In the following, we make use of the insights from
Section 3 to design pulse sequences which induce complete enenatiomer-selective excitation despite the degeneracy of rotational states. All results presented in this section are obtained by numerically solving the time-dependent Schrödinger Equation (
7). In
Section 4.1, we design sequences of linearly polarized pulses to achieve complete enantiomer selectivity. As mentioned in
Section 3, the construction of closed cycles for complete enantiomer selection is particularly difficult in the presence of degenerate initial states if only linearly polarized pulses are used. We therefore consider the most simple rotational subsystem, i.e. the manifold of
and
, in this subsection. Rotational subsystems with larger
J will be considered in in
Section 4.2, where synchronized circularly polarized pulses are applied. The effects of initial rotational temperature and of the pulse duration are discussed in
Section 4.3.
4.1. Pulse Design Using Linearly Polarized Fields: Combination of Three-Wave Mixing Cycles
The smallest subsystem with three rotational levels consists of the state with
, i.e., the non-degenerate rotational ground state denoted by
, and two excited rotational levels with
, namely
and
, as shown in
Figure 4.
In a typical three-wave mixing pulse sequence, the first pulse creates a 50-50-coherence between the ground state and one of the excited states. The second (twist) pulse transfers the complete excited state population to the second excited state. Finally, the third pulse induces the separation by causing constructive interference for one enantiomer and destructive interference for the other in the respective rotational state. Starting with the rotational ground state, interaction with such a sequence of three pulses, polarized in
x-,
y-, and
z- direction and with frequencies
,
and
induces complete enantio-selective population transfer [
24]. However, if the initial condition is given by
i.e., if the three degenerate states of level
are populated, such a three-wave mixing pulse sequence cannot induce complete enantiomer selective excitation. This can be rationalized with the help of
Figure 4. A three-wave mixing cycle can be realized by three different combinations of
x-,
y- and
z-polarized microwave pulses, as seen in panels (i)–(iii). Here, the transitions induced by
x-,
y-, and
z-polarized fields are marked by dashed, dotted, and solid lines, respectively, with the colors indicating the frequencies of the corresponding fields. The degenerate initial states are indicated by black circles. The three-wave mixing cycle (i) only affects the initial state with
, while the cycles (ii) and (iii) transfer the population from the initial states with
. Thus, only one or two of the initially populated states are part of each closed three-level cycle and therefore only part of the initial population can be selectively transferred to different rotational states.
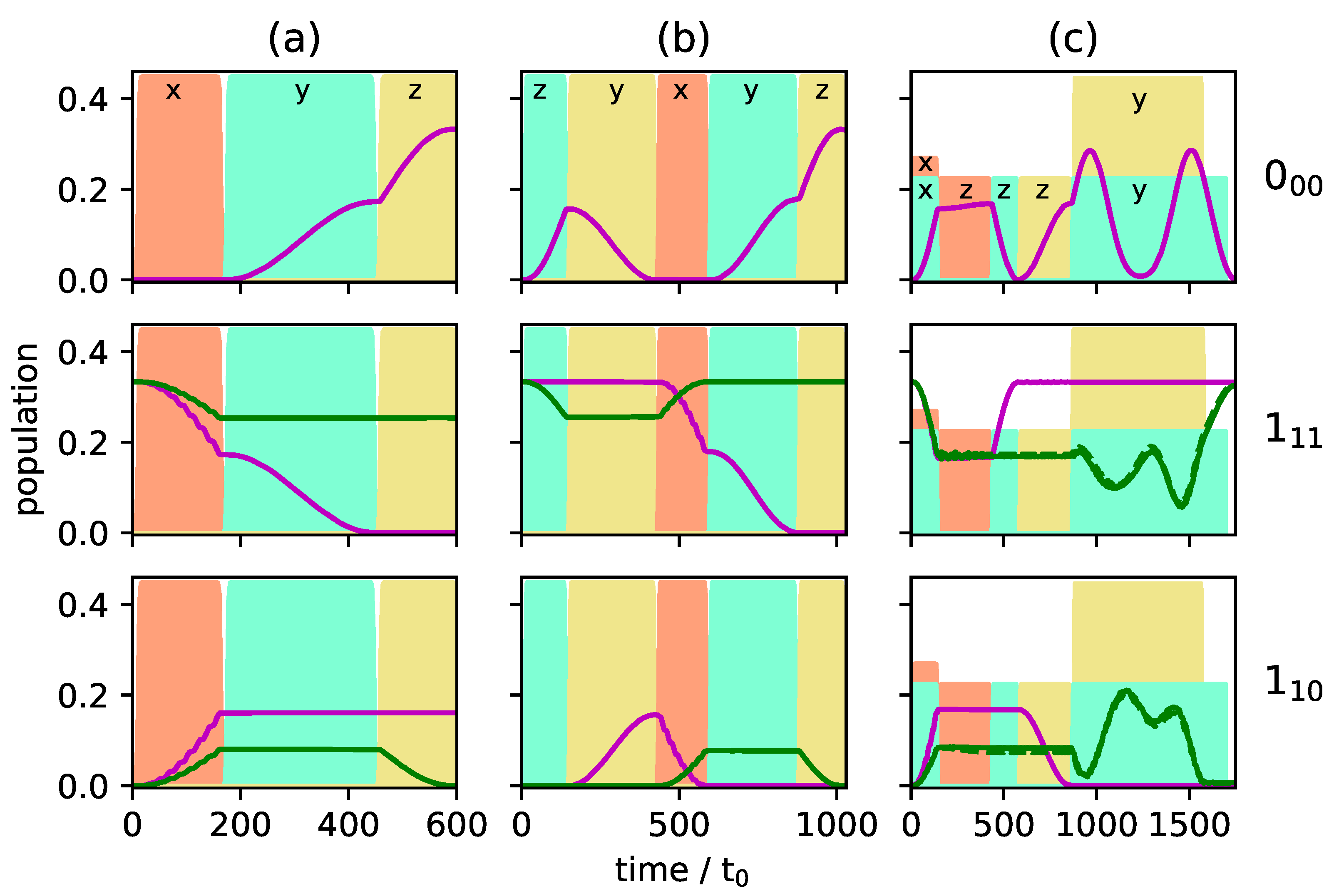
Figure 5a and
Figure 6a present the population transfer for the three-wave mixing cycle depicted in
Figure 4i with
Figure 5a showing the time-dependent population of each rotational level averaged over the corresponding
M-states for the two enantiomers (depicted by solid blue and dashed red lines), whereas the population of the individual
M-states is shown in
Figure 6a for one of the enantiomers. Cycle (i) results in complete enantiomer-selective population transfer for the initial state
, whereas the initial states
are affected only by the first pulse, depicted in dashed orange lines in
Figure 4i. They are not part of a complete three-wave mixing cycle and thus no enantiomer-separation occurs for these initial states. As a result, the ground state
is populated only by a single enantiomer at final time, as shown in the top panel of
Figure 5a. In the first excited state, no enantiomer-separation is observed at all (middle panel), while only partial enantio-selectivity occurs in the second excited state, as shown in the bottom panel.
We quantify the overall selectivity by
where
is the normalized population difference between the two enantiomers
and
in the three energy levels
. For excitation with the three-wave mixing cycle (i),
. The same amount of selectivity can be obtained by cycles (ii) and (iii).
A better selectivity can be expected if cycle (i) is combined with cycles (ii) or (iii) since all initial states are then addressed by a closed cycle. As an example, we combine cycles (i) and (ii). The resulting population transfer is depicted in
Figure 5b and
Figure 6b where the pulse sequence starts with cycle (ii). The first pulse (dashed turquoise lines in
Figure 4ii) affects only the initial states
and creates a coherence between the states
and
. The second pulse (solid yellow line) transfers the ground state population to the second excited state. The third pulse, depicted by dashed orange lines in
Figure 4ii, closes the cycle and, at the same time, acts as the first pulse of cycle (i). By combination of the two three-wave mixing cycles, complete separation of the enantiomers is achieved in levels
and
, while the selectivity in level
is still incomplete. Overall, the selectivity increases to
. Although all initial states are part of closed cycles, complete enantiomer-selectivity cannot be obtained. This is due to the population transfer being induced by
x- or
y-polarized fields. As shown in
Figure 2, an
x-polarized field does not induce a Rabi oscillation between two levels, but rather spreads the population over all
M-states. Thus, part of the population of the initial state
leaks from the cycle connecting
,
and
. The same holds for the population from the initial state
.
Enantiomer-selective population transfer can be completed by adding the third excitation scheme from
Figure 4. The resulting rotational dynamics is shown in
Figure 5c and
Figure 6c. The corresponding pulse sequence consists of seven pulses: three pulses with
x-
y-, and
z-polarization are resonant with the transition
, two pulses,
x- and
z-polarized, are resonant with
and two pulses,
y- and
z-polarized are resonant with
. In order to obtain complete selectivity, it is important to properly synchronize the three cycles by using overlapping pulses and adjusting the field strengths. At
, both enantiomers are assumed to populate only level
. At the end of the pulse sequence, enantiomer
(blue lines) populates level
, while enantiomer
(red lines) exists only in rotational states belonging to level
. The rotational ground state is empty. By combining all three three-wave mixing cycles
is obtained, i.e., almost 100% enantiomer-selectivity. Note that a systematic optimization of the pulse parameters will allow to push the enantiomer-selectivity even closer to 100%.
The sequence achieving essentially complete enantiomer-selectivity, cf.
Figure 5c and
Figure 6c, consists of seven different microwave fields. It is constructed by combining all the different three-wave mixing schemes that exist for this rotational subsystem. This should be compared to Ref. [
27], where enantiomer-selective excitation for the same rotational subsystem has been identified by means of a controllability study. In particular, five different fields, i.e., fields with different combinations of frequency and polarization, were found to be sufficient for the system to be enantiomer-selective controllable [
27]. While the minimal number of different fields is given by controllability analysis, the actual pulse shapes or sequence of pulses has to be determined by other means. In Ref. [
27], a sequence of 12 individual pulses (using five different combinations of frequency and polarization) has been shown to yield complete enantiomer-selectivity. Controllability analysis on the one hand, and pulse design resulting from knowledge of the rotational dynamics are thus two complementary approaches to achieve a complete enantiomer-selective excitation (or any other desired target) in the presence of degeneracies in the rotational spectrum. The pulse sequence constructed here, while simpler than the pulse sequence found in Ref. [
27], is challenging for current microwave experiments due to the need to carefully adjust the field intensities of overlapping pulses. Moreover, there is no automatic way to transfer these results to rotational subsystems with larger
J. To overcome these limitations, we consider in the following a different excitation strategy, namely the use of circularly polarized fields.
4.2. Pulse Design with Circularly Polarized Fields
As discussed Section III, replacing the linear polarizations along
x and
y by circular polarizations prevents the spread of the initial population over the
M-manifold. An excitation scheme using left- and right-circularly polarized pulses together with a
z-polarized pulse has already been proposed in Ref. [
27], where this combination of microwave fields was proven to lead to complete enantiomer-selectivity. Here, we extend this strategy to a set of rotational states for which microwave three-wave mixing was demonstrated experimentally [
15], namely the
,
and
states of carvone, depicted in
Figure 7.
We assume that initially, all degenerate states of the lowest rotational level
are equally populated, i.e.,
c.f. the black circles in
Figure 7, whereas levels
and
are empty. Such an initial condition can be realized by choosing the two excited rotational states in a vibrational state
[
24,
26] or by depleting excited rotational levels by laser excitation [
17].
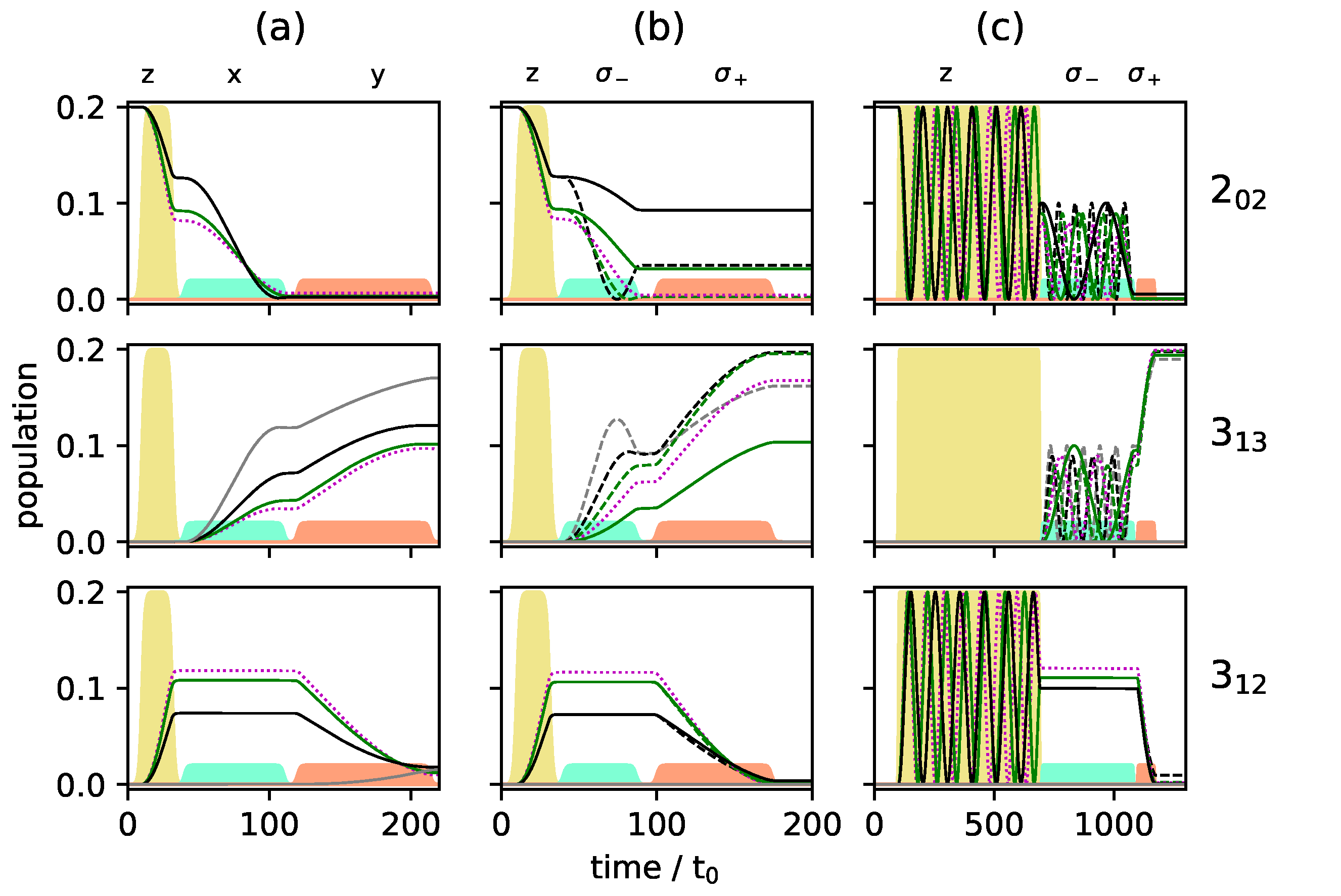
Figure 8 and
Figure 9 show the population transfer for different pulse sequences, with the average population of the three rotational levels for the two enantiomers plotted in
Figure 8, while
Figure 9 shows the population of the individual
M-states for a single enantiomer. In panel (a) of
Figure 8 and
Figure 9, we show, for reference, the population transfer for a three-wave mixing scheme with
x-,
y- and
z-polarized fields which corresponds to an enantiomer-selectivity of
, with enantiomeric excess of enantiomer (+) and (−) in the levels
and
, respectively, and level
empty. Using
and
-polarized fields instead of the
x- and
y-polarized ones confines each of the initial states
with
to a single 3-level cycle, as seen in
Figure 7. We take the first pulse, as before, to be a
z-polarized
-pulse, i.e., the pulse duration is chosen such that 50% of the population (averaged over the
M-states) is in level
and 50% remains in the ground state
. The second and third pulses are
and
-polarized, respectively, with pulse durations determined as in a standard three-wave mixing scheme. The resulting average population is shown in
Figure 8b, and the population of the individual
M-states can be seen in
Figure 9b. The excitation scheme leads to almost complete enantiomer-selection for the levels
and
. However, about 20% of the population of both enantiomers remains in the lowest level
. This is already a clear advantage compared to the standard three-wave mixing scheme with only linearly polarized fields. One could, for example, obtain a purified sample by extracting the population of either level
or
, each corresponding to a single enantiomer. The overall incomplete enantiomer-selectivity can be rationalized as follows: The first pulse leads to an average 50/50-coherence between the levels
and
, c.f.
Figure 8b. However, as also illustrated in
Figure 1, the Rabi frequencies of the two-level transitions with different
are different, with transitions between
states having the largest and transitions between
states having the smallest Rabi frequency. This results in population transfer of more than 50% for states with
and
and less than 50% for
, c.f.
Figure 9b.
One can account for the different Rabi frequencies by adjusting the pulse duration such that every single
M-state undergoes a
-population transfer. To achieve that, one has to wait for several Rabi cycles until all individual cycles are synchronized. Similarly, the complete population transfer between the states
and
has to be synchronized. The pulse durations were determined by a simple parameter optimization using the NLopt software package [
31]. Pulse shapes and maximal field strengths were kept fixed. The resulting rotational dynamics is displayed in
Figure 8c and
Figure 9c. The fully synchronized three-wave mixing scheme with circularly polarized fields leads to almost complete enantiomer-selective excitation—one enantiomer is entirely transferred to level
(dashed red lines in
Figure 8), while the second enantiomer ends up in level
(blue lines in
Figure 8). There is no population left in level
and the overall enantiomer-selectivity amounts to
, limited—according to Equations (
14)–(
16)—by the Rabi frequencies for the different transitions differing by irrational factors.
While it is possible to synchronize the transitions such that a 50/50-coherence is obtained for all individual transitions with arbitrary accuracy, this comes at the expense of longer pulse durations. Eventually, the pulse durations are limited by the coherence time of the experiment, determined, e.g., by collisions of the molecules in the sample with background gas. In practice, it is thus always necessary to find a compromise between accurate synchronization and pulse duration. We discuss the relation between accuracy and pulse duration in
Section 4.3. At the same time, we inspect another factor relevant in an experimental implementation. Most of the current microwave three-wave mixing experiments are carried out for rotational states with thermal population. In
Section 4.3, we thus discuss also the effects of the initial temperature.
4.3. Rotational Temperature and Synchronization Time
So far, we have investigated the rotational dynamics of chiral asymmetric top molecules assuming that only the lowest rotational state of the relevant subsystem is initially populated. Typical microwave three-wave mixing experiments are carried out with thermal samples of chiral molecules [
15,
16]. Then, the initial rotational temperature of the molecules has to be factored in. The initial density operator is given by
where the rotational levels are occupied according to the Boltzmann distribution,
with
Q defined by
and
,
and
. In molecular beam experiments, rotational temperatures in the range of 1 - 10 K can be achieved, which result in non-neglibile initial thermal population of all rotational levels involved in the three-wave mixing cycle.
Figure 10 shows the maximal selectivity for the excitation schemes (a)–(c) in
Figure 8 in a semi-logarithmic plot for initial temperatures between
T = 10 mK and 10 K. For
K, thermal occupation of the upper two levels becomes negligible and the selectivity approaches its maximal value, whereas it gets exponentially reduced for rotational temperatures
T > 0.1 K. Importantly, the modified excitation scheme using synchronized circularly polarized pulses (
Figure 8c) results in a larger selectivity than standard three-wave mixing with linearly polarized fields (
Figure 8a) and three-wave mixing with circularly polarized fields without synchronization of the individual three-level systems (
Figure 8b) for every temperature.
The maximal enantiomer-selectivity that can be achieved in practice with the optimal scheme employing synchronized circularly polarized fields also depends on how accurate the individual Rabi cycles are synchronized. In the following, we discuss the relation between accuracy and the pulse duration in more detail. Since we consider resonant excitation, the population of the lowest states is given by
, where the Rabi frequency
, given in Equation (
12), depends on
M. Consider the first pulse in excitation scheme (c), which is a
z-polarized field that drives a
transition, i.e., the
M-dependence of the Rabi frequency is given in Equation (
14). As shown in
Figure 1b, the first pulse simultaneously drives
two-level transitions. In the bottom panel of
Figure 9c, it can be seen that after the first pulse, the 50/50-coherence is reached for all M-stats within an accuracy of
. Since the Rabi frequencies for different
M-states differ by irrational numbers, a 50/50-coherence can be obtained with arbitrary accuracy by increasing the pulse duration.
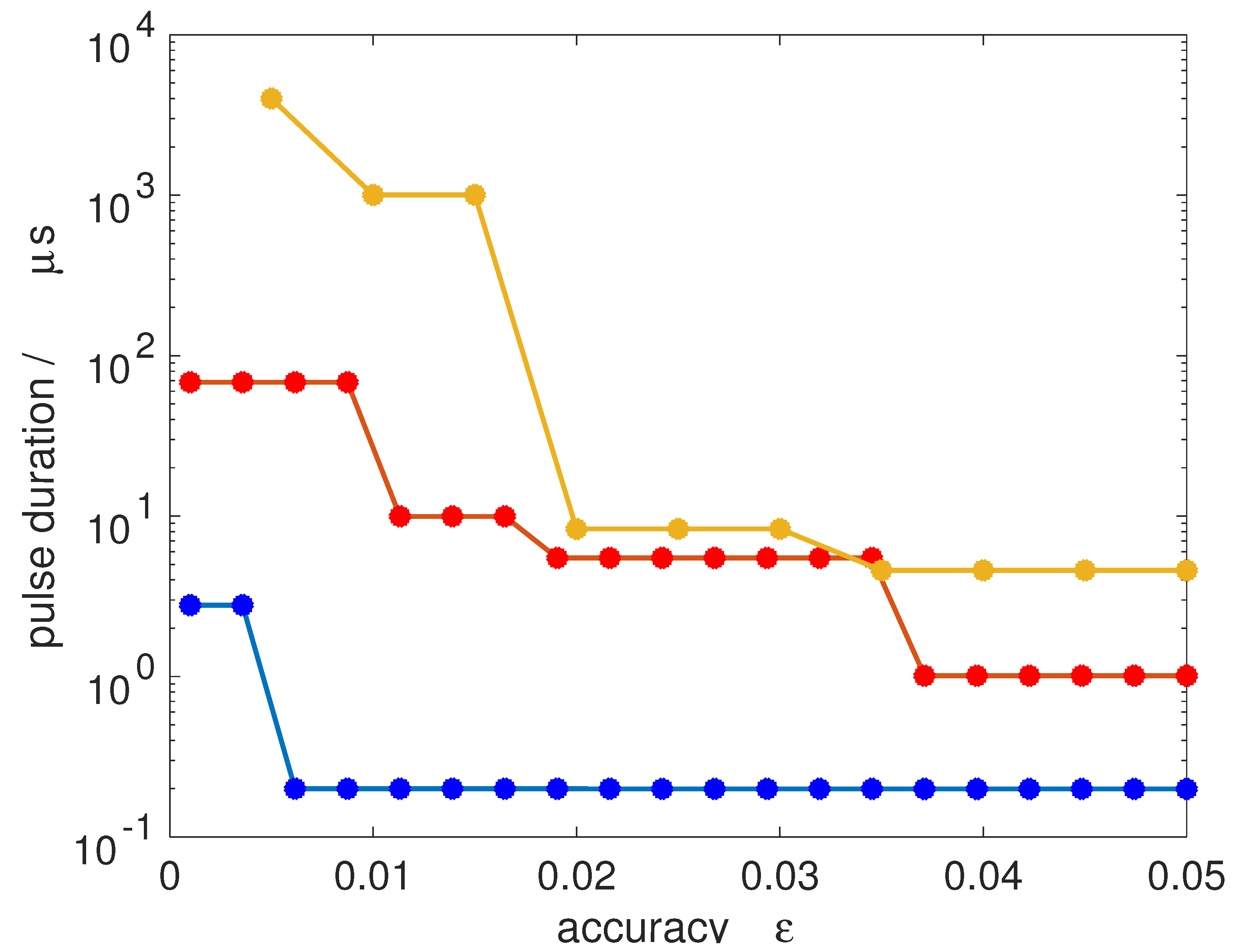
In
Figure 11, we show the pulse duration required to achieve a 50/50-coherence for all
M-states within an accuracy
defined by
. Transitions from
to
(blue dots),
to
(orange dots) and
to
(yellow dots) are considered, assuming a microwave field with an intensity of 10 W/cm
2, comparable to field intensities used in current microwave experiments [
15]. A jump in the pulse duration occurs whenever the number of Rabi cycles has to be increased to obtain a smaller value of
. Since the degeneracy increases with increasing
J, the pulse duration required for very accurate synchronization increases. For a rotational subsystem with initial state
, a 50/50-coherence with an accuracy below one per cent can be achieved within less than 1 μs. Starting with rotational states with
, one per cent accuracy already requires a pulse duration of about 10 μs. This is of the same order as typical coherence times in current microwave experiments. Of course, the absolute value of the pulse duration can in principle be reduced by employing stronger pulses.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
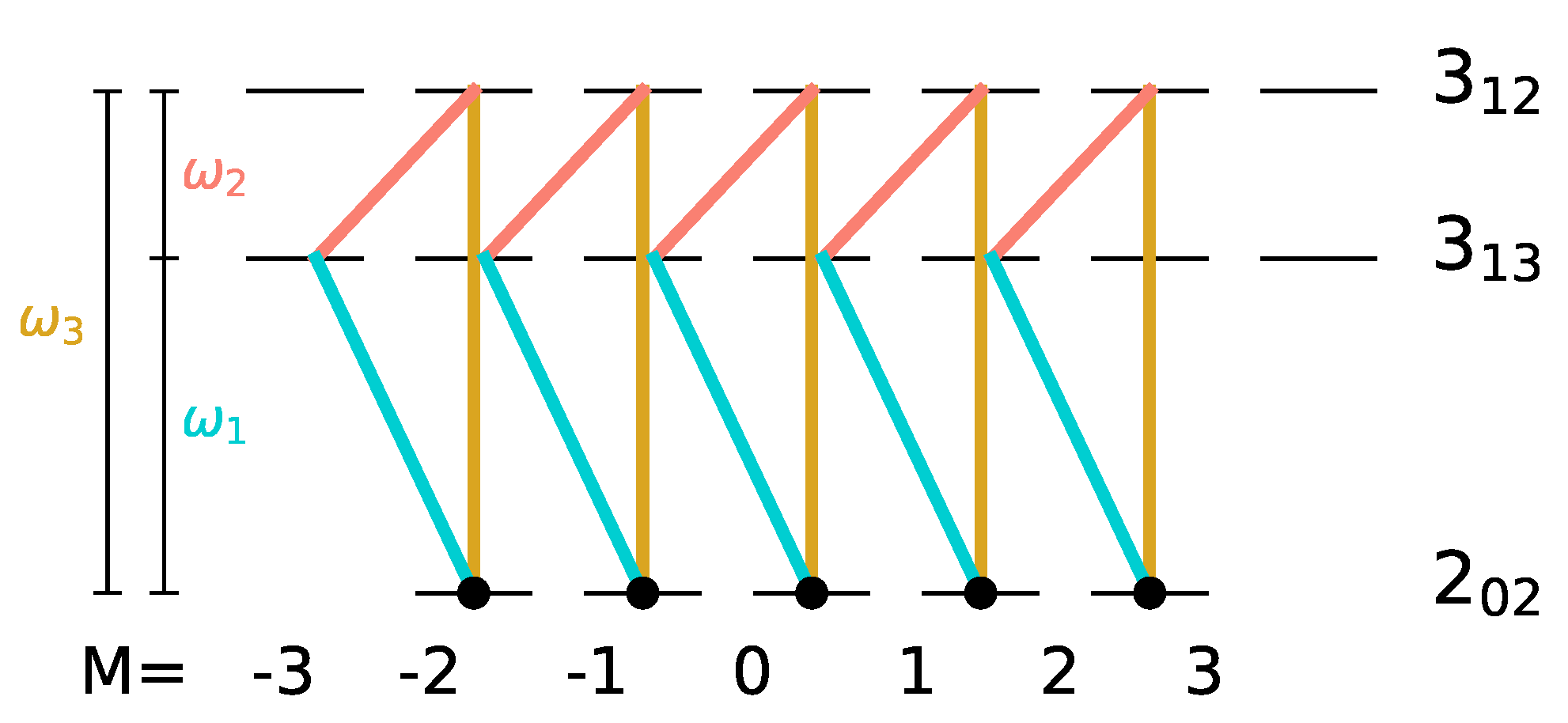{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










