
The Au12 Gold Cluster: Preference for a Non-Planar Structure

 , and
, and
Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Equilibrium Structures
3.2. Electronic Properties
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schmid, G. Nanoclusters–building blocks for future nanoelectronic devices? Adv. Eng. Mater. 2001, 3, 737–743. [Google Scholar] [CrossRef]
- Loth, S.; Baumann, S.; Lutz, C.P.; Eigler, D.M.; Heinrich, A.J. Bistability in atomic-scale antiferromagnets. Science 2012, 335, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Agasti, S.S.; Kim, C.; Li, X.; Rotello, V.M. Gold nanoparticles in chemical and biological sensing. Chem. Rev. 2012, 112, 2739–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, L.A.; Mackey, M.A.; Dreaden, E.C.; El-Sayed, M.A. The optical, photothermal, and facile surface chemical properties of gold and silver nanoparticles in biodiagnostics, therapy, and drug delivery. Arch. Toxicol. 2014, 88, 1391–1417. [Google Scholar] [CrossRef] [Green Version]
- Teles, J.H.; Brode, S.; Chabanas, M. Cationic gold (I) complexes: Highly efficient catalysts for the addition of alcohols to alkynes. Angew. Chem. Int. Ed. Engl. 1998, 37, 1415–1418. [Google Scholar] [CrossRef]
- Veenboer, R.M.; Dupuy, S.; Nolan, S.P. Stereoselective gold (I)-catalyzed intermolecular hydroalkoxlation of alkynes. ACS Catal. 2015, 5, 1330–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, M.; Hashmi, A.S.K. Heterocycles from gold catalysis. Chem. Commun. 2011, 47, 6536–6544. [Google Scholar] [CrossRef]
- Collings, B.A.; Athanassenas, K.; Rayner, D.M.; Hackett, P.A. Optical spectroscopy of Ag7, Ag9+, and Ag9. A test of the photodepletion method. Chem. Phys. Lett. 1994, 227, 490–495. [Google Scholar] [CrossRef]
- Krückeberg, S.; Dietrich, G.; Lützenkirchen, K.; Schweikhard, L.; Walther, C.; Ziegler, J. The dissociation channels of silver clusters Agn+, 3 ≤ n ≤ 20. Int. J. Mass Spectrom. 1996, 155, 141–148. [Google Scholar] [CrossRef]
- Shayeghi, A.; Götz, D.A.; Johnston, R.L.; Schäfer, R. Optical absorption spectra and structures of Ag6+ and Ag8+. Eur. Phys. J. D 2015, 69, 152. [Google Scholar] [CrossRef]
- Yang, X.; Cai, W.; Shao, X. Structural variation of silver clusters from Ag13 to Ag160. J. Phys. Chem. A 2007, 111, 5048. [Google Scholar] [CrossRef] [PubMed]
- Fournier, R. Theoretical study of the structure of silver clusters. J. Chem. Phys. 2001, 115, 2165–2177. [Google Scholar] [CrossRef] [Green Version]
- Bonačić-Koutecky, V.; Veyret, V.; Mitrić, R. Ab initio study of the absorption spectra of (n = 5–8) clusters. J. Chem. Phys. 2001, 115, 10450–10460. [Google Scholar] [CrossRef]
- Chen, M.; Dyer, J.E.; Li, K.; Dixon, D.A. Prediction of structures and atomization energies of small silver clusters,(Ag) n, n < 100. J. Phys. Chem. A 2013, 117, 8298–8313. [Google Scholar] [PubMed]
- Jin, Y.; Tian, Y.; Kuang, X.; Zhang, C.; Lu, C. Ab initio search for global minimum structures of pure and boron doped silver clusters. J. Phys. Chem. A 2015, 119, 6738–6745. [Google Scholar] [CrossRef]
- Fernández, E.M.; Soler, J.M.; Garzón, I.L.; Balbás, L.C. Trends in the structure and bonding of noble metal clusters. Phys. Rev. B 2004, 70, 165403. [Google Scholar] [CrossRef]
- Li, X.-B.; Wang, H.-Y.; Yang, X.-D.; Zhu, Z.-H.; Tang, Y.-J. Size dependence of the structures and energetic and electronic properties of gold clusters. J. Chem. Phys. 2007, 126, 084505. [Google Scholar] [CrossRef]
- Deka, A.; Deka, R.C. Structural and electronic properties of stable Aun (n = 2–13) clusters: A density functional study. J. Mol. Struct. 2008, 870, 83–93. [Google Scholar] [CrossRef]
- Assadollahzadeh, B.; Schwerdtfeger, P. A systematic search for minimum structures of small gold clusters Aun (n = 2–20) and their electronic properties. J. Chem. Phys. 2009, 131, 064306. [Google Scholar] [CrossRef]
- Zanti, G.; Peeters, D. DFT Study of Bimetallic Palladium−Gold Clusters PdnAum of Low Nuclearities (n + m ≤ 14). J. Phys. Chem. A. 2010, 114, 10345–10356. [Google Scholar] [CrossRef]
- Mukhamedzyanova, D.F.; Ratmanova, N.K.; Pichugina, D.A.; Kuz’menko, N.E. A Structural and Stability Evaluation of Au12 from an Isolated Cluster to the Deposited Material. J. Phys. Chem. C 2012, 116, 11507–11518. [Google Scholar] [CrossRef]
- Kinaci, A.; Narayanan, B.; Sen, F.G.; Davis, M.J.; Gray, S.K.; Sankaranarayanan, S.K.R.S.; Chan, M.K.Y. Unraveling the Planar-Globular Transition in Gold Nanoclusters through Evolutionary Search. Sci. Rep. 2016, 6, 34974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nhat, P.V.; Si, N.T.; Leszczynski, J.; Tho, N.M. Another look at structure of gold clusters Aun from perspective of phenomenological shell model. Chem. Phys. 2017, 493, 140–148. [Google Scholar] [CrossRef]
- Goldsmith, B.R.; Florian, J.; Liu, J.-X.; Gruene, P.; Lyon, J.T.; Rayner, D.M.; Fielicke, A.; Scheffler, M.; Ghiringhelli, L.M. Two-to-three dimensional transition in neutral gold clusters: The crucial role of van der Waals interactions and temperature. Phys. Rev. Mater. 2019, 3, 016002. [Google Scholar] [CrossRef] [Green Version]
- Persaud, R.R.; Chen, M.; Dixon, D.A. Prediction of Structures and Atomization Energies of Coinage Metals, (M)n, n < 20: Extrapolation of Normalized Clustering Energies to Predict the Cohesive Energy. J. Phys. Chem. 2020, 124, 1775–1786. [Google Scholar] [CrossRef]
- Johansson, M.P.; Warnke, I.; Le, A.; Furche, F. At What Size Do Neutral Gold Clusters Turn Three-Dimensional? J. Phys. Chem. C 2014, 118, 29370–29377. [Google Scholar] [CrossRef]
- Nhat, P.V.; Si, N.T.; Hang, N.T.N.; Nguyen, M.T. The lowest-energy structure of the gold cluster Au10: Planar vs. nonplanar? Phys. Chem. Chem. Phys. 2022, 24, 42–47. [Google Scholar] [CrossRef]
- Baker-Austin, C.; McArthur, J.V.; Tuckfield, R.C.; Najarro, M.; Lindell, A.H.; Gooch, J.; Stepanauskas, R. Antibiotic resistance in the shellfish pathogen Vibrio parahaemolyticus isolated from the coastal water and sediment of Georgia and South Carolina, USA. J. Food Prot. 2008, 71, 2552–2558. [Google Scholar] [CrossRef]
- Rittby, M.; Bartlett, R.J. An open-shell spin-restricted coupled cluster method: Application to ionization potentials in nitrogen. J. Phys. Chem. 1988, 92, 3033–3036. [Google Scholar] [CrossRef]
- Knowles, P.J.; Hampel, C.; Werner, H.J. Coupled cluster theory for high spin, open shell reference wave functions. J. Chem. Phys. 1993, 99, 5219–5227. [Google Scholar] [CrossRef]
- Hossain, S.; Nair, L.V.; Inoue, J.; Koyama, Y.; Kurashige, W.; Negishi, Y. Ligand-Protected Gold Clusters, in Ligand; IntechOpen: London, UK, 2018. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Constantin, L.A.; Sun, J. Workhorse Semilocal Density Functional for Condensed Matter Physics and Quantum Chemistry. Phys. Rev. Lett. 2009, 103, 026403. [Google Scholar] [CrossRef] [PubMed]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradient-approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Peterson, K.A.; Puzzarini, C. Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements. Theor. Chem. Acc. 2005, 114, 283–296. [Google Scholar] [CrossRef]
- Figgen, D.; Rauhut, G.; Dolg, M.; Stoll, H. Energy-consistent pseudopotentials for group 11 and 12 atoms: Adjustment to multi-configuration Dirac–Hartree–Fock data. Chem. Phys. 2005, 311, 227–244. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general-purpose quantum chemistry program package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Werner, H.J.; Knowles, P.J.; Manby, F.R.; Black, J.A.; Doll, K.; Heßelmann, A.; Kats, D.; Köhn, A.; Korona, T.; Kreplin, D.A. The Molpro quantum chemistry package. J. Chem. Phys. 2020, 152, 144107. [Google Scholar] [CrossRef] [Green Version]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Györffy, W.; Kats, D.; Korona, T.; Lindh, R.; et al. The Molpro quantum chemistry package. J. Chem. Phys. 2020, 152, 144107. [Google Scholar] [CrossRef] [Green Version]
- Zhumagulov, Y.V.; Kashurnikov, V.; Krasavin, A. Calculation of electron density of states for ensemble of gold nanoclusters. J. Phys. Conf. Ser. 2017, 936, 012015. [Google Scholar] [CrossRef]
- Du, J.; Sun, X.; Jiang, G. Hydrogen storage capability of cagelike Li3B12 clusters. J. Appl. Phys. 2020, 127, 054301. [Google Scholar] [CrossRef]
- Tho, N.H.; Bui, T.Q.; Si, N.T.; Nhat, P.V.; Nhung, N.T.A. Structural characteristics and chemical reactivity of gold-based clusters Aun (n = 16, 17) toward lone pairs. J. Mol. Model. 2022, 28, 54. [Google Scholar] [CrossRef] [PubMed]
- Idrobo, J.C.; Walkosz, W.; Yip, S.F.; Öğüt, S.; Wang, J.; Jellinek, J. Static polarizabilities and optical absorption spectra of gold clusters (Au n, n = 2–14 and 20) from first principles. J. Phys. Rev. B 2007, 76, 205422. [Google Scholar] [CrossRef] [Green Version]
- Liao, M.-S.; Bonifassi, P.; Leszczynski, J.; Ray, P.C.; Huang, M.-J.; Watts, J.D. Structure, bonding, and linear optical properties of a series of silver and gold nanorod clusters: DFT/TDDFT studies. J. Phys. Chem. A 2010, 114, 12701–12708. [Google Scholar] [CrossRef]
- Lecoultre, S.; Rydlo, A.; Félix, C.; Buttet, J.; Gilb, S.; Harbich, W. UV–visible absorption of small gold clusters in neon: Au n (n = 1–5 and 7–9). J. Chem. Phys. 2011, 134, 074302. [Google Scholar] [CrossRef] [Green Version]
- Koppen, J.V.; Hapka, M.; Szczęśniak, M.M.; Chałasiński, G. Optical absorption spectra of gold clusters Aun (n = 4, 6, 8, 12, 20) from long-range corrected functionals with optimal tuning. J. Chem. Phys. 2012, 137, 114302. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
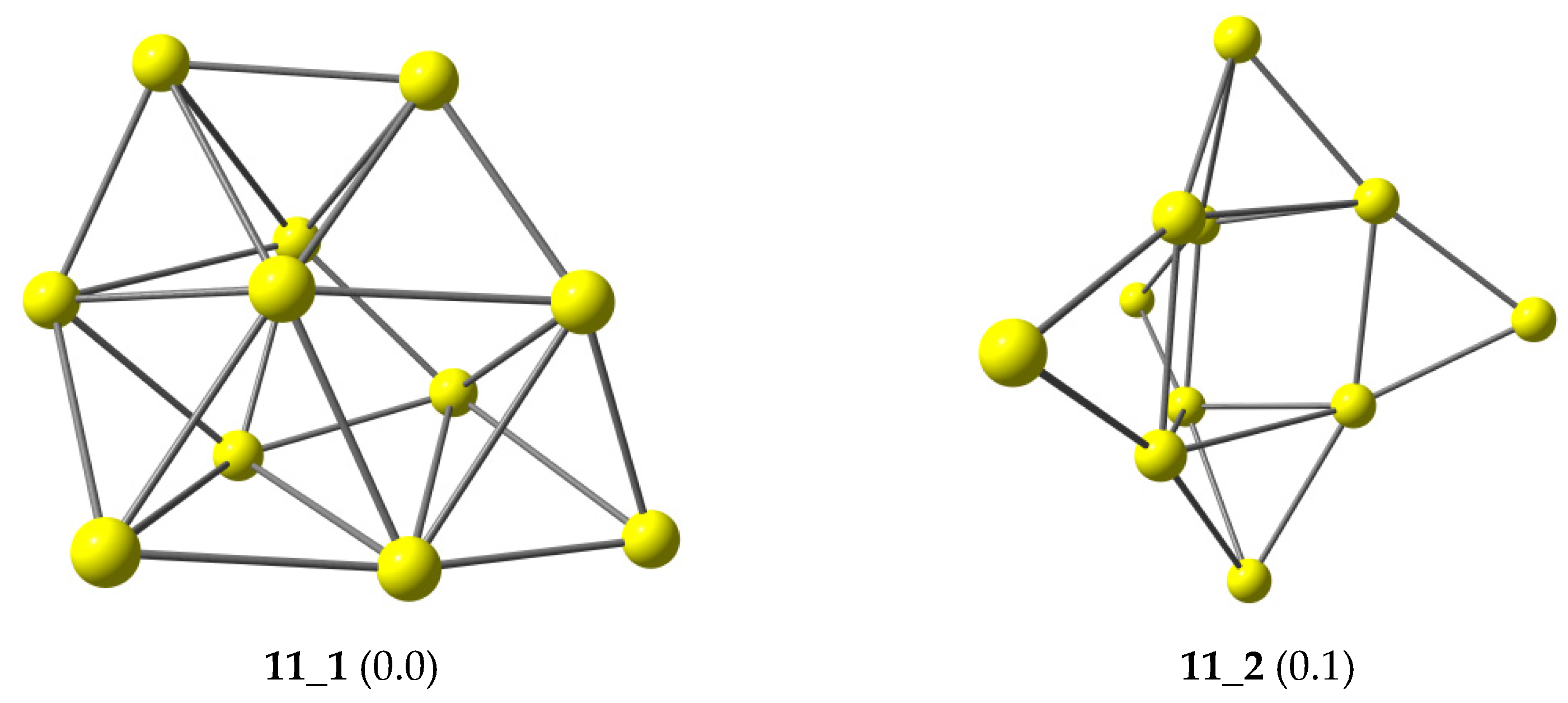{kind=link}
{kind=link}
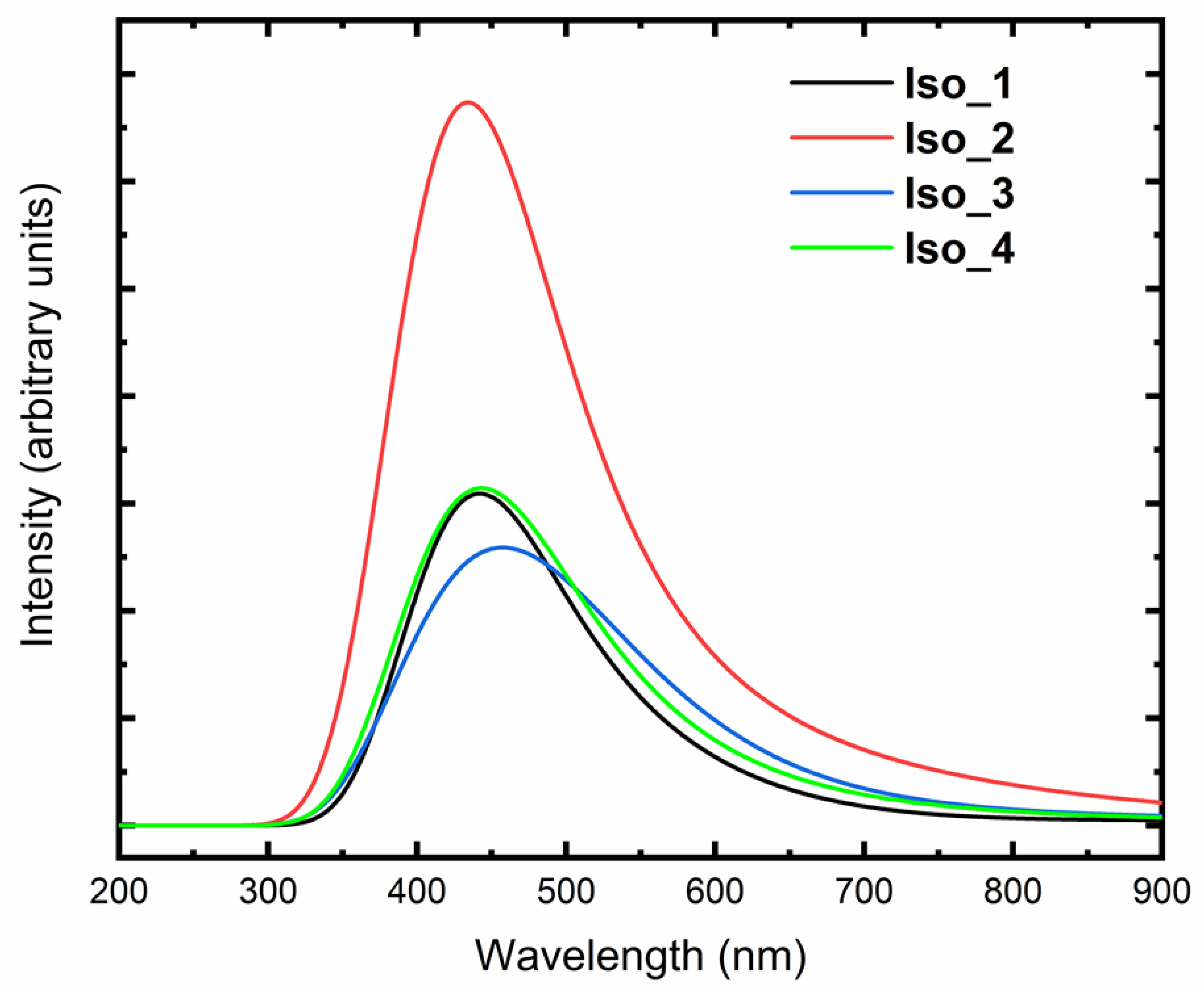{kind=link}
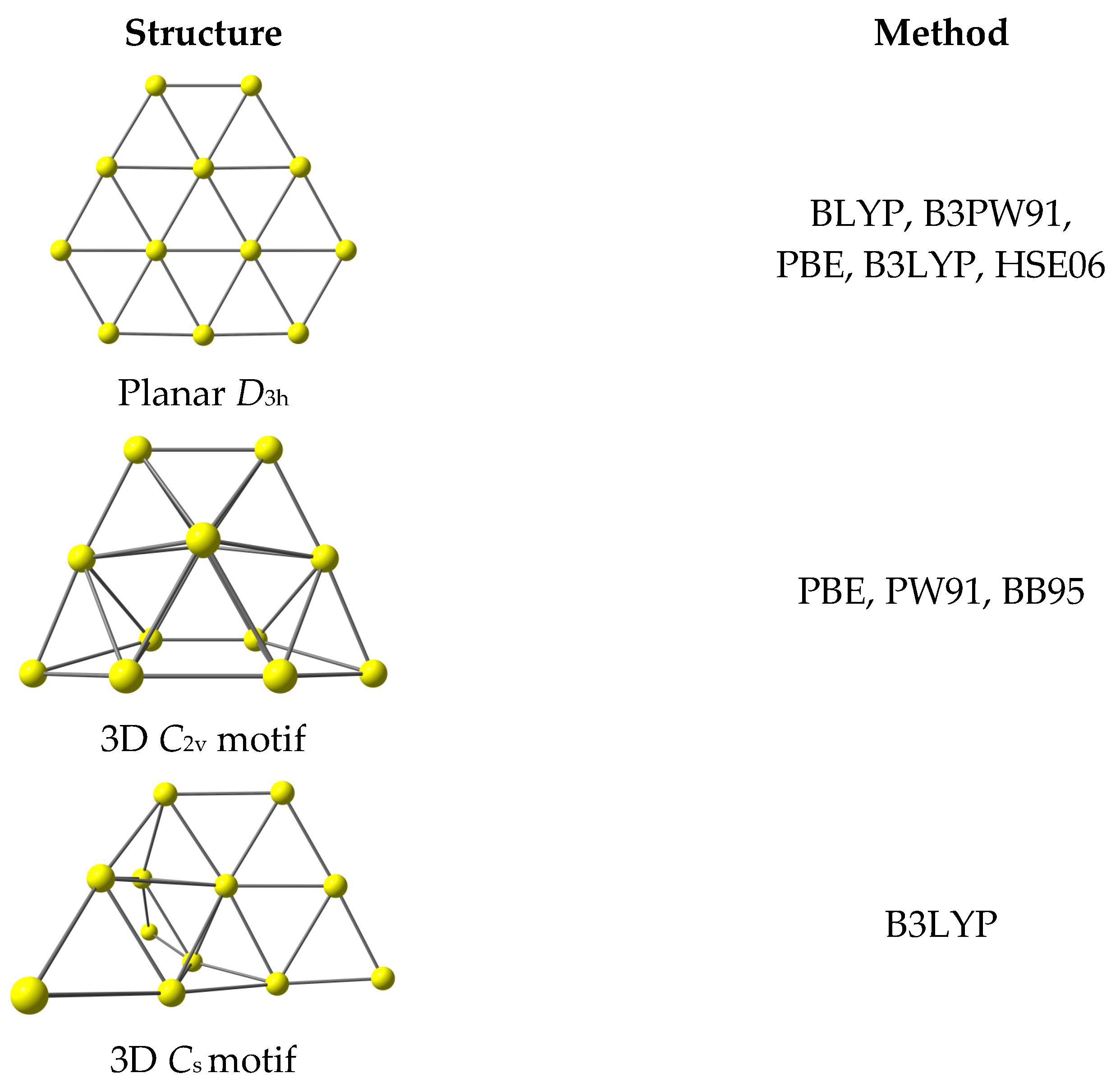{kind=link}
| Isomer | TPSS | PBE | LC-BLYP | revTPSS | CCSD(T) | |
|---|---|---|---|---|---|---|
| aVDZ-PP | VDZ-PP | VTZ-PP | ||||
| Iso_1 | 0.10 | 0.36 | 0.13 | 0.00 | 0.00 | 0.00 |
| Iso_2 | 0.00 | 0.00 | 0.22 | 0.17 | 0.52 | 0.04 |
| Iso_3 | 0.38 | 0.74 | 0.78 | 0.18 | 0.11 | 0.25 |
| Iso_4 | 0.37 | 0.41 | 0.00 | 0.52 | 0.41 | 0.33 |
| IEv, eV | EAv, eV | Energy of S0 → S1 Transition, eV | |
|---|---|---|---|
| Iso_1 | 7.01 | 2.80 | 1.06 |
| Iso_2 | 7.07 | 2.90 | 1.08 |
| Iso_3 | 7.09 | 2.78 | 1.24 |
| Iso_4 | 6.94 | 2.79 | 0.95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nhat, P.V.; Si, N.T.; Anh, N.N.K.; Duong, L.V.; Nguyen, M.T. The Au12 Gold Cluster: Preference for a Non-Planar Structure. Symmetry 2022, 14, 1665. https://doi.org/10.3390/sym14081665
Nhat PV, Si NT, Anh NNK, Duong LV, Nguyen MT. The Au12 Gold Cluster: Preference for a Non-Planar Structure. Symmetry. 2022; 14(8):1665. https://doi.org/10.3390/sym14081665
Chicago/Turabian StyleNhat, Pham Vu, Nguyen Thanh Si, Nguyen Ngoc Khanh Anh, Long Van Duong, and Minh Tho Nguyen. 2022. "The Au12 Gold Cluster: Preference for a Non-Planar Structure" Symmetry 14, no. 8: 1665. https://doi.org/10.3390/sym14081665