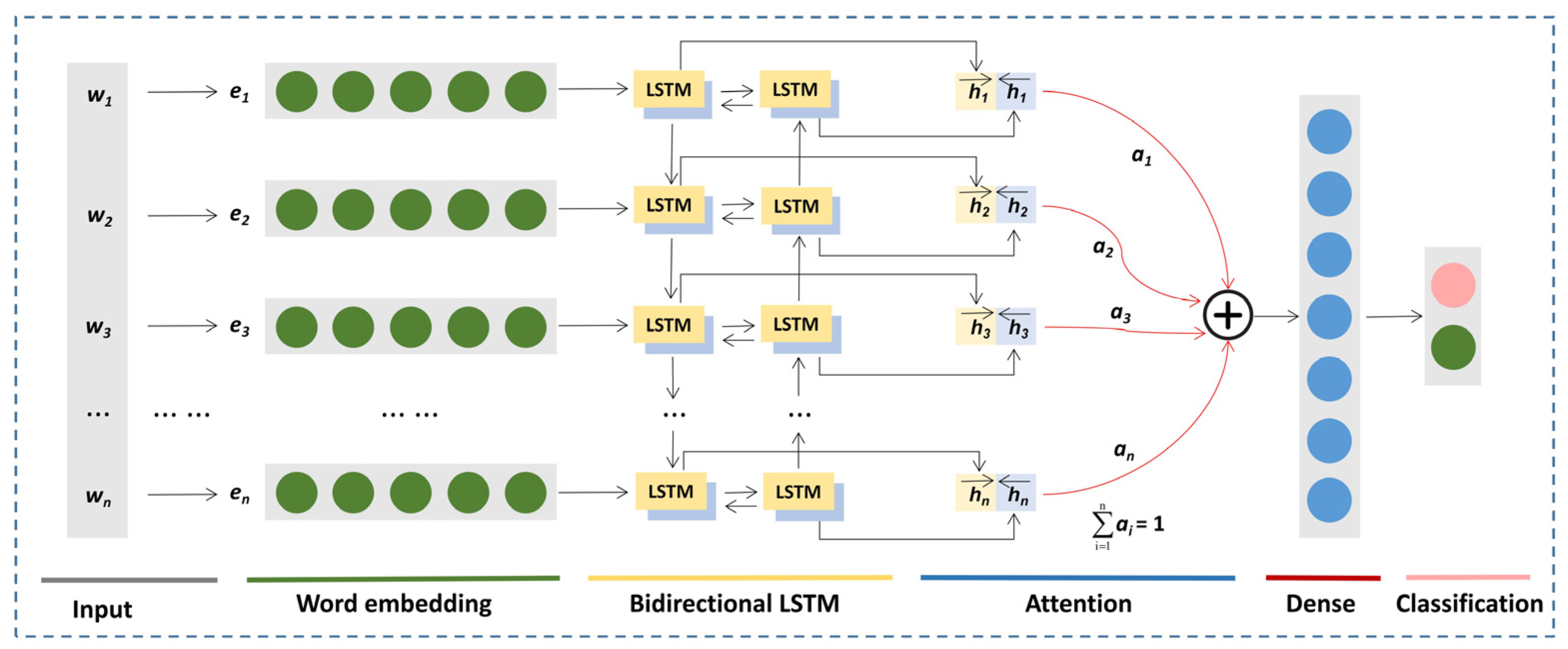
2.3.2. BiLSTM-Attention
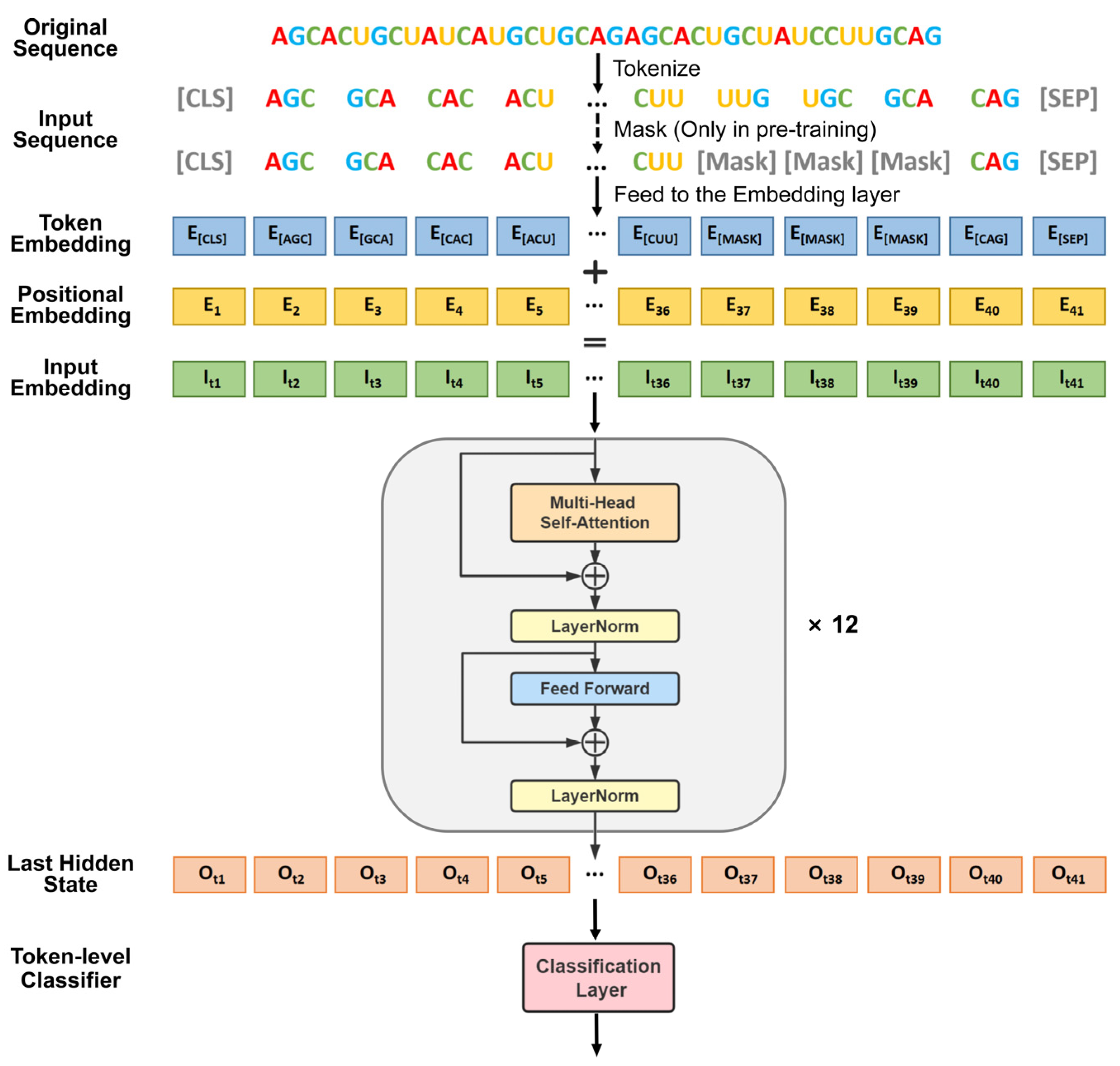
2.3.3. Fine-Tuned DNABERT
2.3.4. Fully Connected Network
2.4. Performance Assessment
3. Results and Discussions
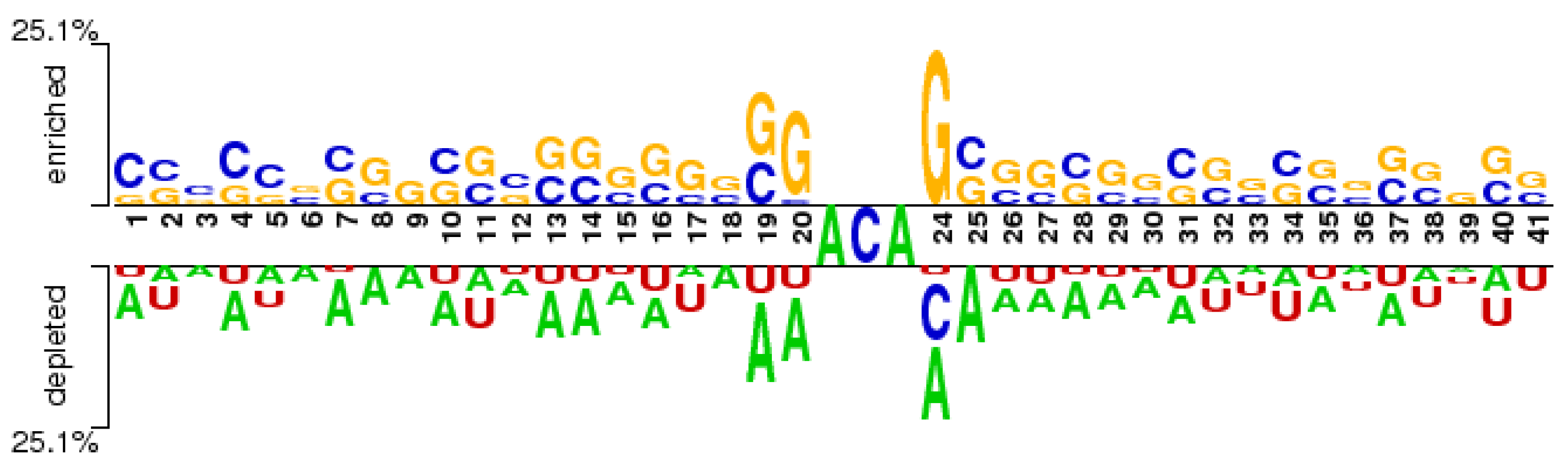
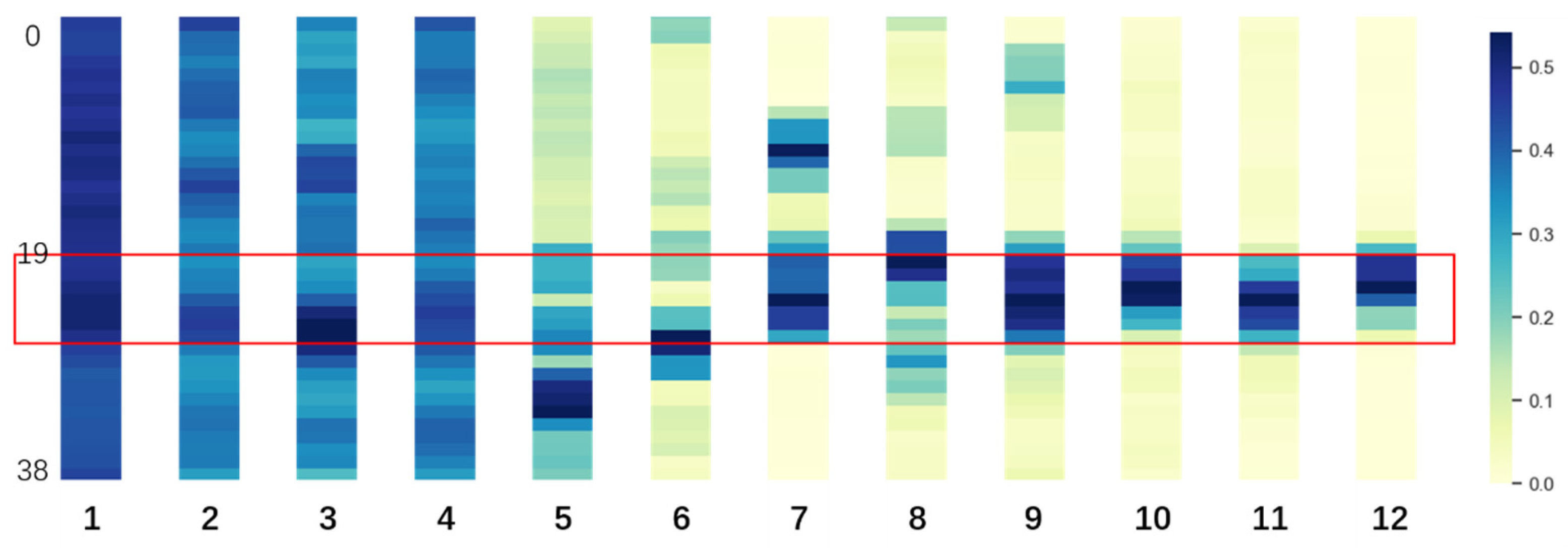
3.1. Fine-Tuned DNABERT Attention Analysis
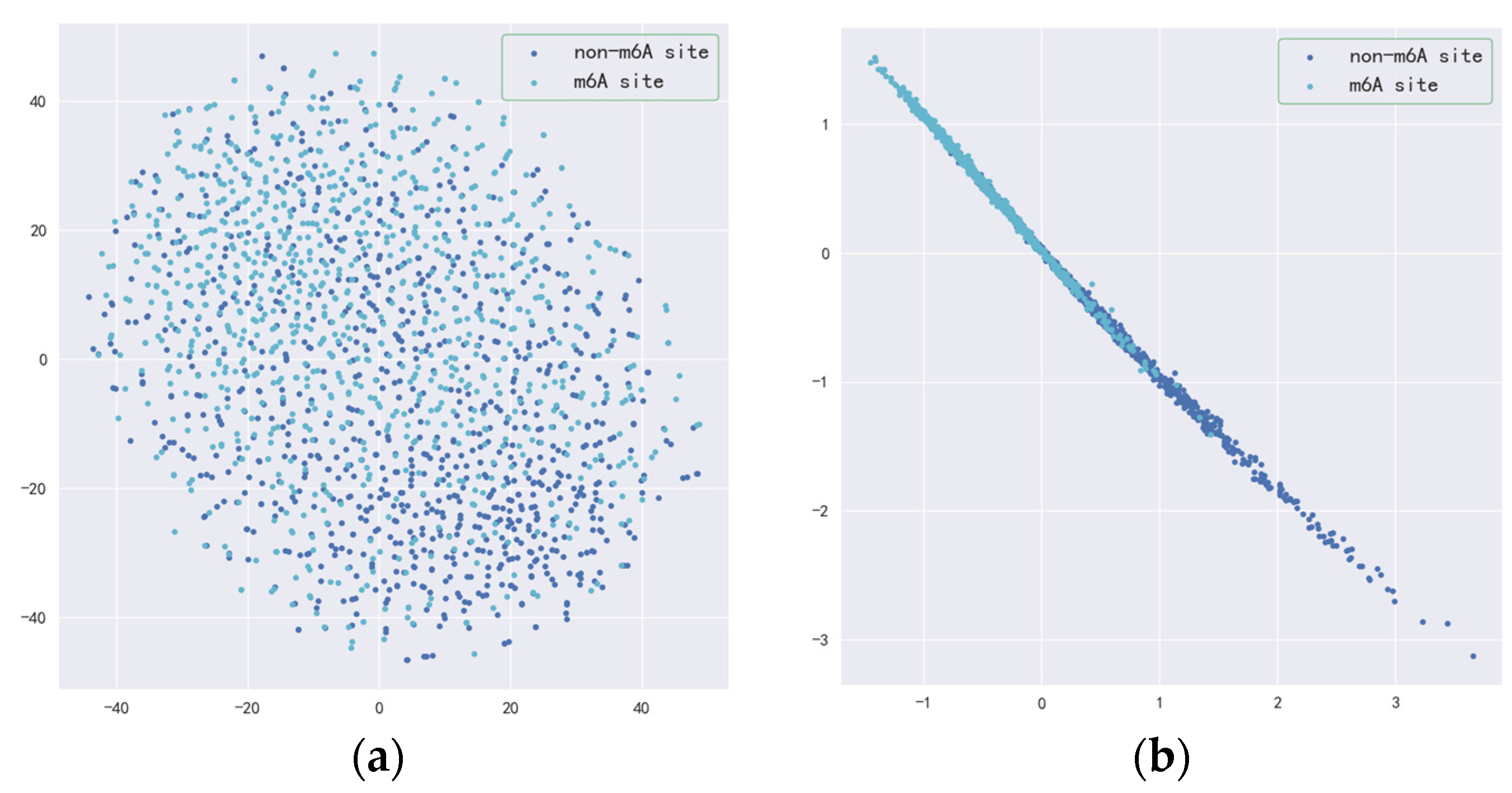
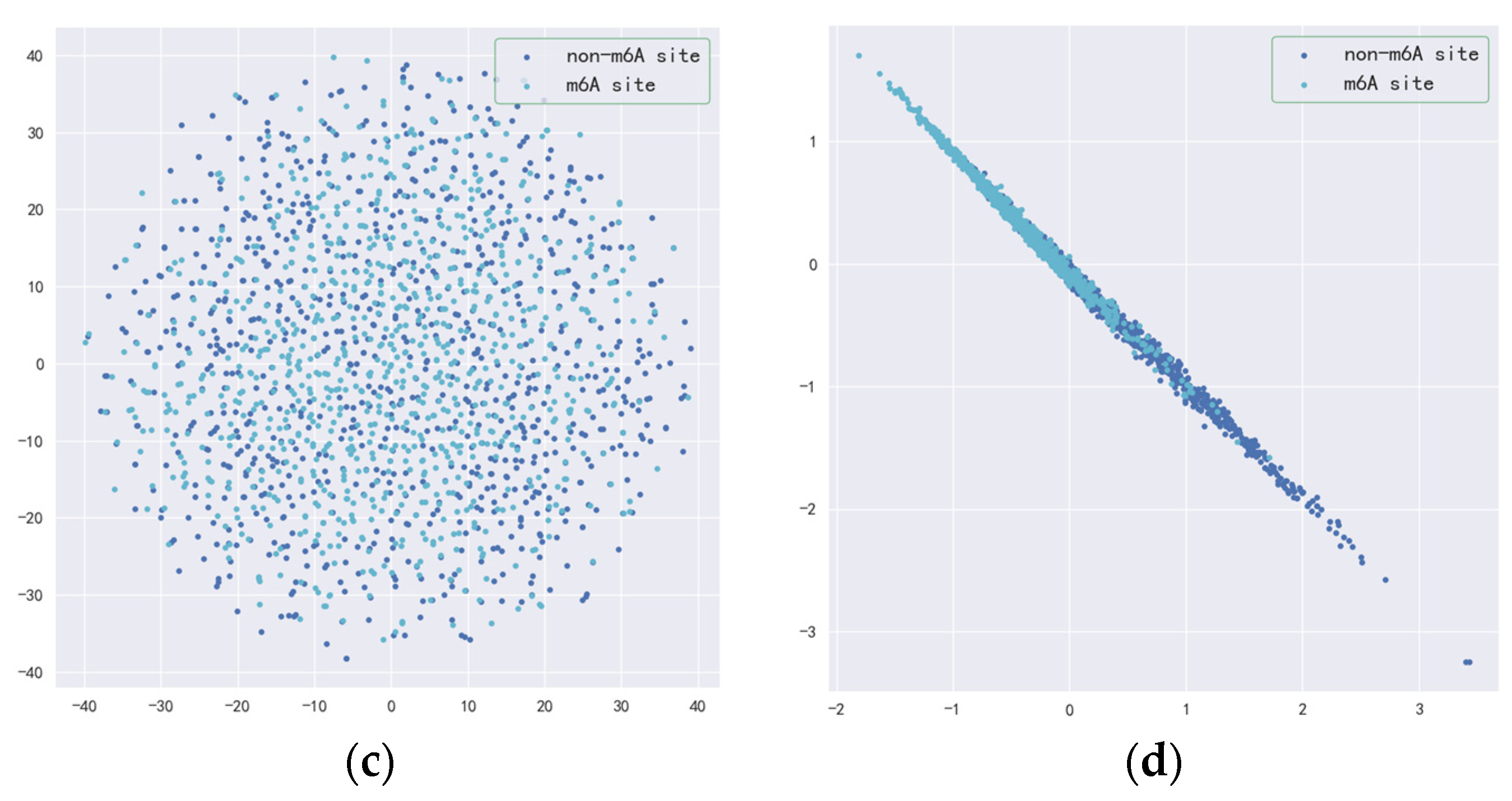
3.2. Validity of Resnet-CBAM and BilSTM-Attention
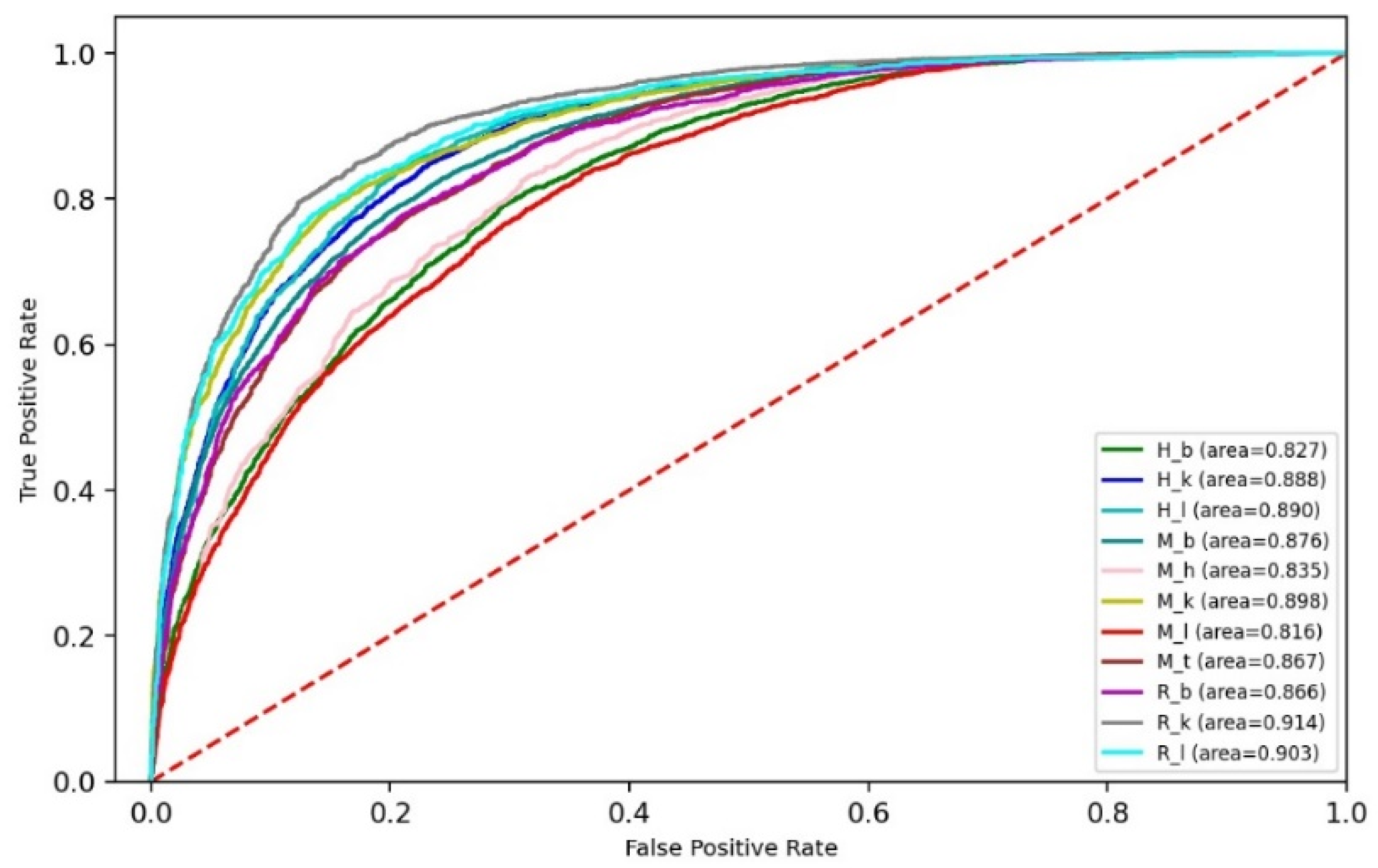
3.3. Performance of Ensemble Models
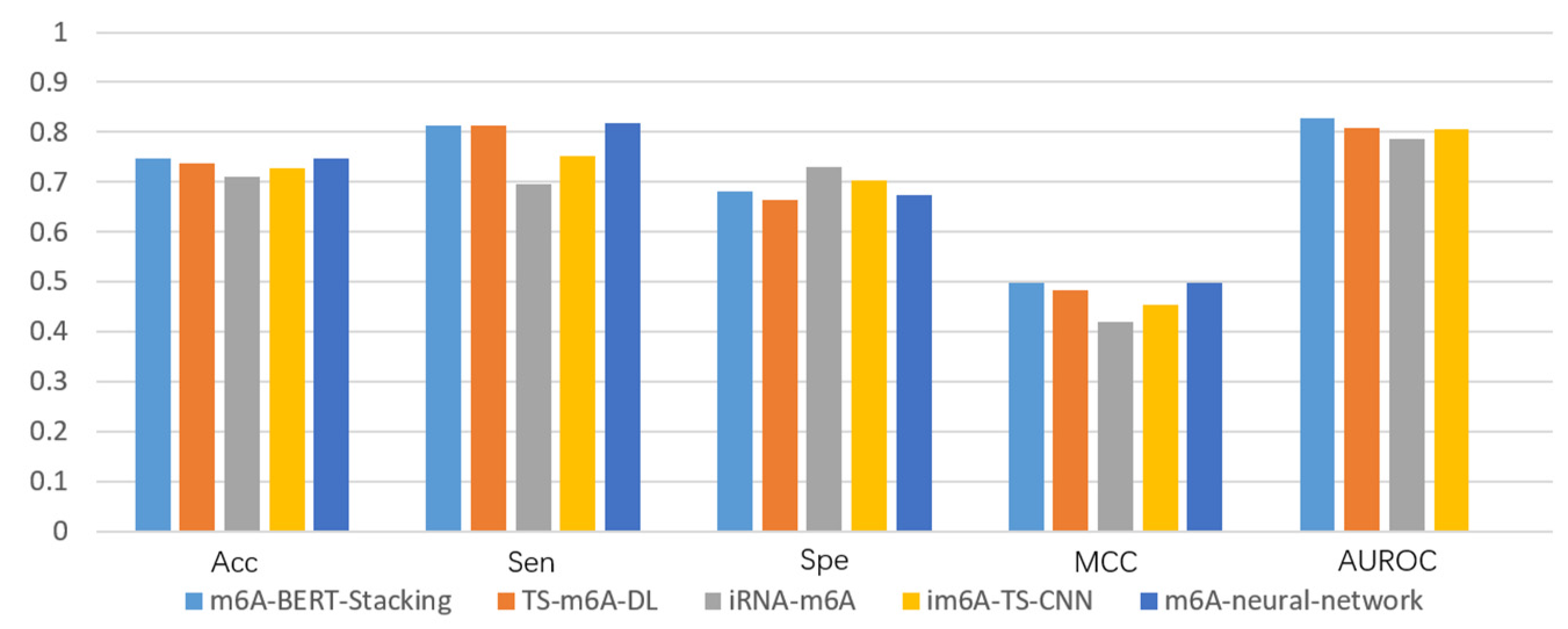
3.4. Performance Comparison with Existing Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Boccaletto, P.; Stefaniak, F.; Ray, A.; Cappannini, A.; Mukherjee, S.; Purta, E.; Kurkowska, M.; Shirvanizadeh, N.; Destefanis, E.; Groza, P.; et al. MODOMICS: A database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022, 50, D231–D235. [Google Scholar] [CrossRef] [PubMed]
- He, P.C.; He, C. m6A RNA methylation: From mechanisms to therapeutic potential. Embo J. 2021, 40, e105977. [Google Scholar] [CrossRef] [PubMed]
- He, L.E.; Li, H.Y.; Wu, A.Q.; Peng, Y.L.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.M.; Luo, H.Z.; Izzo, F.; Pickering, B.F.; Nguyen, D.; Myers, R.; Schurer, A.; Gourkanti, S.; Bruning, J.C.; Vu, L.P.; et al. m6A RNA Methylation Maintains Hematopoietic Stem Cell Identity and Symmetric Commitment. Cell Rep. 2019, 28, 1703–1716. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Wei, Z.; Zhang, Q.; Wu, X.; Rong, R.; Lu, Z.; Su, J.; de Magalhaes, J.P.; Rigden, D.J.; Meng, J. WHISTLE: A high-accuracy map of the human N6-methyladenosine (m6A) epitranscriptome predicted using a machine learning approach. Nucleic Acids Res. 2019, 47, e41. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3 ‘ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Chen, K.; Lu, Z.; Wang, X.; Fu, Y.; Luo, G.-Z.; Liu, N.; Han, D.; Dominissini, D.; Dai, Q.; Pan, T.; et al. High-Resolution N6-Methyladenosine (m6A) Map Using Photo-Crosslinking-Assisted m6A Sequencing. Angew. Chem. Int. Ed. 2015, 54, 1587–1590. [Google Scholar] [CrossRef] [Green Version]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef]
- Zhou, Y.; Zeng, P.; Li, Y.-H.; Zhang, Z.; Cui, Q. SRAMP: Prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016, 44, e91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Feng, P.M.; Ding, H.; Lin, H.; Chou, K.C. iRNA-Methyl: Identifying N6-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015, 490, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Q.; Liu, Z.; Shen, H.B.; Yu, D.J. TargetM6A: Identifying N6-Methyladenosine Sites From RNA Sequences via Position-Specific Nucleotide Propensities and a Support Vector Machine. IEEE Trans. Nanobioscience 2016, 15, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xing, P.W.; Zou, Q. Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Sci. Rep. 2017, 7, 40242. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.F.; Yan, R.X. RFAthM6A: A new tool for predicting m6A sites in Arabidopsis thaliana. Plant Mol. Biol. 2018, 96, 327–337. [Google Scholar] [CrossRef]
- Wei, L.Y.; Chen, H.R.; Su, R. M6APred-EL: A Sequence-Based Predictor for Identifying N6-methyladenosine Sites Using Ensemble Learning. Mol. Ther. Nucleic Acids 2018, 12, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.W.; Zhang, Y.; Ning, Q.; Zhang, H.R.; Ji, J.C.; Yin, M.H. Identifying N6-methyladenosine sites using extreme gradient boosting system optimized by particle swarm optimizer. J. Theor. Biol. 2019, 467, 39–47. [Google Scholar] [CrossRef]
- Govindaraj, R.G.; Subramaniyam, S.; Manavalan, B. Extremely-randomized-tree-based Prediction of N6-Methyladenosine Sites in Saccharomyces cerevisiae. Curr. Genom. 2020, 21, 26–33. [Google Scholar] [CrossRef]
- Zhang, Z.W.; Wang, L.D. Using Chou’s 5-steps rule to identify N6-methyladenine sites by ensemble learning combined with multiple feature extraction methods. J. Biomol. Struct. Dyn. 2022, 40, 796–806. [Google Scholar] [CrossRef]
- Luo, Z.; Lou, L.; Qiu, W.; Xu, Z.; Xiao, X. Predicting N6-Methyladenosine Sites in Multiple Tissues of Mammals through Ensemble Deep Learning. Int. J. Mol. Sci. 2022, 23, 15490. [Google Scholar] [CrossRef]
- Zhang, L.; Qin, X.; Liu, M.; Xu, Z.; Liu, G. DNN-m6A: A Cross-Species Method for Identifying RNA N6-methyladenosine Sites Based on Deep Neural Network with Multi-Information Fusion. Genes 2021, 12, 354. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Xing, P.; Wei, L.; Liu, B. Gene2vec: Gene subsequence embedding for prediction of mammalian N6-methyladenosine sites from mRNA. Rna 2019, 25, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazari, I.; Tahir, M.; Tayara, H.; Chong, K.T. iN6-Methyl (5-step): Identifying RNA N6-methyladenosine sites using deep learning mode via Chou’s 5-step rules and Chou’s general PseKNC. Chemom. Intell. Lab. Syst. 2019, 193, 103811. [Google Scholar] [CrossRef]
- Tahir, M.; Hayat, M.; Chong, K.T. Prediction of N6-methyladenosine sites using convolution neural network model based on distributed feature representations. Neural Netw. 2020, 129, 385–391. [Google Scholar] [CrossRef]
- Wang, H.; Zhao, S.; Cheng, Y.; Bi, S.; Zhu, X. MTDeepM6A-2S: A two-stage multi-task deep learning method for predicting RNA N6-methyladenosine sites of Saccharomyces cerevisiae. Front. Microbiol. 2022, 13, 999506. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, S.Y.; Zhang, Y.; Bi, S.D.; Zhu, X.L. A brief review of machine learning methods for RNA methylation sites prediction. Methods 2022, 203, 399–421. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, P.; Li, F.Y.; Wang, Y.N.; Smith, A.I.; Webb, G.I.; Akutsu, T.; Baggag, A.; Bensmail, H.; Song, J.N. Comprehensive review and assessment of computational methods for predicting RNA post-transcriptional modification sites from RNA sequences. Brief. Bioinform. 2020, 21, 1676–1696. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Yu, Z.M.; Yu, B.; Wang, X.; Gao, H.L.; Sun, J.Q.; Li, S.Y. StackRAM: A cross-species method for identifying RNA N6-methyladenosine sites based on stacked ensemble. Chemom. Intell. Lab. Syst. 2022, 222, 104495. [Google Scholar] [CrossRef]
- Rehman, M.U.; Hong, K.J.; Tayara, H.; Chong, K.T. m6A-NeuralTool: Convolution Neural Tool for RNA N6-Methyladenosine Site Identification in Different Species. IEEE Access 2021, 9, 17779–17786. [Google Scholar] [CrossRef]
- Abbas, Z.; Tayara, H.; Zou, Q.; Chong, K.T. TS-m6A-DL: Tissue-specific identification of N6-methyladenosine sites using a universal deep learning model. Comput. Struct. Biotechnol. J. 2021, 19, 4619–4625. [Google Scholar] [CrossRef]
- Liu, K.W.; Cao, L.; Du, P.F.; Chen, W. im6A-TS-CNN: Identifying the N6-Methyladenine Site in Multiple Tissues by Using the Convolutional Neural Network. Mol. Ther. Nucleic Acids 2020, 21, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Dao, F.Y.; Lv, H.; Yang, Y.H.; Zulfiqar, H.; Gao, H.; Lin, H. Computational identification of N6-methyladenosine sites in multiple tissues of mammals. Comput. Struct. Biotechnol. J. 2020, 18, 1084–1091. [Google Scholar] [CrossRef]
- Qiang, X.L.; Chen, H.R.; Ye, X.C.; Su, R.; Wei, L.Y. M6AMRFS: Robust Prediction of N6-Methyladenosine Sites With Sequence-Based Features in Multiple Species. Front. Genet. 2018, 9, 495. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; He, N.N.; Chen, Y.; Chen, Z.; Li, L. BERMP: A cross-species classifier for predicting m6A sites by integrating a deep learning algorithm and a random forest approach. Int. J. Biol. Sci. 2018, 14, 1669–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, C.; Jin, D.; Wang, X.; Zhao, Q. Tissue specific prediction of N6-methyladenine sites based on an ensemble of multi-input hybrid neural network. Biocell 2022, 46, 1105–1121. [Google Scholar] [CrossRef]
- Rogers, A.; Kovaleva, O.; Rumshisky, A. A Primer in BERTology: What We Know About How BERT Works. Trans. Assoc. Comput. Linguist. 2020, 8, 842–866. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Ji, Y.R.; Zhou, Z.H.; Liu, H.; Davuluri, R.V. DNABERT: Pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics 2021, 37, 2112–2120. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, Z.; Yang, Y.; Wong, K.-C.; Li, X. Genome-wide identification and characterization of DNA enhancers with a stacked multivariate fusion framework. PLoS Comput. Biol. 2022, 18, e1010779. [Google Scholar] [CrossRef]
- Jin, J.; Yu, Y.; Wang, R.; Zeng, X.; Pang, C.; Jiang, Y.; Li, Z.; Dai, Y.; Su, R.; Zou, Q.; et al. iDNA-ABF: Multi-scale deep biological language learning model for the interpretable prediction of DNA methylations. Genome Biol. 2022, 23, 219. [Google Scholar] [CrossRef]
- Yamada, K.; Hamada, M. Prediction of RNA-protein interactions using a nucleotide language model. Bioinform. Adv. 2022, 2, vbac023. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Amerifar, S.; Norouzi, M.; Ghandi, M. A tool for feature extraction from biological sequences. Brief. Bioinform. 2022, 23, bbac108. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, W.; Guo, F.; Xu, L.; Zhang, L.J.P. 6mA-Pred: Identifying DNA N6-methyladenine sites based on deep learning. PeerJ 2021, 9, e10813. [Google Scholar] [CrossRef] [PubMed]
- Friedel, M.; Nikolajewa, S.; Suehnel, J.; Wilhelm, T. DiProDB: A database for dinucleotide properties. Nucleic Acids Res. 2009, 37, D37–D40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.-Y.; Xu, J.; Wang, J.; Zhou, Y.-K.; Chen, W.; Du, P.-F. KNIndex: A comprehensive database of physicochemical properties for k-tuple nucleotides. Brief. Bioinform. 2021, 22, bbaa284. [Google Scholar] [CrossRef]
- He, K.M.; Zhang, X.Y.; Ren, S.Q.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Seattle, WA, USA, 27–30 June 2016; pp. 770–778. [Google Scholar]
- Woo, S.H.; Park, J.; Lee, J.Y.; Kweon, I.S. CBAM: Convolutional block attention module. In Proceedings of the 15th European Conference on Computer Vision (ECCV), Munich, Germany, 8–14 September 2018; pp. 3–19. [Google Scholar]
- Van Houdt, G.; Mosquera, C.; Napoles, G. A review on the long short-term memory model. Artif. Intell. Rev. 2020, 53, 5929–5955. [Google Scholar] [CrossRef]
- Schuster, M.; Paliwal, K.K. Bidirectional recurrent neural networks. IEEE Trans. Signal Process. 1997, 45, 2673–2681. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Shi, W.; Tian, J.; Qi, Z.Y.; Li, B.C.; Hao, H.W.; Xu, B. Attention-Based Bidirectional Long Short-Term Memory Networks for Relation Classification. In Proceedings of the 54th Annual Meeting of the Association-for-Computational-Linguistics (ACL), Berlin, Germany, 7–12 August 2016; pp. 207–212. [Google Scholar]
- Acheampong, F.A.; Nunoo-Mensah, H.; Chen, W. Transformer models for text-based emotion detection: A review of BERT-based approaches. Artif. Intell. Rev. 2021, 54, 5789–5829. [Google Scholar] [CrossRef]
- Vacic, V.; Iakoucheva, L.M.; Radivojac, P. Two Sample Logo: A graphical representation of the differences between two sets of sequence alignments. Bioinformatics 2006, 22, 1536–1537. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tool | Classifier | Feature Encoding Scheme | Species | Data Scale | URL Accessibility |
|---|---|---|---|---|---|
| M6AMRFS [33] | XGBoost | dinucleotide binary, localPSDF | S. cerevisiae | 2614 | accessible |
| H. sapiens | 2260 | ||||
| Musculus | 1450 | ||||
| A. thaliana | 2000 | ||||
| BERMP [34] | RF, GRU, LR | ENAC | Mammalian | 736,023 | accessible |
| S. cerevisiae | 2614 | ||||
| A. thaliana | 5036 | ||||
| StackRAM [28] | LightGBM, SVM | binary encoding, chemical property, NF, PSTNP, KNF, pseDNC | S. cerevisiae | 2614 | inaccessible |
| H. sapiens | 2260 | ||||
| A. thaliana | 788 | ||||
| im6A-TS-CNN [31] | CNN | one-hot-encoding | Human | 47,248 | inaccessible |
| Mouse | 92,070 | ||||
| Cat | 30,184 | ||||
| iRNA-m6A [32] | SVM | physical–chemical property, mono-nucleotide binary encoding, NCP | Human | 47,248 | accessible |
| Mouse | 92,067 | ||||
| Cat | 30,184 | ||||
| m6A-NeuralTool [29] | CNN, SVM, NB | one-hot-encoding | S. cerevisiae | 6540 | accessible |
| A. thaliana | 4200 | ||||
| Mus musculus | 1450 | ||||
| H. sapiens | 2260 | ||||
| TS-m6A-DL [30] | CNN | one-hot-encoding | Human | 47,248 | accessible |
| Mouse | 92,070 | ||||
| Cat | 30,184 | ||||
| m6A-neural-network [35] | CNN, BiGRU | one-hot-encoding, sequence features, KNF | Human | 47,248 | inaccessible |
| Mouse | 92,070 | ||||
| Cat | 30,184 |
| Species | Tissues | Name | Training Dataset | Independent Dataset | ||
|---|---|---|---|---|---|---|
| Positive | Negative | Positive | Negative | |||
| Rat | Brain | R_b | 2352 | 2352 | 2351 | 2351 |
| Kidney | R_k | 3433 | 3433 | 3432 | 3432 | |
| Liver | R_l | 1762 | 1762 | 1762 | 1762 | |
| Mouse | Brain | M_b | 8025 | 8025 | 8025 | 8025 |
| Heart | M_h | 2201 | 2201 | 2200 | 2200 | |
| Kidney | M_k | 3953 | 3953 | 3952 | 3952 | |
| Liver | M_l | 4133 | 4133 | 4133 | 4133 | |
| Testis | M_t | 4707 | 4707 | 4706 | 4706 | |
| Human | Brain | H_b | 4605 | 4605 | 4604 | 4604 |
| Kidney | H_k | 4574 | 4574 | 4573 | 4573 | |
| Liver | H_l | 2634 | 2634 | 2634 | 2634 | |
| Name | Methods | Acc | Sen | Spe | MCC | AUROC |
|---|---|---|---|---|---|---|
| H_b | Our model | 0.747 | 0.812 | 0.681 | 0.498 | 0.827 |
| TS-m6A-DL | 0.738 | 0.812 | 0.664 | 0.482 | 0.809 | |
| iRNA-m6A | 0.711 | 0.695 | 0.73 | 0.42 | 0.785 | |
| im6A-TS-CNN | 0.727 | 0.752 | 0.702 | 0.454 | 0.806 | |
| m6A-neural-network | 0.746 | 0.818 | 0.674 | 0.497 | / | |
| H_k | Our model | 0.806 | 0.838 | 0.775 | 0.614 | 0.888 |
| TS-m6A-DL | 0.802 | 0.804 | 0.799 | 0.604 | 0.88 | |
| iRNA-m6A | 0.778 | 0.771 | 0.784 | 0.56 | 0.857 | |
| im6A-TS-CNN | 0.792 | 0.8 | 0.785 | 0.585 | 0.873 | |
| m6A-neural-network | 0.798 | 0.823 | 0.773 | 0.597 | / | |
| H_l | Our model | 0.815 | 0.857 | 0.773 | 0.632 | 0.89 |
| TS-m6A-DL | 0.805 | 0.82 | 0.79 | 0.611 | 0.878 | |
| iRNA-m6A | 0.79 | 0.782 | 0.799 | 0.58 | 0.868 | |
| im6A-TS-CNN | 0.799 | 0.848 | 0.75 | 0.601 | 0.881 | |
| m6A-neural-network | 0.809 | 0.841 | 0.777 | 0.62 | / | |
| M_b | Our model | 0.792 | 0.806 | 0.775 | 0.582 | 0.876 |
| TS-m6A-DL | 0.787 | 0.829 | 0.746 | 0.577 | 0.872 | |
| iRNA-m6A | 0.783 | 0.772 | 0.794 | 0.57 | 0.861 | |
| im6A-TS-CNN | 0.785 | 0.862 | 0.707 | 0.577 | 0.872 | |
| m6A-neural-network | 0.792 | 0.829 | 0.758 | 0.589 | / | |
| M_h | Our model | 0.757 | 0.831 | 0.684 | 0.521 | 0.835 |
| TS-m6A-DL | 0.75 | 0.793 | 0.707 | 0.502 | 0.823 | |
| iRNA-m6A | 0.713 | 0.705 | 0.721 | 0.43 | 0.788 | |
| im6A-TS-CNN | 0.736 | 0.758 | 0.714 | 0.472 | 0.816 | |
| m6A-neural-network | 0.753 | 0.803 | 0.703 | 0.509 | / | |
| M_k | Our model | 0.819 | 0.814 | 0.824 | 0.638 | 0.898 |
| TS-m6A-DL | 0.807 | 0.842 | 0.773 | 0.616 | 0.889 | |
| iRNA-m6A | 0.793 | 0.784 | 0.803 | 0.59 | 0.87 | |
| im6A-TS-CNN | 0.808 | 0.805 | 0.81 | 0.615 | 0.886 | |
| m6A-neural-network | 0.814 | 0.842 | 0.786 | 0.629 | / | |
| M_l | Our model | 0.736 | 0.786 | 0.686 | 0.474 | 0.816 |
| TS-m6A-DL | 0.72 | 0.78 | 0.66 | 0.443 | 0.791 | |
| iRNA-m6A | 0.688 | 0.678 | 0.699 | 0.38 | 0.762 | |
| im6A-TS-CNN | 0.716 | 0.756 | 0.676 | 0.433 | 0.793 | |
| m6A-neural-network | 0.73 | 0.753 | 0.707 | 0.461 | / | |
| M_t | Our model | 0.78 | 0.772 | 0.789 | 0.561 | 0.867 |
| TS-m6A-DL | 0.764 | 0.842 | 0.686 | 0.535 | 0.843 | |
| iRNA-m6A | 0.735 | 0.722 | 0.751 | 0.47 | 0.818 | |
| im6A-TS-CNN | 0.762 | 0.835 | 0.689 | 0.529 | 0.847 | |
| m6A-neural-network | 0.769 | 0.816 | 0.722 | 0.541 | / | |
| R_b | Our model | 0.783 | 0.773 | 0.793 | 0.566 | 0.866 |
| TS-m6A-DL | 0.772 | 0.813 | 0.732 | 0.547 | 0.854 | |
| iRNA-m6A | 0.751 | 0.739 | 0.765 | 0.5 | 0.827 | |
| im6A-TS-CNN | 0.77 | 0.781 | 0.758 | 0.539 | 0.852 | |
| m6A-neural-network | 0.775 | 0.797 | 0.752 | 0.55 | / | |
| R_k | Our model | 0.838 | 0.848 | 0.828 | 0.676 | 0.914 |
| TS-m6A-DL | 0.832 | 0.852 | 0.813 | 0.666 | 0.908 | |
| iRNA-m6A | 0.814 | 0.802 | 0.828 | 0.63 | 0.897 | |
| im6A-TS-CNN | 0.827 | 0.849 | 0.806 | 0.655 | 0.908 | |
| m6A-neural-network | 0.834 | 0.848 | 0.82 | 0.669 | / | |
| R_l | Our model | 0.82 | 0.844 | 0.796 | 0.64 | 0.903 |
| TS-m6A-DL | 0.81 | 0.854 | 0.765 | 0.622 | 0.885 | |
| iRNA-m6A | 0.799 | 0.777 | 0.823 | 0.6 | 0.876 | |
| im6A-TS-CNN | 0.802 | 0.845 | 0.759 | 0.607 | 0.885 | |
| m6A-neural-network | 0.815 | 0.841 | 0.788 | 0.63 | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Cheng, X.; Song, C.; Liu, T. M6A-BERT-Stacking: A Tissue-Specific Predictor for Identifying RNA N6-Methyladenosine Sites Based on BERT and Stacking Strategy. Symmetry 2023, 15, 731. https://doi.org/10.3390/sym15030731
Li Q, Cheng X, Song C, Liu T. M6A-BERT-Stacking: A Tissue-Specific Predictor for Identifying RNA N6-Methyladenosine Sites Based on BERT and Stacking Strategy. Symmetry. 2023; 15(3):731. https://doi.org/10.3390/sym15030731
Chicago/Turabian StyleLi, Qianyue, Xin Cheng, Chen Song, and Taigang Liu. 2023. "M6A-BERT-Stacking: A Tissue-Specific Predictor for Identifying RNA N6-Methyladenosine Sites Based on BERT and Stacking Strategy" Symmetry 15, no. 3: 731. https://doi.org/10.3390/sym15030731