
Molecular Descriptors—Spectral Property Relations for Characterizing Molecular Interactions in Binary and Ternary Solutions, Excited State Dipole Moment Estimation
 ,
,
Abstract
:1. Introduction
2. Theoretical Bases
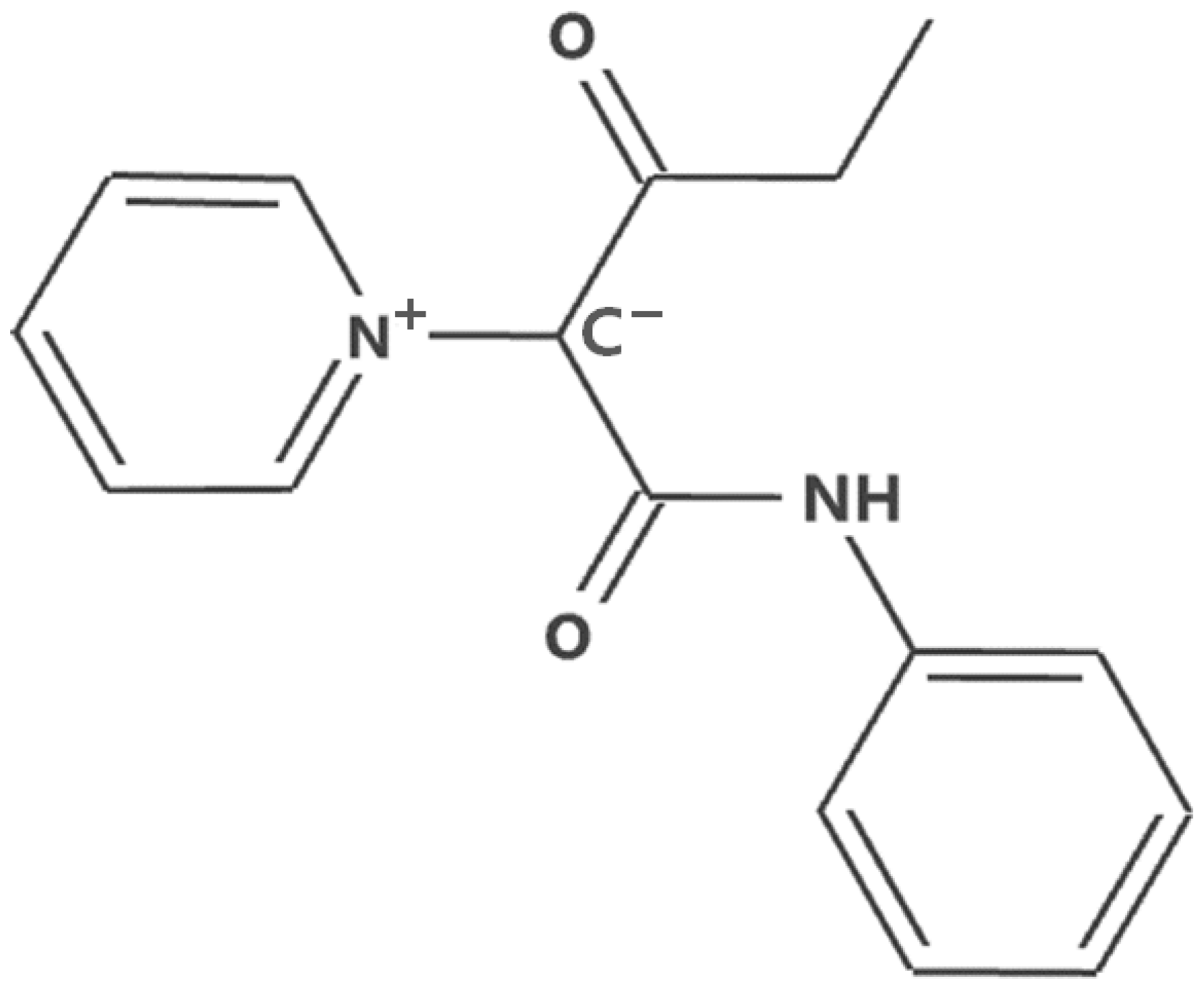
3. Materials and Methods
4. Results and Discussions
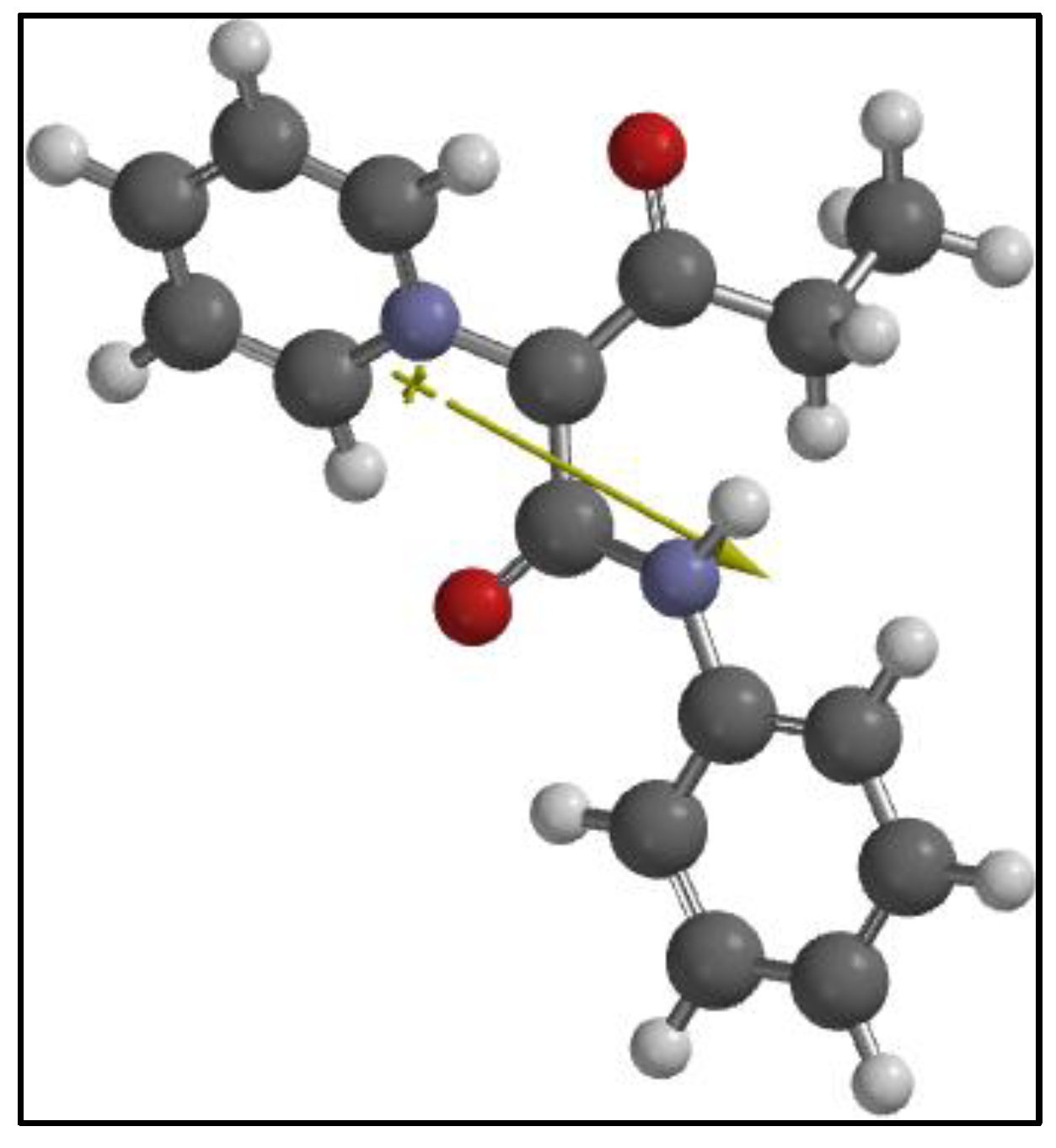
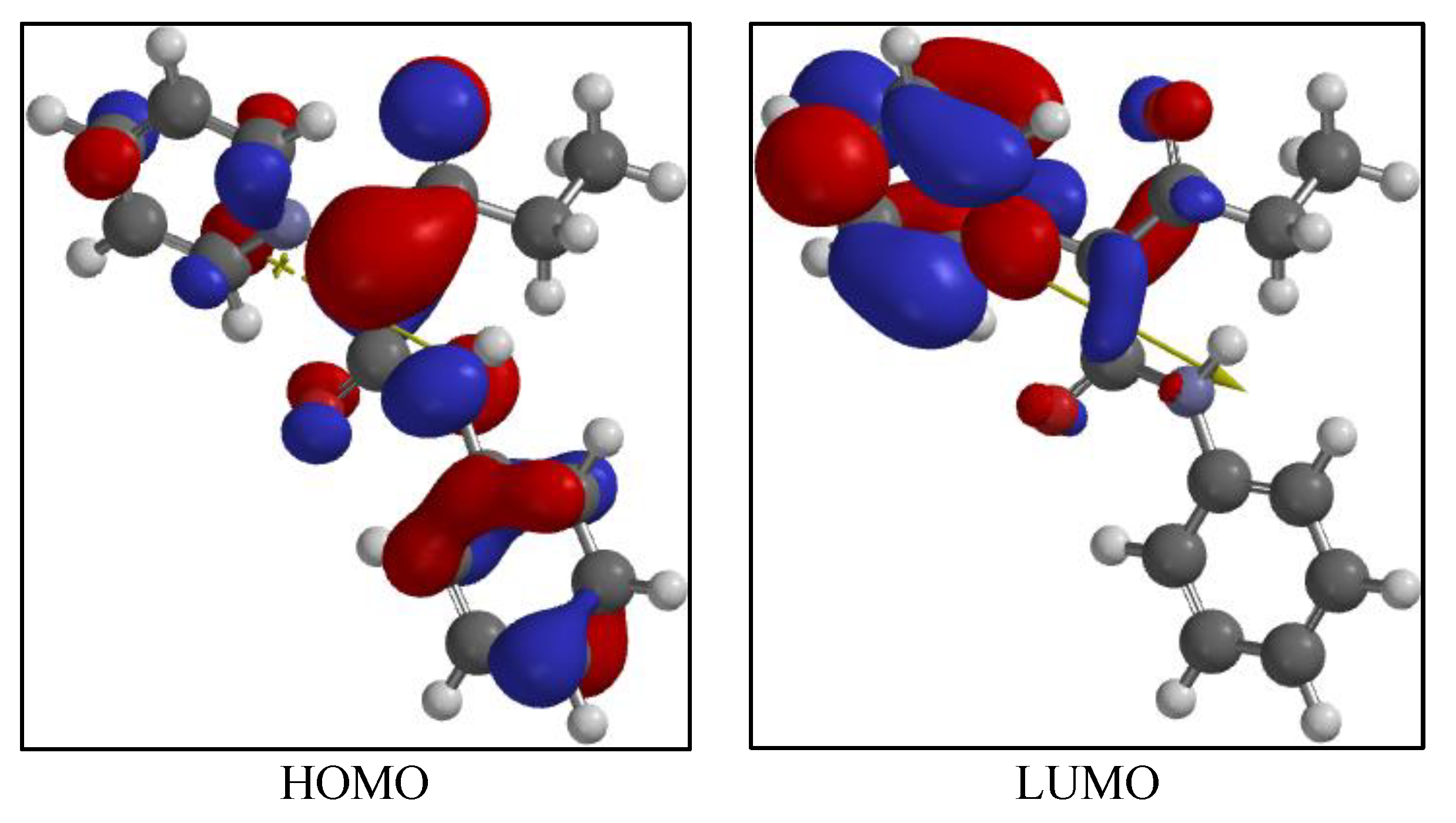
4.1. Computational Results
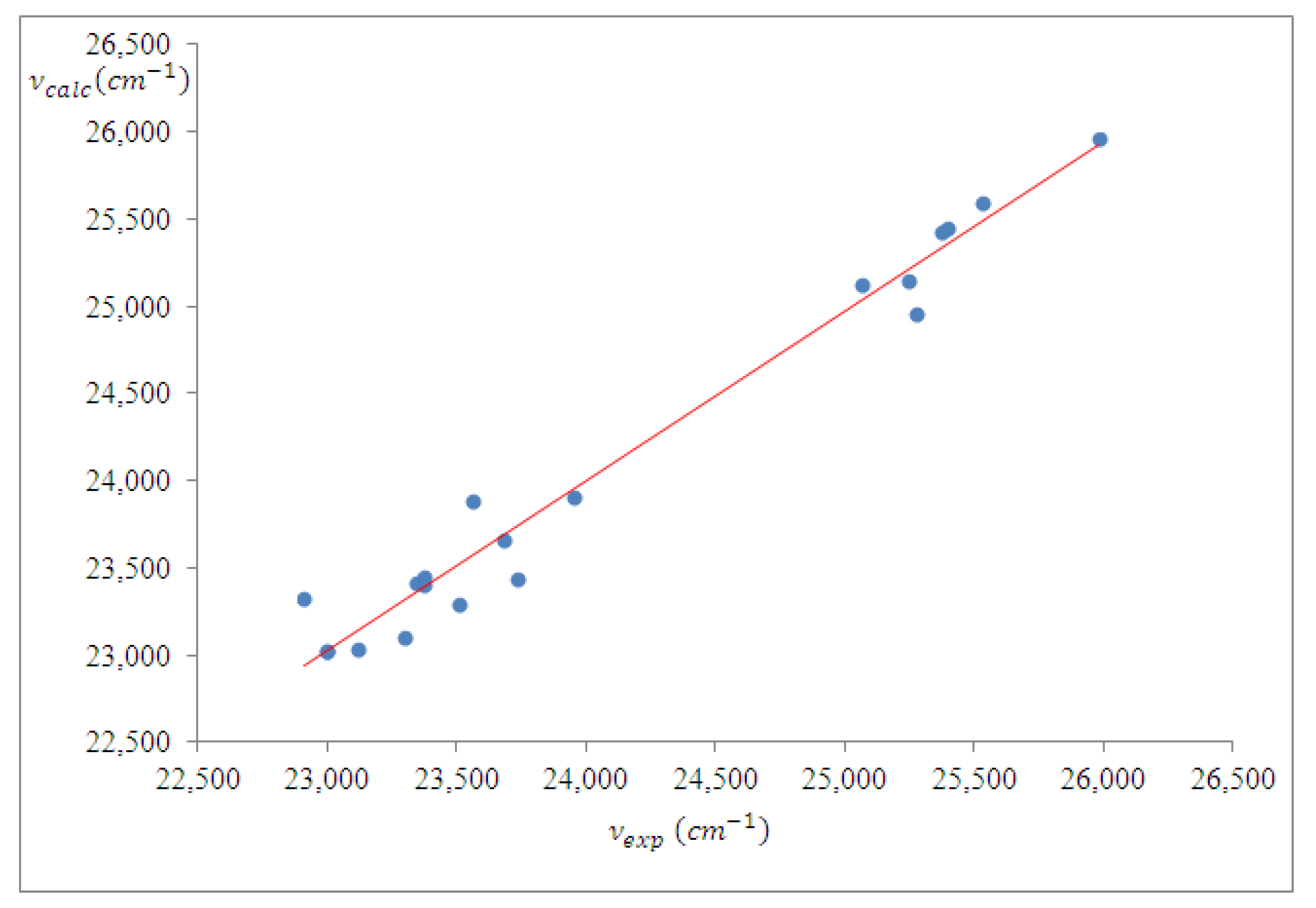
4.2. Spectral Analysis
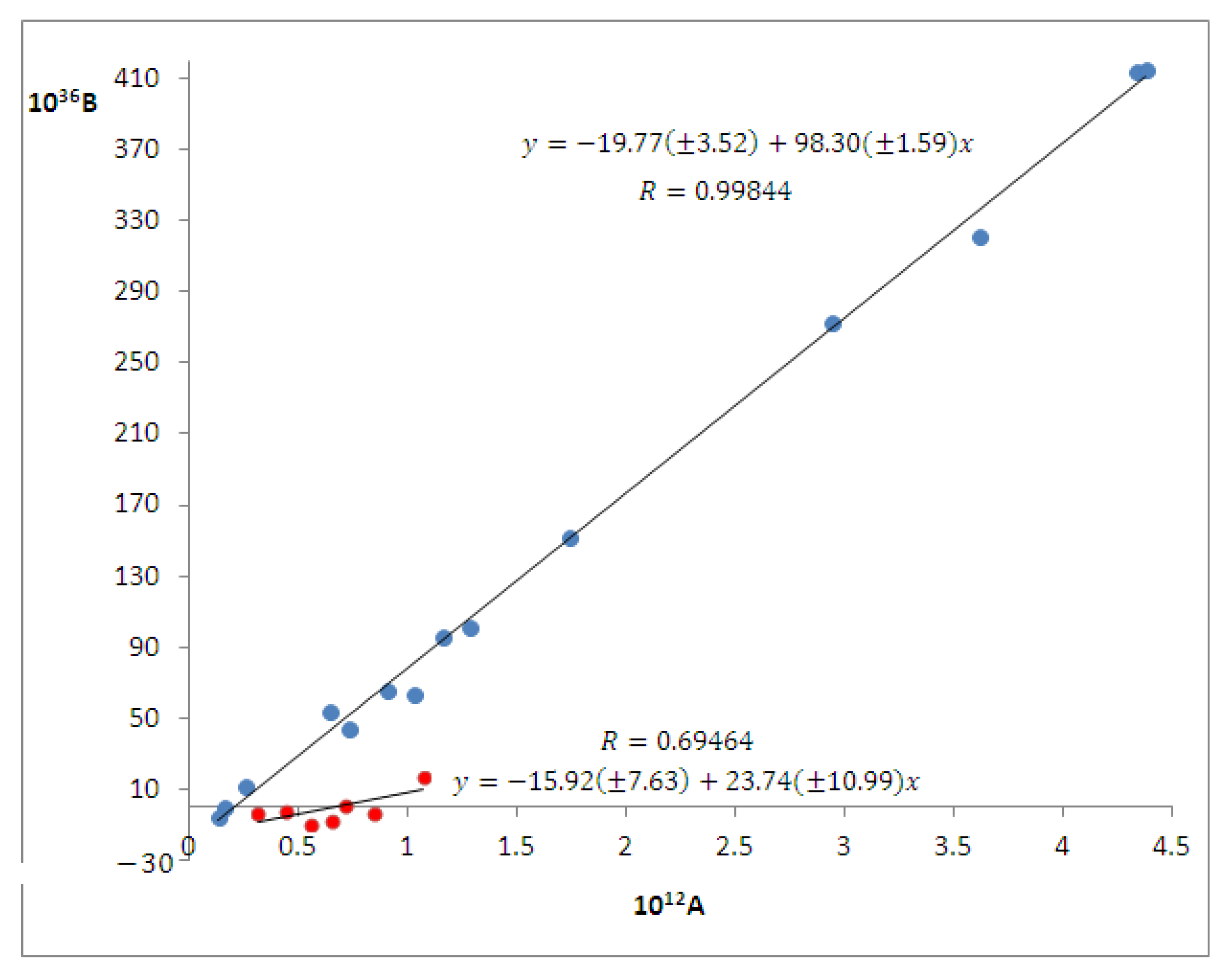
4.2.1. Binary Solutions—Solvent Influence on the Wavenumber in the Maximum of the ICT Visible Band of PCAnM
+ (327 ± 234)β + (1792 ± 203)α
4.2.2. Dipole Moment of PCAnM Computed Based on Takehiro Abe’s Model
4.2.3. Excited State Dipole Moment of PCAnM Estimated Based on Variational Model
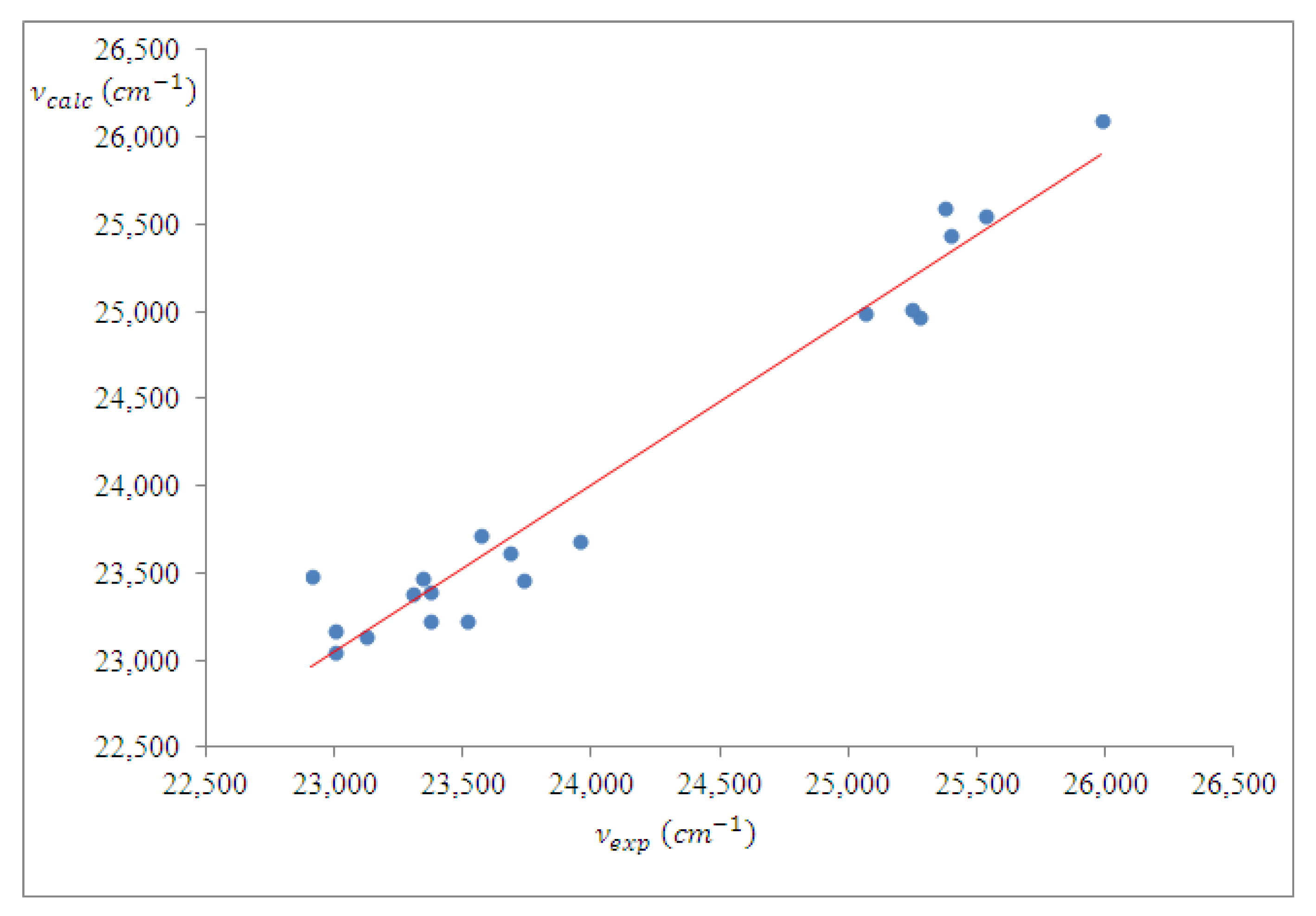
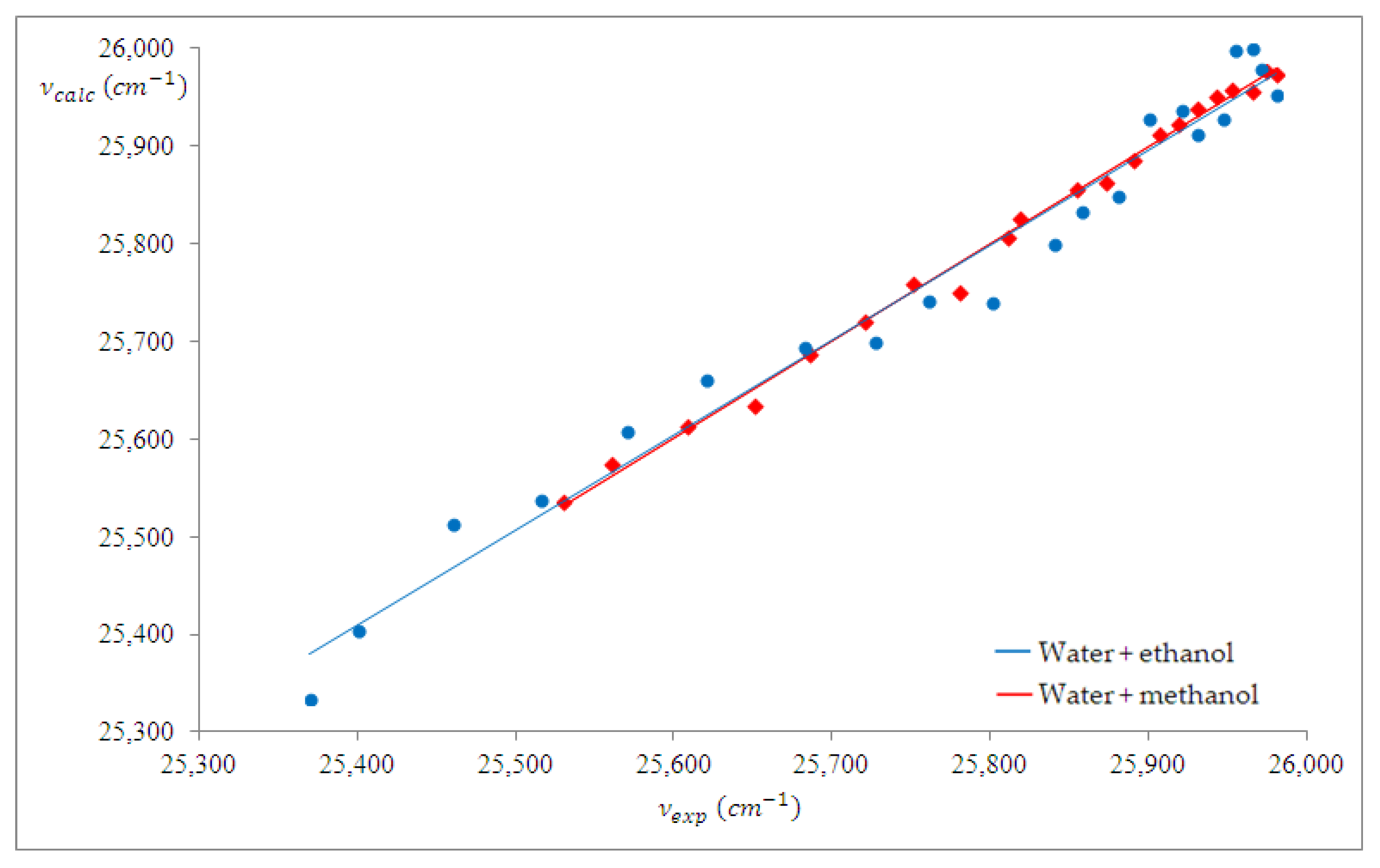
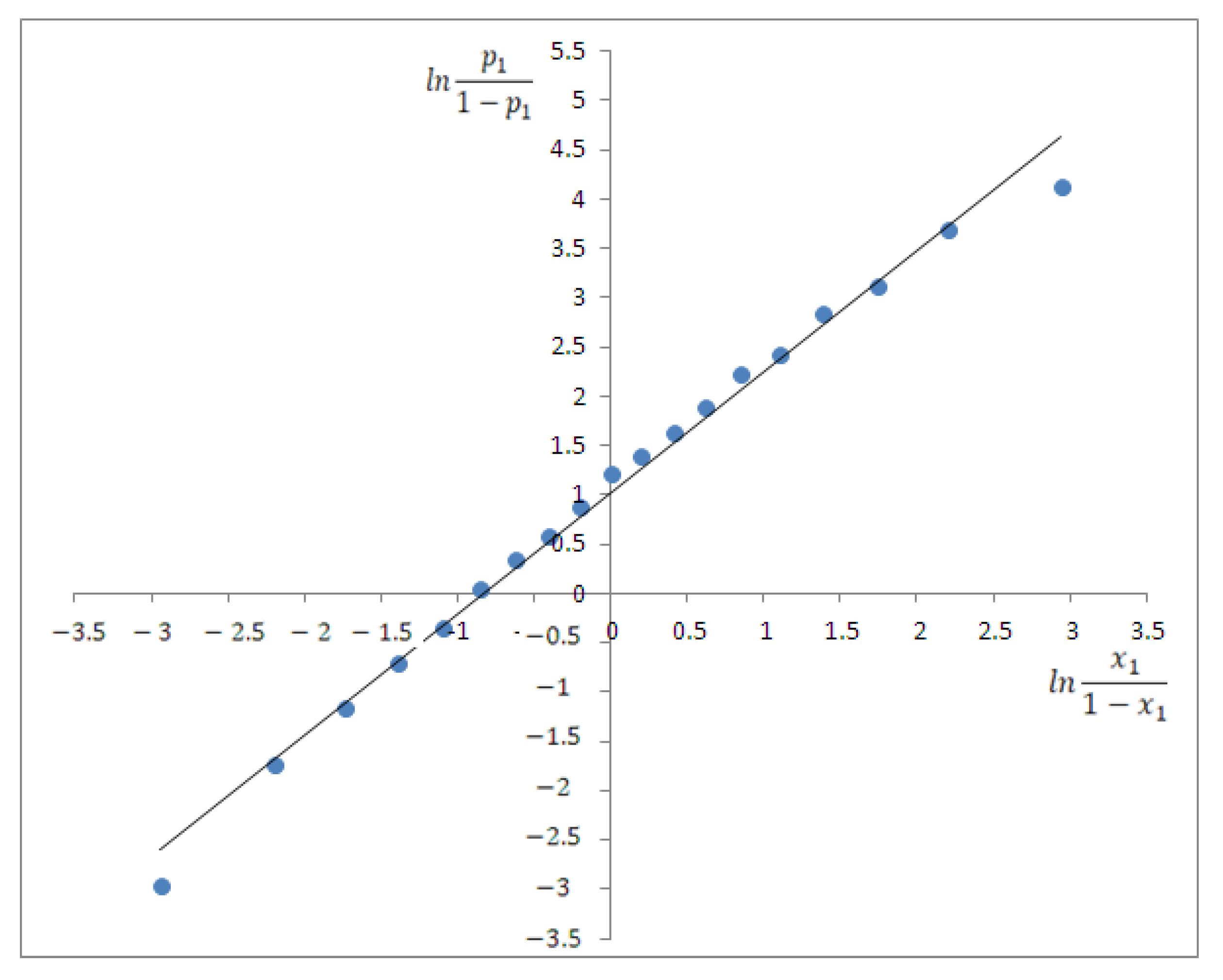
4.3. Spectral Analysis of Ternary Solutions
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Johnson, A.W. Ylid Chemistry; Academic Press: New York, NY, USA, 1966. [Google Scholar]
- Zugravescu, I.; Petrovanu, M. N-Ylid Chemistry; McGraw Hill: New York, NY, USA, 1976. [Google Scholar]
- Dorohoi, D.O. Electronic spectra of N-ylids. J. Mol. Struct. 2004, 704, 31–43. [Google Scholar] [CrossRef]
- Kröhnke, F. Specific synthesis of pyridines and oligopyridines. Synthesis 1976, 1976, 1–24. [Google Scholar] [CrossRef]
- Seifi, M.; Bahonar, S.; Ebrahimipour, S.Y.; Simpson, J.; Dusek, M.; Eigner, V.; Sheibani, H. Combination of pyridinium and isoquinolinium ylides with phenylisocyanate and isothiocyanates: Synthesis, characterization, and X-ray crystal structures of mesoionic monosubstituted 3-oxo-propanamides or thioamides. Aust. J. Chem. 2015, 68, 1577–1582. [Google Scholar] [CrossRef]
- Sar, S.; Guha, S.; Prabakar, T.; Maiti, D.; Sen, S. Blue light-emitting diode-mediated in situ generation of pyridinium and isoquinolinium ylides from aryl diazoesters: Their application in the synthesis of diverse dihydroindilozine. J. Org. Chem. 2021, 86, 11736–11747. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.; van Hende, E.; Claessens, S.; de Kimpe, N. Pyridinium ylids in heterocyclic synthesis. Curr. Org. Chem. 2011, 15, 1340–1362. [Google Scholar] [CrossRef]
- Pawda, A.; Austin, D.J.; Precedo, L.; Zhi, L. Cycloaddition reactions of pyridinium and related azomethine ylides. J. Org. Chem. 1993, 58, 1144–1150. [Google Scholar] [CrossRef]
- Allgäuer, D.S.; Mayer, P.; Mayr, H. Nucleophilicity parameters of pyridinium ylides and their use in mechanistic analyses. J. Am. Chem. Soc. 2013, 135, 15216–15224. [Google Scholar] [CrossRef] [PubMed]
- Bonte, S.; Ghenea, I.O.; Dinica, R.; Baussanne, I.; Demeunynck, M. Investigation of the pyridinium ylide-alkyne cycloaddition as a fluorogenic coupling reaction. Molecules 2016, 21, 332. [Google Scholar] [CrossRef]
- Dorohoi, D.O.; Creanga, D.E.; Dimitriu, D.G.; Morosanu, A.C.; Gritco-Todirascu, A.; Mariciuc, G.G.; Puica Melniciuc, N.; Ardelean, E.; Cheptea, C. Computational and spectral means for characterizing the intermolecular interactions in solutions and for estimating excited state dipole moment of solute. Symmetry 2020, 12, 1299. [Google Scholar] [CrossRef]
- Morosanu, A.C.; Gritco-Todirascu, A.; Creanga, D.E.; Dorohoi, D.O. Computational and solvatochromic study of pyridinium-acetyl-benzoyl-methylid (PABM). Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 189, 307–315. [Google Scholar] [CrossRef]
- Albrecht, M.; Yulikov, M.; Kohn, T.; Jeschke, G.; Adams, J.; Schmidt, A. Pyridinium salts and ylides as partial structures of photoresponsive Merrifield resins. J. Mater. Chem. 2010, 20, 3025–3034. [Google Scholar] [CrossRef]
- Schwalm, R.; Bottcher, A.; Koch, H. MID UV resist materials containing pyridinium ylides. Proc. SPIE 1988, 0920, 21–26. [Google Scholar] [CrossRef]
- Streith, J. The photochemistry of N-iminopyridinium ylides in retrospect. From a simple concept to some applications. CHIMIA 1991, 45, 65–76. [Google Scholar] [CrossRef]
- Taylor, L.D.; Haubs, M.K.-J. Polymeric Pyridinium Ylide and Products from Same. European Patent EP0164100A2, 14 April 1993. [Google Scholar]
- Babusca, D.; Morosanu, A.C.; Dimitriu, D.G.; Dorohoi, D.O.; Cheptea, C. Spectroscopic and quantum-chemical study of molecular interactions of iso-quinolinium ylids in polar solutions. Mol. Cryst. Liq. Cryst. 2020, 698, 87–97. [Google Scholar] [CrossRef]
- Pop, V.; Dorohoi, D.O.; Holban, V. Molecular interactions in binary solutions of 4-aminophthalimide and 3-p-cumyl-pyridazinium-acetyl-benzoyl-methylid. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1994, 50, 2281–2289. [Google Scholar] [CrossRef]
- Onsager, L. Electric moments of molecules in liquids. J. Am. Chem. Soc. 1936, 58, 1486–1493. [Google Scholar] [CrossRef]
- Fleming, I. Frontier Orbitals and Organic Chemical Reactions; John Wiley & Sons Chichester: Hoboken, NJ, USA, 1976. [Google Scholar]
- Hitchcock, S.A.; Pennington, L.D. Structure-brain exposure relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef] [PubMed]
- Young, D. Computational Chemistry: A Practical Guide for Appling Techniques to Real World Problems; John Wiley & Sons: New York, NY, USA, 2001; pp. 322–331. [Google Scholar]
- Gosav, S.; Praisler, M.; Dorohoi, D.O.; Popa, G. Structure-activity correlations for illicit amphetamines using ANN and constitutional descriptors. Talanta 2006, 70, 922–928. [Google Scholar] [CrossRef]
- Gosav, S.; Praisler, M.; Dorohoi, D.O. ANN expert system screening for illicit amphetamines using molecular descriptors. J. Mol. Struct. 2007, 834–836, 188–194. [Google Scholar] [CrossRef]
- McRae, E.G. Theory of solvent effects on molecular electronic spectra. Frequency shifts. J. Phys. Chem. 1957, 61, 562–572. [Google Scholar] [CrossRef]
- Abe, T. Theory of solvent effects on molecular electronic spectra. Frequency shifts. Bull. Chem. Soc. Jpn. 1965, 38, 1314–1318. [Google Scholar] [CrossRef]
- Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Dorohoi, D.O. Physics of Liquid State; Gama: Iasi, Romania, 1993; ISBN 973-95120-8-9. (In Romanian) [Google Scholar]
- Kamlet, M.J.; Abboud, J.L.M.; Abraham, M.H.; Taft, R.W. Linear solvation energy relationships. 23. A comprehensive collection of the solvatochromic parameters π*, α, and β, and some methods for simplifying the generalized solvatochromic equation. J. Org. Chem. 1983, 48, 2877–2887. [Google Scholar] [CrossRef]
- Sıdır, İ.; Sıdır, Y.G. Investigation of the interactions of E-4-methoxycinnamic acid with solvents: Solvatochromism, electric dipole moment and pH effect. J. Mol. Struct. 2018, 249, 1161–1171. [Google Scholar] [CrossRef]
- Dorohoi, D.O.; Dimitriu, D.G.; Dimitriu, M.; Closca, V. Specific interactions in N-ylid solutions, studied by nuclear magnetic resonance and electronic absorption spectroscopy. J. Mol. Struct. 2013, 1044, 79–86. [Google Scholar] [CrossRef]
- Kawski, A. On the estimation of excited state dipole moments from solvatochromic shifts of absorption and fluorescence spectra. Z. Naturforsch. A 2002, 57, 255–262. [Google Scholar] [CrossRef]
- Kawski, A.; Bojarski, P. Comments on the determination of excited state dipole moment of molecules using the method of solvatochromism. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 82, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Gahlaut, R.; Tewari, N.; Bridhkoti, J.P.; Joshi, N.K.; Joshi, H.C.; Pant, S. Determination of ground and excited states dipole moments of some naphthols using solvatochromic shift method. J. Mol. Liq. 2011, 163, 141–146. [Google Scholar] [CrossRef]
- Zakerhamidi, M.S.; Moghadam, M.; Ghanadzadeh, A.; Hosseini, S. Anisotropic and isotropic solvent effects on the dipole moment and photophysical properties of rhodamine dyes. J. Lumin. 2012, 132, 931–937. [Google Scholar] [CrossRef]
- Dorohoi, D.O. Excited state molecular parameters determined by spectral means. Ukr. J. Phys. 2018, 63, 701–708. [Google Scholar] [CrossRef]
- Pop, V.; Dorohoi, D.O.; Delibas, M. Considerations on the statistic model of the intermolecular interactions in ternary solutions. An. Stiint. Univ. Al. I. Cuza Iasi 1986, 32, 79–84. [Google Scholar]
- Dorohoi, D.O.; Avadanei, M.; Postolache, M. Characterization of the solvation spheres of some dipolar spectrally active molecules in binary solvents. Optoelectron. Adv. Mater. 2008, 2, 51–55. [Google Scholar]
- Avadanei, M.; Dorohoi, D.O. Interaction energy in pairs of pyridazinium ylide-solvent molecules estimated by spectral means within the cell ternary solution model. Ukr. J. Phys. 2012, 57, 118–123. [Google Scholar] [CrossRef]
- Closca, V.; Puica-Melniciuc, N.; Dorohoi, D.O. Specific interactions in hydroxyl ternary solutions of three carbanion monosubstituted 4-tolyl-1,2,4-triazol-1-ium-4-R-phenacylids studied by visible electronic absorption spectra. J. Mol. Struct. 2014, 200, 431–438. [Google Scholar] [CrossRef]
- Pavel, C.M.; Ambrosi, E.; Dimitriu, D.G.; Dorohoi, D.O. Complex formation and micro-heterogeneity in water-alcohol binary mixtures investigated by solvatochromic study. Eur. Phys. J. Spec. Top. 2023, 232, 415–425. [Google Scholar] [CrossRef]
- Hehre, W.; Ohlinger, S.; Klunzinger, P.; Deppmeier, B.; Driessen, A.; Johnson, J.; Ohsan, P. Spartan’14 for Windows, Macintosh and Linux. Tutorial and User’s Guide; Wavefunction Inc.: Irvine, CA, USA, 2014. [Google Scholar]
- Lin, C.Y.; George, M.W.; Gill, P.M.W. EDF2—A density functional for predicting molecular vibrational frequencies. Aust. J. Chem. 2004, 57, 365–370. [Google Scholar] [CrossRef]
- Marenich, A.V.; Olson, R.M.; Kelly, C.P.; Truhlar, G.D.; Ramer, J.C. Self-Consistent Reaction Field model for aqueous and Nonaqueous solutions based on accurate polarized partial charges. J. Chem. Theory Comput. 2007, 3, 2011–2033. [Google Scholar] [CrossRef] [PubMed]
- Kamlet-Taft Solvent Parameters. Available online: https://www.stenutz.eu/chem/kamlettaft.php (accessed on 3 October 2023).
- Buhvestov, U.; Rived, F.; Ràfols, C.; Bosch, E.; Rosés, M. Solute-solvent and solvent-solvent interactions in binary solvent mixtures. Part 7. Comparison of the enhancement of the water structure in alcohol-water mixtures measured by solvatochromic indicators. J. Phys. Org. Chem. 1998, 11, 185–192. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Descriptor | Vacuum | Water | Ethanol | Toluene |
|---|---|---|---|---|
| Energy (au) | −878.644 | −878.668 | −878.676 | −878.668 |
| EHOMO (eV) | −5.12 | −5.27 | −5.19 | −5.02 |
| ELUMO (eV) | −1.98 | −1.80 | −1.77 | −1.79 |
| (eV) | 3.14 | 3.47 | 3.42 | 3.23 |
| Dipole moment (D) | 3.43 | 5.61 | 5.63 | 4.67 |
| Polarizability (Å3) | 63.71 | 63.71 | 63.71 | 63.71 |
| Area (Å2) | 299.43 | 303.68 | 303.14 | 300.51 |
| Volume (Å3) | 284.53 | 285.50 | 285.34 | 284.82 |
| PSA (Å2) | 30.005 | 32.002 | 31.678 | 30.686 |
| HBD Count | 1 | 1 | 1 | 1 |
| HBA Count | 4 | 4 | 4 | 4 |
| No. | Solvent | f(n) | f(ε) | π* | α | β | ν (cm−1) |
|---|---|---|---|---|---|---|---|
| 1 | Dioxane | 0.300 | 0.286 | 0.55 | 0.00 | 0.37 | 23,300 |
| 2 | Benzene | 0.295 | 0.299 | 0.59 | 0.00 | 0.10 | 22,995 |
| 3 | o-Xylene | 0.292 | 0.302 | 0.41 | 0.00 | 0.11 | 22,995 |
| 4 | Toluene | 0.297 | 0.302 | 0.54 | 0.00 | 0.11 | 23,120 |
| 5 | Trichloroethylene | 0.282 | 0.448 | 0.53 | 0.00 | 0.50 | 22,910 |
| 6 | Chloroform | 0.267 | 0.552 | 0.69 | 0.20 | 0.10 | 23,680 |
| 7 | Anisole | 0.300 | 0.524 | 0.73 | 0.00 | 0.32 | 23,340 |
| 8 | Cyclohexanol | 0.276 | 0.824 | 0.45 | 0.66 | 0.84 | 25,060 |
| 9 | Chlorobenzene | 0.307 | 0.605 | 0.71 | 0.00 | 0.07 | 23,370 |
| 10 | Dichloromethane | 0.256 | 0.727 | 0.82 | 0.20 | 0.10 | 23,560 |
| 11 | n-Butyl alcohol | 0.242 | 0.833 | 0.47 | 0.84 | 0.84 | 25,370 |
| 12 | Benzyl alcohol | 0.311 | 0.804 | 0.98 | 0.60 | 0.52 | 25,270 |
| 13 | n-Propyl alcohol | 0.240 | 0.866 | 0.52 | 0.84 | 0.90 | 25,390 |
| 14 | Ethanol | 0.221 | 0.895 | 0.86 | 0.86 | 0.75 | 25,370 |
| 15 | Methanol | 0.203 | 0.909 | 0.60 | 0.98 | 0.66 | 25,530 |
| 16 | Iso-Butyl alcohol | 0.240 | 0.852 | 0.40 | 0.69 | 0.84 | 25,240 |
| 17 | Iso-Amyl acetate | 0.241 | 0.589 | 0.71 | 0.00 | 0.07 | 23,510 |
| 18 | Ethyl acetate | 0.228 | 0.625 | 0.55 | 0.00 | 0.45 | 23,730 |
| 19 | n-Butyl acetate | 0.240 | 0.577 | 0.46 | 0.00 | 0.45 | 23,370 |
| 20 | Water | 0.206 | 0.964 | 1.09 | 1.17 | 0.47 | 26,410 |
| 21 | Acetone | 0.222 | 0.868 | 0.62 | 0.08 | 0.48 | 23,950 |
| 22 | Formamide | 0.267 | 0.973 | 0.97 | 0.97 | 0.71 | 25,190 |
| No. | Solvent | Computed from Equation (18) | Computed from Equation (19) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C1f(ε) (cm−1) | C1f(ε) (%) | C4α (cm−1) | C4α (%) | Pπ* (cm−1) | Pπ* (%) | Bβ (cm−1) | Bβ (%) | Aα (cm−1) | Aα (%) | ||
| 1 | Dioxane | 360 | 100 | 0 | 0 | 404 | 72 | 155 | 28 | 0 | 0 |
| 2 | Benzene | 376 | 100 | 0 | 0 | 433 | 83 | 91 | 17 | 0 | 0 |
| 3 | o-Xylene | 380 | 100 | 0 | 0 | 301 | 75 | 100 | 25 | 0 | 0 |
| 4 | Toluene | 380 | 100 | 0 | 0 | 1125 | 42 | 1541 | 58 | 0 | 0 |
| 5 | Trichloroethylene | 563 | 100 | 0 | 0 | 1258 | 46 | 1505 | 54 | 0 | 0 |
| 6 | Chloroform | 694 | 66 | 358 | 34 | 506 | 52 | 91 | 9 | 380 | 39 |
| 7 | Anisole | 659 | 100 | 0 | 0 | 536 | 65 | 292 | 35 | 0 | 0 |
| 8 | Cyclohexanol | 1034 | 47 | 1183 | 53 | 330 | 12 | 766 | 29 | 1594 | 59 |
| 9 | Chlorobenzene | 760 | 100 | 0 | 0 | 521 | 80 | 133 | 20 | 0 | 0 |
| 10 | Dichloromethane | 914 | 72 | 358 | 28 | 602 | 56 | 91 | 9 | 380 | 35 |
| 11 | n-Butyl alcohol | 1258 | 46 | 1505 | 54 | 345 | 13 | 766 | 28 | 1594 | 59 |
| 12 | Benzyl alcohol | 1256 | 54 | 1075 | 46 | 719 | 31 | 474 | 20 | 1139 | 49 |
| 13 | n-Propyl alcohol | 1089 | 42 | 1505 | 58 | 382 | 14 | 821 | 29 | 1594 | 57 |
| 14 | Ethanol | 1125 | 42 | 1541 | 58 | 631 | 22 | 684 | 23 | 1632 | 55 |
| 15 | Methanol | 1143 | 39 | 1756 | 61 | 440 | 15 | 609 | 21 | 1860 | 64 |
| 16 | Iso-Butyl alcohol | 1071 | 46 | 1236 | 54 | 294 | 12 | 766 | 33 | 1310 | 55 |
| 17 | Iso-Amyl acetate | 740 | 100 | 0 | 0 | 521 | 89 | 64 | 11 | 0 | 0 |
| 18 | Ethyl acetate | 786 | 100 | 0 | 0 | 404 | 50 | 410 | 50 | 0 | 0 |
| 19 | n-Butyl acetate | 725 | 100 | 0 | 0 | 338 | 45 | 410 | 55 | 0 | 0 |
| 20 | Water | 1212 | 37 | 2097 | 63 | 800 | 23 | 429 | 13 | 2221 | 64 |
| 21 | Acetone | 1091 | 88 | 143 | 12 | 455 | 44 | 438 | 41 | 152 | 15 |
| 22 | Formamide | 1223 | 41 | 1738 | 59 | 712 | 22 | 648 | 20 | 1841 | 58 |
| No. | Solvent | μg (D) | αg (Å3) | Ig (eV) | M (g/mol) | ρ (g/cm3) | ν (cm−1) |
|---|---|---|---|---|---|---|---|
| 1 | Dioxane | 0 | 9.44 | 9.52 | 88.11 | 1.417 | 23,300 |
| 2 | Benzene | 0 | 10.44 | 9.25 | 78.11 | 0.868 | 22,995 |
| 3 | o-Xylene | 0.64 | 14.25 | 8.56 | 106.17 | 0.880 | 22,995 |
| 4 | Toluene | 0.38 | 12.4 | 8.72 | 92.14 | 0.867 | 23,120 |
| 5 | Trichloroethylene | 0.80 | 9.75 | 9.45 | 131.4 | 1.460 | 22,910 |
| 6 | Chloroform | 1.15 | 8.23 | 11.50 | 119.38 | 1.446 | 23,680 |
| 7 | Anisole | 1.38 | 13.10 | 8.20 | 108.14 | 0.995 | 23,340 |
| 8 | Cyclohexanol | 1.46 | 11.94 | 10.0 | 100.16 | 0.962 | 25,060 |
| 9 | Chlorobenzene | 1.50 | 13.0 | 9.07 | 112.56 | 1.110 | 23,370 |
| 10 | Dichloromethane | 1.60 | 6.66 | 11.32 | 84.93 | 1.330 | 23,560 |
| 11 | n-Butyl alcohol | 1.66 | 8.88 | 9.99 | 74.12 | 0.810 | 25,370 |
| 12 | Benzyl alcohol | 1.67 | 11.89 | 8.26 | 108.14 | 1.044 | 25,270 |
| 13 | n-Propyl alcohol | 1.68 | 6.67 | 10.52 | 60.09 | 0.803 | 25,390 |
| 14 | Ethanol | 1.69 | 5.06 | 10.70 | 46.07 | 0.789 | 25,370 |
| 15 | Methanol | 1.70 | 3.21 | 10.85 | 32.04 | 0.792 | 25,527 |
| 16 | Iso-Butyl alcohol | 1.76 | 9.07 | 10.12 | 74.12 | 0.802 | 25,240 |
| 17 | Iso-Amyl acetate | 1.77 | 15.18 | 9.90 | 130.18 | 0.884 | 23,510 |
| 18 | Ethyl acetate | 1.78 | 9.70 | 10.11 | 88.11 | 0.902 | 23,730 |
| 19 | n-Butyl acetate | 1.84 | 13.42 | 10.00 | 116.16 | 0.883 | 23,370 |
| 20 | Water | 1.85 | 1.5 | 12.59 | 18 | 1.000 | 26,413 |
| 21 | Acetone | 2.80 | 6.27 | 9.89 | 58.08 | 0.971 | 23,950 |
| 22 | Formamide | 3.73 | 4.08 | 10.20 | 45.04 | 1.133 | 25,190 |
| φ (Degree) | μe (D) | αe (Å3) |
|---|---|---|
| 0 | 4.069 | 63.78 |
| 8 | 4.1098 | 63.74 |
| 10 | 4.1329 | 63.73 |
| 10.5 | 4.1396 | 63.71 |
| 11 | 4.1466 | 63.70 |
| 15 | 4.2134 | 63.63 |
| 20 | 4.3334 | 63.50 |
| 30 | 4.7068 | 62.52 |
| 40 | 5.3354 | 62.31 |
| 50 | 6.3904 | 60.77 |
| No. | α | β | ν (cm−1) | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | 0.000 | - | 0.51 | 0.83 | 0.98 | 0.000 | - | 25,370 |
| 2 | 0.050 | −2.94 | 0.54 | 0.83 | 0.97 | 0.049 | −2.97 | 25,400 |
| 3 | 0.100 | −2.20 | 0.57 | 0.84 | 0.96 | 0.148 | −1.75 | 25,460 |
| 4 | 0.150 | −1.74 | 0.60 | 0.83 | 0.94 | 0.238 | −1.16 | 25,515 |
| 5 | 0.200 | −1.39 | 0.63 | 0.83 | 0.93 | 0.329 | −0.72 | 25,570 |
| 6 | 0.250 | −1.10 | 0.65 | 0.83 | 0.93 | 0.410 | −0.36 | 25,620 |
| 7 | 0.300 | −0.85 | 0.68 | 0.82 | 0.92 | 0.511 | 0.05 | 25,682 |
| 8 | 0.350 | −0.62 | 0.70 | 0.81 | 0.91 | 0.584 | 0.34 | 25,726 |
| 9 | 0.400 | −0.41 | 0.73 | 0.80 | 0.91 | 0.639 | 0.57 | 25,760 |
| 10 | 0.450 | −0.20 | 0.75 | 0.79 | 0.89 | 0.705 | 0.87 | 25,800 |
| 11 | 0.500 | 0.00 | 0.77 | 0.79 | 0.90 | 0.770 | 1.21 | 25,840 |
| 12 | 0.550 | 0.20 | 0.80 | 0.78 | 0.89 | 0.799 | 1.38 | 25,857 |
| 13 | 0.600 | 0.41 | 0.82 | 0.77 | 0.89 | 0.836 | 1.63 | 25,880 |
| 14 | 0.650 | 0.62 | 0.85 | 0.77 | 0.89 | 0.869 | 1.89 | 25,900 |
| 15 | 0.700 | 0.85 | 0.90 | 0.74 | 0.88 | 0.901 | 2.21 | 25,920 |
| 16 | 0.750 | 1.10 | 0.94 | 0.71 | 0.86 | 0.918 | 2.42 | 25,930 |
| 17 | 0.800 | 1.39 | 1.00 | 0.67 | 0.87 | 0.944 | 2.83 | 25,946 |
| 18 | 0.850 | 1.74 | 1.06 | 0.64 | 0.90 | 0.957 | 3.11 | 25,954 |
| 19 | 0.900 | 2.20 | 1.11 | 0.59 | 0.97 | 0.975 | 3.68 | 25,965 |
| 20 | 0.950 | 2.94 | 1.13 | 0.54 | 1.11 | 0.984 | 4.12 | 25,970 |
| 21 | 1.000 | - | 1.13 | 0.50 | 1.26 | 1.000 | - | 25,980 |
| No. | α | β | ν (cm−1) | |||||
|---|---|---|---|---|---|---|---|---|
| 1 | 0.000 | - | 0.58 | 0.74 | 1.14 | 0.000 | - | 25,530 |
| 2 | 0.050 | −2.94 | 0.61 | 0.74 | 1.13 | 0.067 | −2.63 | 25,560 |
| 3 | 0.100 | −2.20 | 0.64 | 0.74 | 1.12 | 0.173 | −1.56 | 25,608 |
| 4 | 0.150 | −1.74 | 0.66 | 0.74 | 1.10 | 0.267 | −1.01 | 25,650 |
| 5 | 0.200 | −1.39 | 0.70 | 0.74 | 1.09 | 0.344 | −0.64 | 25,685 |
| 6 | 0.250 | −1.10 | 0.73 | 0.74 | 1.07 | 0.422 | −0.31 | 25,720 |
| 7 | 0.300 | −0.85 | 0.76 | 0.74 | 1.06 | 0.489 | −0.04 | 25,750 |
| 8 | 0.350 | −0.62 | 0.78 | 0.72 | 1.04 | 0.556 | 0.22 | 25,780 |
| 9 | 0.400 | −0.41 | 0.82 | 0.72 | 1.04 | 0.622 | 0.50 | 25,810 |
| 10 | 0.450 | −0.20 | 0.85 | 0.71 | 1.02 | 0.640 | 0.58 | 25,818 |
| 11 | 0.500 | 0.00 | 0.88 | 0.70 | 1.03 | 0.720 | 0.94 | 25,854 |
| 12 | 0.550 | 0.20 | 0.91 | 0.68 | 1.02 | 0.760 | 1.15 | 25,872 |
| 13 | 0.600 | 0.41 | 0.95 | 0.66 | 1.01 | 0.800 | 1.39 | 25,890 |
| 14 | 0.650 | 0.62 | 0.98 | 0.65 | 1.01 | 0.836 | 1.63 | 25,906 |
| 15 | 0.700 | 0.85 | 1.01 | 0.63 | 1.01 | 0.862 | 1.83 | 25,918 |
| 16 | 0.750 | 1.10 | 1.04 | 0.61 | 1.02 | 0.889 | 2.08 | 25,930 |
| 17 | 0.800 | 1.39 | 1.06 | 0.59 | 1.06 | 0.916 | 2.40 | 25,942 |
| 18 | 0.850 | 1.74 | 1.09 | 0.56 | 1.09 | 0.938 | 2.71 | 25,952 |
| 19 | 0.900 | 2.20 | 1.11 | 0.54 | 1.09 | 0.967 | 3.37 | 25,965 |
| 20 | 0.950 | 2.94 | 1.12 | 0.52 | 1.19 | 0.986 | 4.30 | 25,974 |
| 21 | 1.000 | - | 1.14 | 0.49 | 1.23 | 1.000 | - | 25,980 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorohoi, D.O.; Gosav, S.; Moroșanu, A.C.; Dimitriu, D.G.; Apreotesei, G.; Gosav, T. Molecular Descriptors—Spectral Property Relations for Characterizing Molecular Interactions in Binary and Ternary Solutions, Excited State Dipole Moment Estimation. Symmetry 2023, 15, 2075. https://doi.org/10.3390/sym15112075
Dorohoi DO, Gosav S, Moroșanu AC, Dimitriu DG, Apreotesei G, Gosav T. Molecular Descriptors—Spectral Property Relations for Characterizing Molecular Interactions in Binary and Ternary Solutions, Excited State Dipole Moment Estimation. Symmetry. 2023; 15(11):2075. https://doi.org/10.3390/sym15112075
Chicago/Turabian StyleDorohoi, Dana Ortansa, Steluta Gosav, Ana Cezarina Moroșanu, Dan Gheorghe Dimitriu, Gabriela Apreotesei, and Teodora Gosav. 2023. "Molecular Descriptors—Spectral Property Relations for Characterizing Molecular Interactions in Binary and Ternary Solutions, Excited State Dipole Moment Estimation" Symmetry 15, no. 11: 2075. https://doi.org/10.3390/sym15112075