
Symmetry Constraints on Spin Order Transfer in Parahydrogen-Induced Polarization (PHIP)


1
Section Biomedical Imaging, Molecular Imaging North Competence Center (MOIN CC), Department of Radiology and Neuroradiology, University Medical Center Schleswig-Holstein (UKSH), Kiel University, Am Botanischen Garten 14, 24118 Kiel, Germany
2
Helmholtz-Institut Mainz, GSI Helmholtzzentrum fuür Schwerionenforschung, 55128 Mainz, Germany
3
Quantum, Atomic, and Neutron Physics (QUANTUM) Institute of Physics, Johannes Gutenberg University, Mainz, 55099 Mainz, Germany
4
International Tomography Center, SB RAS, 3A Institutskaya st., 630090 Novosibirsk, Russia
*
Authors to whom correspondence should be addressed.
Symmetry 2022, 14(3), 530; https://doi.org/10.3390/sym14030530
Submission received: 13 December 2021
/
Revised: 11 February 2022
/
Accepted: 27 February 2022
/
Published: 4 March 2022
(This article belongs to the Special Issue Symmetry Principles in the Nuclear Magnetic Resonance)
Abstract
:It is well known that the association of parahydrogen (pH2) with an unsaturated molecule or a transient metalorganic complex can enhance the intensity of NMR signals; the effect is known as parahydrogen-induced polarization (PHIP). During recent decades, numerous methods were proposed for converting pH2-derived nuclear spin order to the observable magnetization of protons or other nuclei of interest, usually 13C or 15N. Here, we analyze the constraints imposed by the topological symmetry of the spin systems on the amplitude of transferred polarization. We find that in asymmetric systems, heteronuclei can be polarized to 100%. However, the amplitude drops to 75% in A2BX systems and further to 50% in A3B2X systems. The latter case is of primary importance for biological applications of PHIP using sidearm hydrogenation (PHIP-SAH). If the polarization is transferred to the same type of nuclei, i.e., 1H, symmetry constraints impose significant boundaries on the spin-order distribution. For AB, A2B, A3B, A2B2, AA’(AA’) systems, the maximum average polarization for each spin is 100%, 50%, 33.3%, 25%, and 0, respectively, (where A and B (or A’) came from pH2). Remarkably, if the polarization of all spins in a molecule is summed up, the total polarization grows asymptotically with ~1.27 and can exceed 2 in the absence of symmetry constraints (where is the number of spins). We also discuss the effect of dipole–dipole-induced pH2 spin-order distribution in heterogeneous catalysis or nematic liquid crystals. Practical examples from the literature illustrate our theoretical analysis.
1. Introduction
Parahydrogen-induced polarization (PHIP) is a cost-efficient and fast method to polarize nuclear spins [1]. PHIP exploits the symmetry of molecular dihydrogen that exists as two nuclear spin isomers: Parahydrogen (pH2) and orthohydrogen (oH2). The nuclear spin state of pH2 is the singlet state, , which is assymetric under the exchange of the nuclear spins. The total wave function of the H2 nuclei is antisymmetric under the exchange of two nuclei (two fermions), so the quantum numbers of the rotational states take even values [2]. oH2 is represented by three nuclear spin states, , , and . These three nuclear spin states are symmetric under spin exchange, hence necessitating odd rotational quantum numbers [2]. This selection is dictated by the generalized Pauli principle, which states that the total wave function of two protons (two fermions with spin-½) is antisymmetric upon permutation [2]. Note, however, that the hydrogen atom, consisting of a proton and an electron (i.e., two fermions), is a boson, hence the total wave function of H2 is symmetric under the exchange of two atoms (further discussion below). Note that here we discuss the interplay between the symmetry of the nuclear spin states and symmetry of rotation–molecular spin states.
Thus, pH2 takes the lowest rotational energy state, which is symmetric, and oH2 takes the next rotational energy state, which is asymmetric. The gap between the lowest two rotational energy levels, i.e., pH2 and oH2, is significant (170.5 K) and can be used to enrich the para-state: 50% pH2 is obtained by cooling H2 to liquid nitrogen temperatures [3] and 99% at about 25 K (e.g., using a two-stage Helium cryo-cooler or liquid Helium) [4,5]. Because pH2 and oH2 can be separated, they are also referred to as nuclear spin isomers (NSIM). Some other molecules also have NSIM, for instance, molecular deuterium [6], ethylene [7], and water [8].
The density matrix for an ensemble of molecules containing N spin-½ nuclei, where spins and B originate from pH2, can be written as follows:
Here, is the identity matrix, i.e., a {} matrix with ones on the diagonal. The individual spin operators in the dot product, , are obtained using the Kronecker (direct) product of the corresponding Pauli matrices (with k = X, Y or Z) and of identity matrices . Here, the numbering of the spins in the molecule is important. For example, for the first spin, the operator is constructed as
There are two primary variants of PHIP: (a) Hydrogenative PHIP (Figure 1A), such as PASADENA (parahydrogen and synthesis allow dramatically enhanced nuclear alignment [9]) and ALTADENA (adiabatic longitudinal transport after dissociation engenders net alignment [10]), and (b) non-hydrogenative PHIP, or SABRE (signal amplification by reversible exchange [11], Figure 1B), where pH2 and the substrate interact via a reversible exchange at a catalyst. Both methods have found applications at high (~T) [12], low ~1 mT [13], ultra-low ~1 μT [14,15], and zero fields [16]. To limit the scope of this paper, however, we focus our discussion on hydrogenative PHIP at high magnetic fields only. It should be noted that a similar analysis for four spin-½ SABRE systems was recently performed [17].
For hydrogenative PHIP, the spin state of the molecule after pH2 addition strongly depends on the coupling regime. Two spins and are considered strongly coupled when the difference of their Larmor precession frequencies, , is much smaller than their mutual indirect spin–spin coupling , i.e., . In the opposite case, the spins are weakly coupled [18]. The frequency of spin or depends on the strength of the magnetic field , chemical shift , and magnetogyric ratio .
In the PASADENA case, upon the addition of pH2 to an asymmetric molecular environment at high fields, 1H spins are weakly coupled. Since individual molecular hydrogenation events are distributed in time over the course of the hydrogenation reaction, the molecules evolve for different times, and the non-stationary X and Y coherences (Equation (1)) are lost. The singlet spin state is averaged to the non-evolving stationary state, the so-called ZZ spin order [1]:
From now on, we will omit operator “hats”.
It is common to transfer pH2-derived spin alignment to proton and X-nuclear magnetization (e.g., 13C, 15N, 19F) for use as an MRI contrast agent [19,20,21], monitoring of chemical and enzymatic reactions [7,22], or for analytical chemistry [23]. Many of such spin-order transformations are represented by unitary transformations of the density matrix:
where is the density matrix at time , before the spin-order transfer (SOT), and is the final density matrix, after the SOT. The unitary evolution operators , also known as propagators, can be found by solving the corresponding Liouville von-Neumann equation,
for a time-dependent Hamiltonian and the initial condition .
2. Methods
2.1. Spin Operators and Observables
The general SOT from the initial spin state to the desired target spin state under the action of the propagator can be written as
where is the final spin state, is the amplitude of the target spin state , and is the difference between and that is not relevant for our considerations. We will use for density matrices with the trace of one and for traceless spin operators (or traceless density matrices).
Since the propagator is unitary, the transformation (6) implies boundaries on the parameter . There is no general way to determine all possible final states for undefined . However, it is possible to obtain boundary conditions for the amplitude and a given and in general.
We define the operator of polarization of a single spin (e.g., A) in a molecule as
and the operator of polarization of spins-½ as
Now it is straightforward to calculate a polarization () of one spin, or the average of many spins, using corresponding spin operators (7) and (8):
Here, is the density matrix of the system at the time of interest .
In the same fashion, the amplitude of the state for after SOT can be evaluated as:
We will use in the following to report the maximum theoretically possible polarization ().
We note that care should be taken when different initial states are analyzed. For example, consider a general operator in the form that consists of two orthogonal operators and (Equation (8)). Here, all diagonal elements of the unitary operator are and the largest element (its absolute value) of is equal to the maximum total spin of N spin-½ system, , times the normalization coefficient , that is, . Since the population of any individual state must be between 0 and 1, additional constraints are imposed on the allowed values of P: . One can see that to avoid non-negative populations, is needed. This means that for the system of two spins with net magnetization, the maximum average polarization per spin can only be 50%. However, if there are other components in the density matrix, larger values of P are possible. For example, the polarization of spins in state is 100%; the corresponding density matrix is for = 2.
2.2. No Symmetry Constraints
The boundaries for the amplitude of the target state after SOT (Equation (6)) are [24]
Here, and are the eigenvalues arranged in vectors for the operators and . The arrows up and down indicate that these eigenvalues are sorted in an ascending or descending order. In general, .
These boundaries arise because we assume all transformations to be unitary, and the initial and final states are given by Hermitian operators [24]. Because and are traceless operators, the boundary parameters often have the same absolute value: unless otherwise noted.
2.3. Symmetry Constraints (SC)
When a system has spin symmetry (i.e., groups of equivalent spins), only the states belonging to the same irreducible representations () of this group of symmetry can be mixed by unitary transformations [17,24,25]. In this case, the boundary conditions can be found as:
where are vectors of eigenvalues of the operator (X = initial or target) that correspond to eigenvectors belonging to the same and sorted in an ascending () or descending () order.
The transformation amplitude is bounded as .
2.4. Eigenvalues in the Case of SC
It is not trivial to define when SCs are present. To find the transformation of a density matrix into a group-symmetrized basis, one needs to construct a symmetry group-specific matrix from orthonormal basis vectors . Each vector must belong to only one irreducible representation . Vectors are written vertically. Let us enumerate these vectors in such a way that all vectors from the same stand next to each other: . Then, the matrix of spin state ( or ) in the new basis can be found as
There are three different situations for :
- is diagonal. When the predefined basis of the group coincides with the eigenstates of the operator , then is diagonal with eigenvalues on the diagonal. All coherences (off-diagonal elements) are zero. To find , one has only to sort and enumerate the eigenvalues inside each :
Here, is an eigenvalue of and is its corresponding eigenvector belonging to irreducible representations .
- 2.
- is-block diagonal. When has coherences only inside the same irreducible representations , then is a -block diagonal matrix
Here, are the corresponding blocks of irreducible representations . Because all vectors from one have the same symmetry, their superposition also has the same symmetry. It means that each block should be additionally diagonalized, and resulting diagonal elements are corresponding eigenvalues .
- 3.
- is not block diagonal. The most general case is when there are off-diagonal elements between different irreducible representations.
In this situation, we will assume that the coherences are averaged to zero during the hydrogenation reaction due to magnetic field inhomogeneity and the different evolution of each hydrogenated molecule. When such off-diagonal elements are removed (), the is “-block diagonal” and equivalent to Equation (15). Hence, the consequent diagonalization and analysis are equivalent and described in “case 2”.
In the following discussion, we use these three methods to find eigenvalues to evaluate when SCs are imposed. A script is available in SI to evaluate for a different number of spins, symmetry, initial, and target spin states (Matlab).
Below, we will discuss some specific cases and demonstrate the effect of symmetry on PHIP and spin order transfer.
2.5. Spin Systems Notations
We use a notation that is slightly different from Pople’s spin-system notation. The main idea is to distinguish the symmetries of the spin system. In addition, we fix X-spin to the target 13C nucleus. Let us consider some examples.
First, for us, “ABC” stands for a system with three chemically nonequivalent spins, and only weak coupling is considered between spins. Second, “A2B” stands for a system with two magnetically equivalent spins A2 (strongly coupled) that are weakly coupled to B and have different chemical shifts with B. Third, according to Pople’s notation, 12C2-ethylene consists of four chemically and magnetically equivalent 1H spins, hence the spin symmetry is A4. However, it does not reflect the permutation group symmetry of ethylene. Hence, we refer to the spin symmetry of 12C2-ethylene as AA’(AA’).
3. Results
Note that the polarization values reported in the following are the upper theoretical limits (as implied by the transformation mathematics).
3.1. Parahydrogen Spin-Order Transfer in a Two Spin-½ System
3.1.1. The Symmetry of AB and A2 Systems
The simplest PHIP system consists of two spin-½ nuclei. If the protons of pH2 after hydrogenation are magnetically and chemically nonequivalent (AB-system)—no symmetry constrains—the spins can be treated separately, and the appropriate basis would be the Zeeman basis:
When the protons are magnetically equivalent, the system is A2 and there are restrictions on the choice of the basis. Here, singlet-triplet (S-T) states should be used:
Among these two systems, only A2 has nontrivial symmetry, which is C2. In Appendix A, we describe all relevant groups of symmetry. The transformation elements of the C2 group are identity transformation or null permutation and the permutation of two protons :
The C2 group (or G12) has two irreducible representations: Even (gerade–“g”) and odd (ungerade–“u”). The singlet state is the only member of the odd irreducible representation = B, while three triplet states are the members of = A. In terms of sets, it can be written as
Tables of characters and decomposition of spin states into irreducible representations are given for A2, A3, and AA’ (AA’) systems in Appendix A.
3.1.2. pH2 to Magnetization in AB Systems
Let us consider the transformation of spin order in an AB system (no symmetry constraints, Equation (11)) to magnetization (Equations (7) and (8)):
This means that the PASADENA spin order () can be transferred to 100% polarization of one spin, or 50% polarization of each spin. In the latter case, the net magnetization can be 50% per spin or zero (21).
The examples of spin-order transfer (SOT) sequences for direct polarization transfers to one spin are Selective Excitation of Polarization using PASADENA (SEPP) [26] and adiabatic-passage spin order conversion (APSOC) [27,28,29]. SOT for polarization transfer to two spins include out of phase echo (OPE) [30], only parahydrogen spectroscopy (OPSY) [30,31], and APSOC [27,28,29].
3.1.3. pH2 to Magnetization in A2 Systems
The situation is different for two magnetically equivalent spins A1 and A2 in an A2 spin system. Symmetry constraints do not allow spin order conversion of into net magnetization:
The only way to transfer polarization is to break the symmetry (A2 → AB) that is happening e.g., during ALTADENA.
3.1.4. Limitation of the Method: ALTADENA Example
One of the first experiments that demonstrated spin order conversion of pH2 was ALTADENA [10], using adiabatic magnetic field variation (AMFV). AMFV-induced spin order transfer in an AB two spin-½ system results in the following transformation of [1]:
The sign ( depends on the relative chemical shift difference and the sign of the J-coupling constant of the spins [1]. It follows that in ALTADENA, both spins acquire maximum polarization of (see Equation (7)), but the total (net) polarization of the molecule is zero.
The Hamiltonian of an AB system is, in general, asymmetric (lacking permutation symmetry). However, at a zero field , it has the same permutation symmetry as the Hamiltonian of A2 system :
This means that the initial and final symmetries of the Hamiltonian (and the spin system) are different and Equation (12) cannot be used for the estimation of (i.e., the symmetry changes during the experiment). We do not introduce a calculation method for such situations.
However, notice that the symmetry of the Hamiltonian (24) changes by introducing a magnetic field, while molecular symmetry does not change. This means that molecular symmetry does not have to coincide with the spin symmetry (or the symmetry of the nuclear spin Hamiltonian). The latter is essential for SOT, but molecular symmetry is essential for spin isomers (discussed below). Thus, there are at least three relevant symmetries: The Hamiltonian, interactions, and the spatial configuration of the molecule.
3.1.5. pH2 on the Surface of a Solid
It was predicted that the spin order of pH2 after chemisorption, i.e., interaction with a surface, could be transformed into net magnetization even when the two spins have the same chemical shifts [32,33]. Note that there are a minimum of two requirements for PHIP via chemisorption: (a) The pH2 nascent protons have to be chemically nonequivalent, and (b) if they split, there is a non-zero chance to reunite again with preserved quantum coherences. The main reason for spin-order conversion is the difference in chemical shifts and the intramolecular dipole–dipole (DD) interaction, which is relevant on the surface. The Hamiltonian of such an AB system at a zero field is
As a result, the state of the dihydrogen after the chemisorption of pH2 is expected to be a superposition of and [33]:
where and are the relative weight of the states and . This result was predicted for two AB spins with the Hamiltonian (Equation (26)) by averaging the pH2-derived initial spin state, , over the hydrogenation period.
We showed before that it is impossible to transfer to the total net magnetization of two spins in an A2 system (Equation (22)). However, it is possible to transfer :
even with symmetry constraints. Note that (compare Equations (21) and (26)).
However, one should bear in mind that the mere feasibility of such a transfer computed using the presented approach does not take into account whether or not there are interactions in the system that can be used for the observation of the resulting spin order. Using solid echo sequences, SOT from to was predicted for chemisorbed pH2 [32,33].
3.2. Transfer of pH2 Spin Order to 1H Magnetization in Multispin Systems
3.2.1. No Symmetry Constraints
Now we will consider the transfer of either or spin orders into total spin magnetization, (Equation (8), in asymmetric N spin-½ systems (Equation (11)).
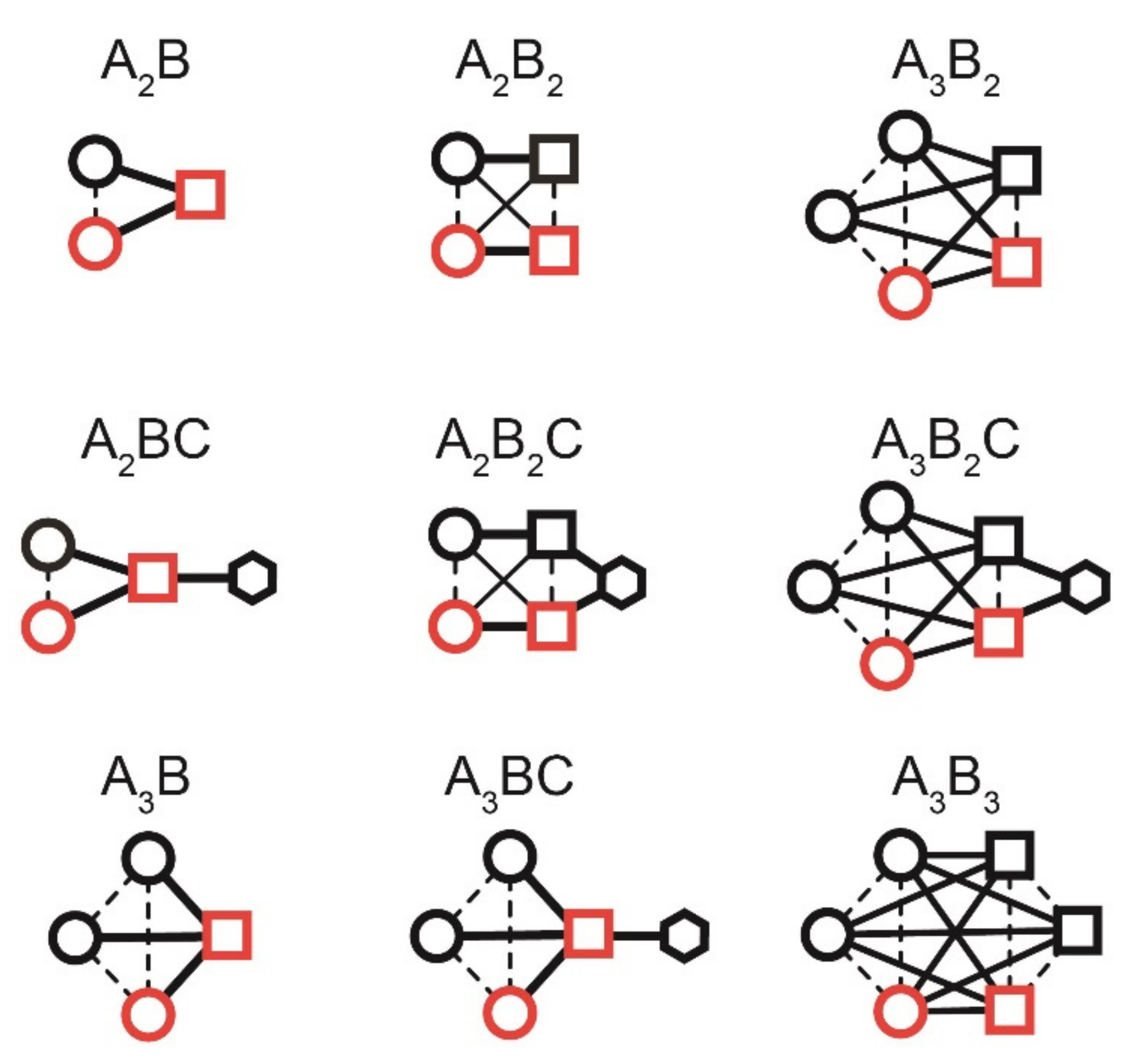
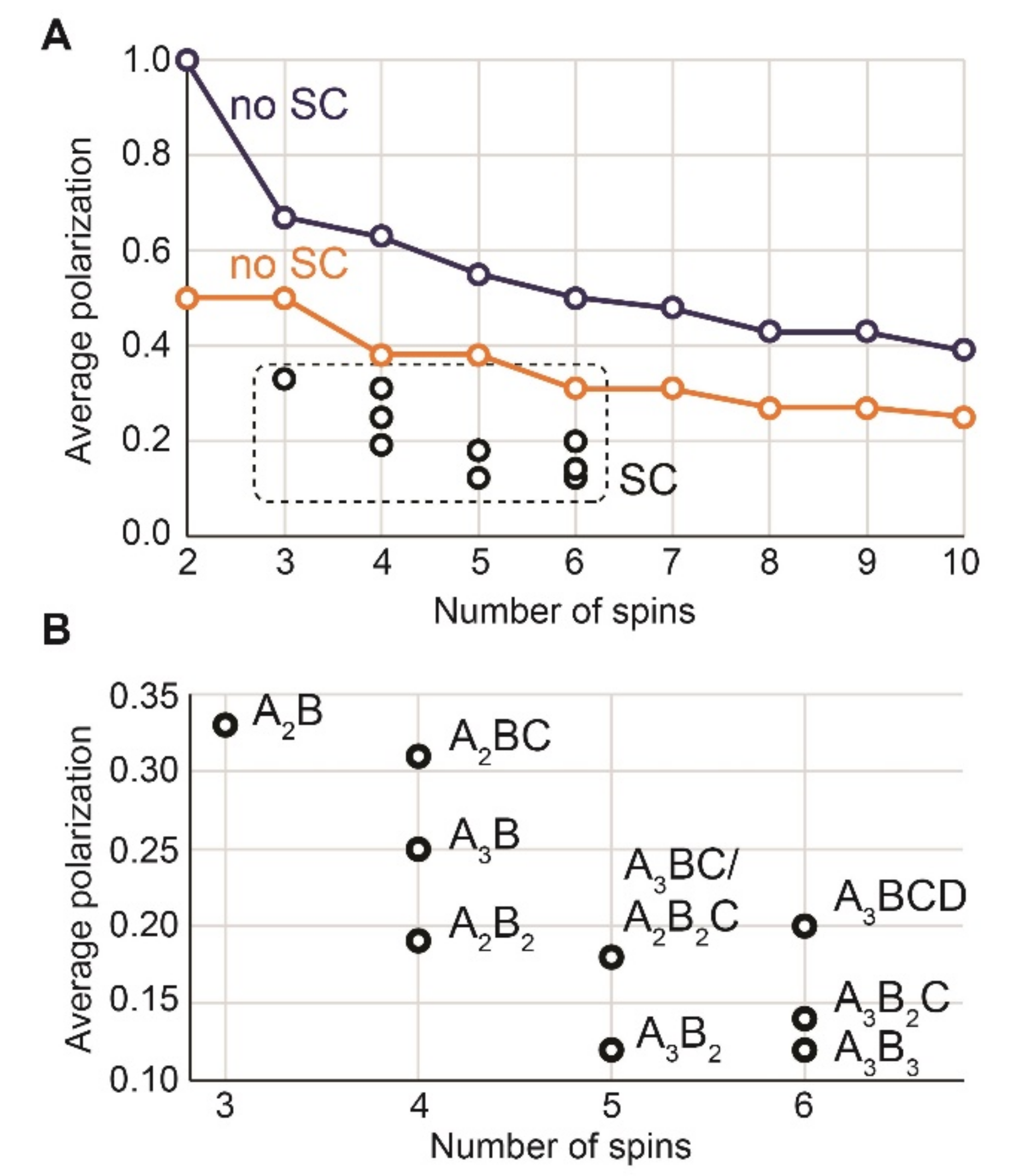
The highest level of polarization (i.e., the average polarization across all coupled spins) is possible if the system is in a pure singlet state rather than in (Figure 2, Figure 3 and Figure 4 and Appendix B, Table A6 and Table A7, and Appendix C for the corresponding analytical calculations for N = 2, 3, and 4).
This situation is achieved when the S-T states are a (stationary) eigenbasis, i.e., when the J-couplings dominate the interactions. This can be achieved by adding pH2 at low fields like in ALTADENA or via strong RF pulses suppressing chemical shift evolution at high fields [34]. Spin order transfer at the low field can be realized using SLIC pulses and was demonstrated for molecular systems with up to five nonequivalent spins [35].
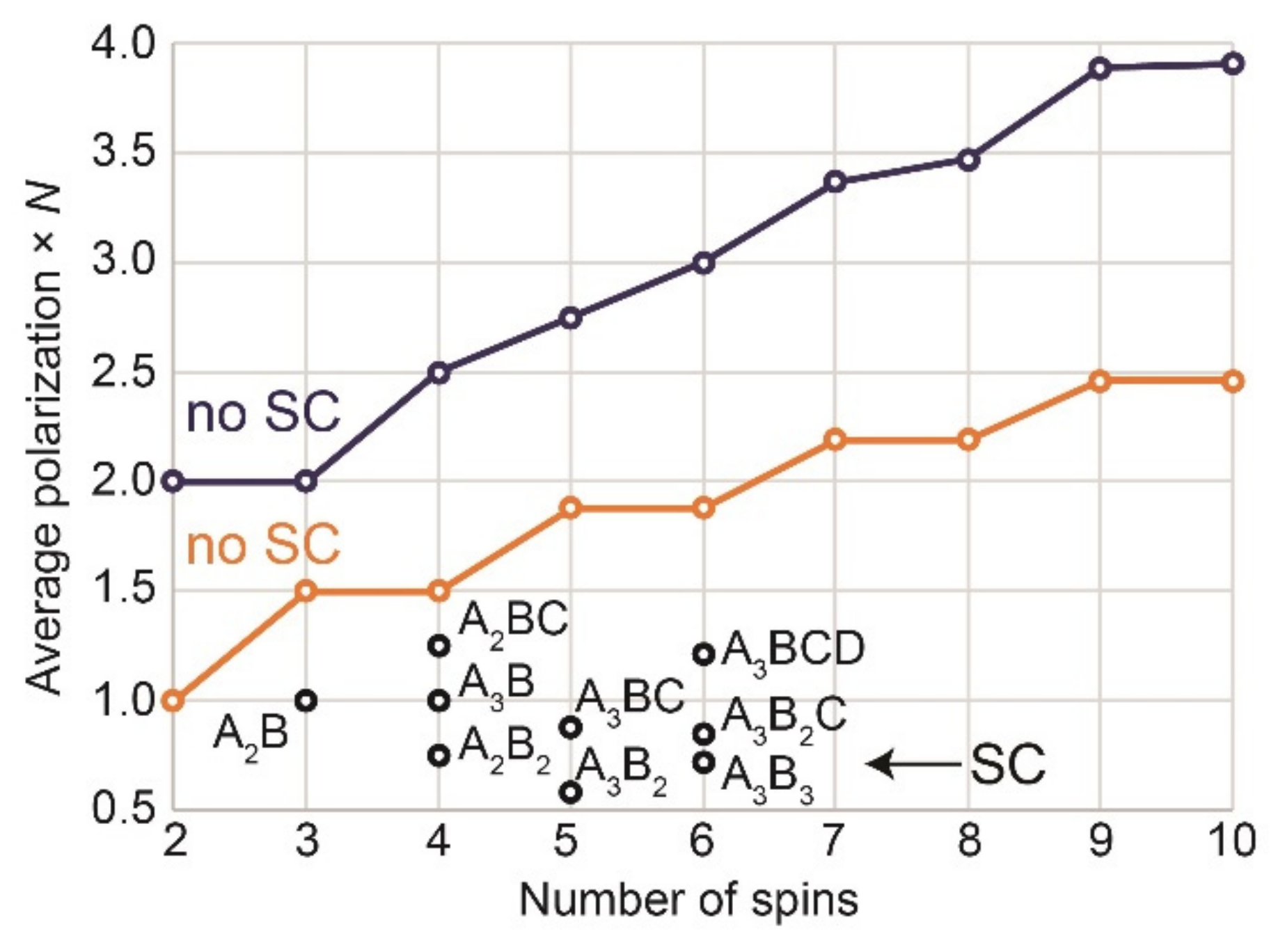
Interestingly, the maximum achievable polarization per molecule increases up to 4 (i.e., the equivalent of 4 spins polarized to 100%) if the system approaches 10 spins and the initial density matrix is (Figure 4); even higher polarizations are possible for a larger number of coupled spins (Appendix C).
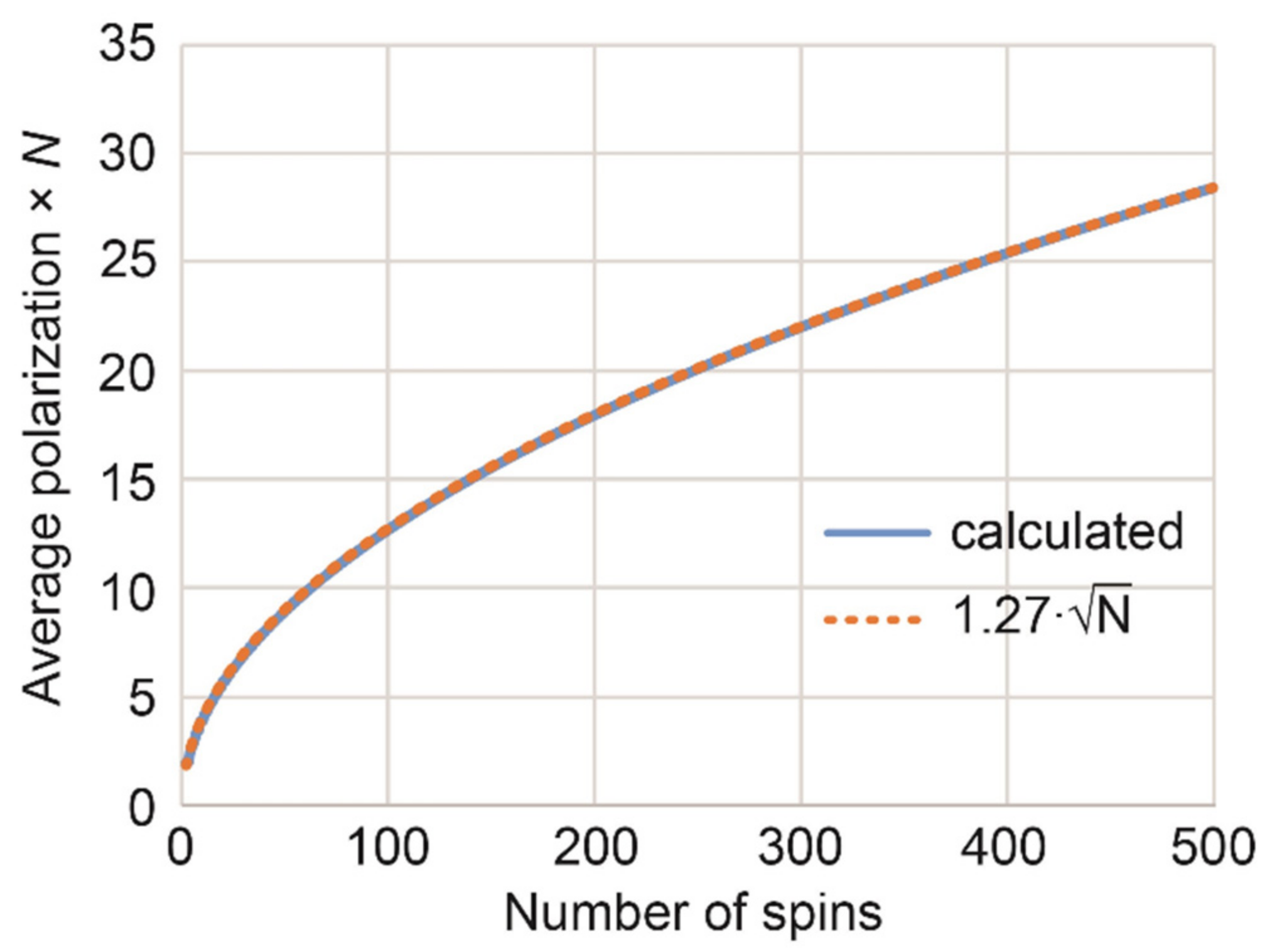
Indeed, in an N-spin system where two spins are polarized with parahydrogen, 1/4 of all states, , are equally populated. A unitary transformation can move that system into the state , which has two spins with = 1 (total polarization per molecule is then 2) and other spins are at equilibrium. Remarkably and nonintuitively, further unitary transformations can convert this into a state with higher angular momentum (or total polarization per molecule). Our calculations show that in the absence of symmetry constraints, the maximum of average spin polarization is approximately 1.27/; this corresponds to a total polarization per molecule of . For example, the sum of all polarizations in one molecule with N = 500 can reach ~29 (Appendix C).
We note that real systems always impose additional symmetry constraints, and calculated values of maximal polarization per molecule are significantly lower (see below).
3.2.2. With Symmetry Constraints
When a spin system topology has any permutation symmetries (Figure 2), the theoretically achievable proton polarization drops significantly. For example, in the A3B2C system of ethanol, the maximum average polarization is only 14.2% if pH2 was added pairwise in positions A and B. If the symmetry constraints are relaxed so that six spins are nonequivalent, the maximum achievable polarization increases to 31.2% for and 50% for , respectively (Figure 3).
Note that some similar systems were experimentally and theoretically studied in the related context of long-lived nuclear singlet spin states [36,37,38]. The process of spin order transfer discussed here is important for LLS because it gives the upper estimates for the maximum amplitude of the LLS conversion to magnetization.
3.2.3. Nuclear Spin Isomers of H2 and Ethylene
Dihydrogen. We discuss nuclear spin isomers of molecules (NSIM) in more detail, starting with H2. The molecular symmetry group of H2 is , while the permutation symmetry group of two spins is only C2. The symmetry includes an infinite number of symmetry elements and is a product of C2 and rotation groups, rotation–reflection, and groups. For the sake of simplicity, and to exemplify NSIM, let us consider the C2 symmetry only. The four nuclear spin states (sp) of H2 can be grouped in two sets A and B (Equation (20) and Appendix A): (3 states, oH2) and (1 state, pH2).
Ethylene. Ethylene is another example of a molecule with different NSIMs. Unlike H2, however, the molecular symmetry group of ethylene is D2h. Although the permutation symmetry group of spins is D2 [39], it is helpful to use molecular symmetry to obtain a connection to corresponding rotational symmetries.
The permutation D2 subgroup consists of one trivial and three nontrivial permutations that correspond to three orthogonal 180° rotations (Figure 5). In the D2 symmetry group (see Appendix A), the 16 spin states (sp) of ethylene are grouped in four sets that correspond to , , , and symmetries; seven states for the A-symmetry set and three states for each of the B-symmetry sets.
The D2h symmetry group includes additional inversion operation () and its combinations with the above-mentioned 180° rotations. In this case, the decomposition of 16 spin states also results in four groups with additional symmetry indices: (seven states), (three states), (three states), and (three states). Seven states of symmetry include five states with total spin 2 () and two states with total spin 0 ( and . Each B group corresponds to three spin states of the same symmetry with the total spin 1.
The parity of spin states is even, and so is the parity of these four groups of symmetry. The rotational wavefunctions of ethylene are all of g symmetry, which leaves only four rotational (rot) symmetries, , , , and .
The total (rotational and nuclear spin) wavefunction should have A-symmetry (either Ag or Au), therefore there are four allowed combinations for ethylene: , , , .
We discuss the ethylene case in detail because it gives very good insight into the problem of polarization transfer between states of different symmetries.
Upon NSIM interconversion, the associated energy changes are much larger than what is normally induced in NMR by RF pulses. An NSIM interconversion inevitably changes both rotational and spin states of ethylene. This is in strong contrast to non-symmetric molecules where the energy of spin flips is comparable to the strength of nuclear spin interactions. The large gap between H2 rotational energies helps to enrich the pH2 state at low temperatures and is responsible for its long lifetime [3,5].
Some of the states belonging to different NSIMs of polyatomic molecules may have similar energies (in contrast to H2 for which this is impossible). Such gateways are the basis of the quantum relaxation theory of NSIM interconversion [40]. For instance [41], for the (J = 23) and (J = 21) states of ethylene (where J is a rotational quantum number), the energy gap is “only” 46 MHz, i.e., within the reach of dipolar couplings. However, the energy of these states is 900 cm−1 (1300 K) above the ground state of ethylene; therefore, their thermal population is low and the NSIM interconversion is also relatively slow despite efficient mixing of these states by the dipole–dipole interaction.
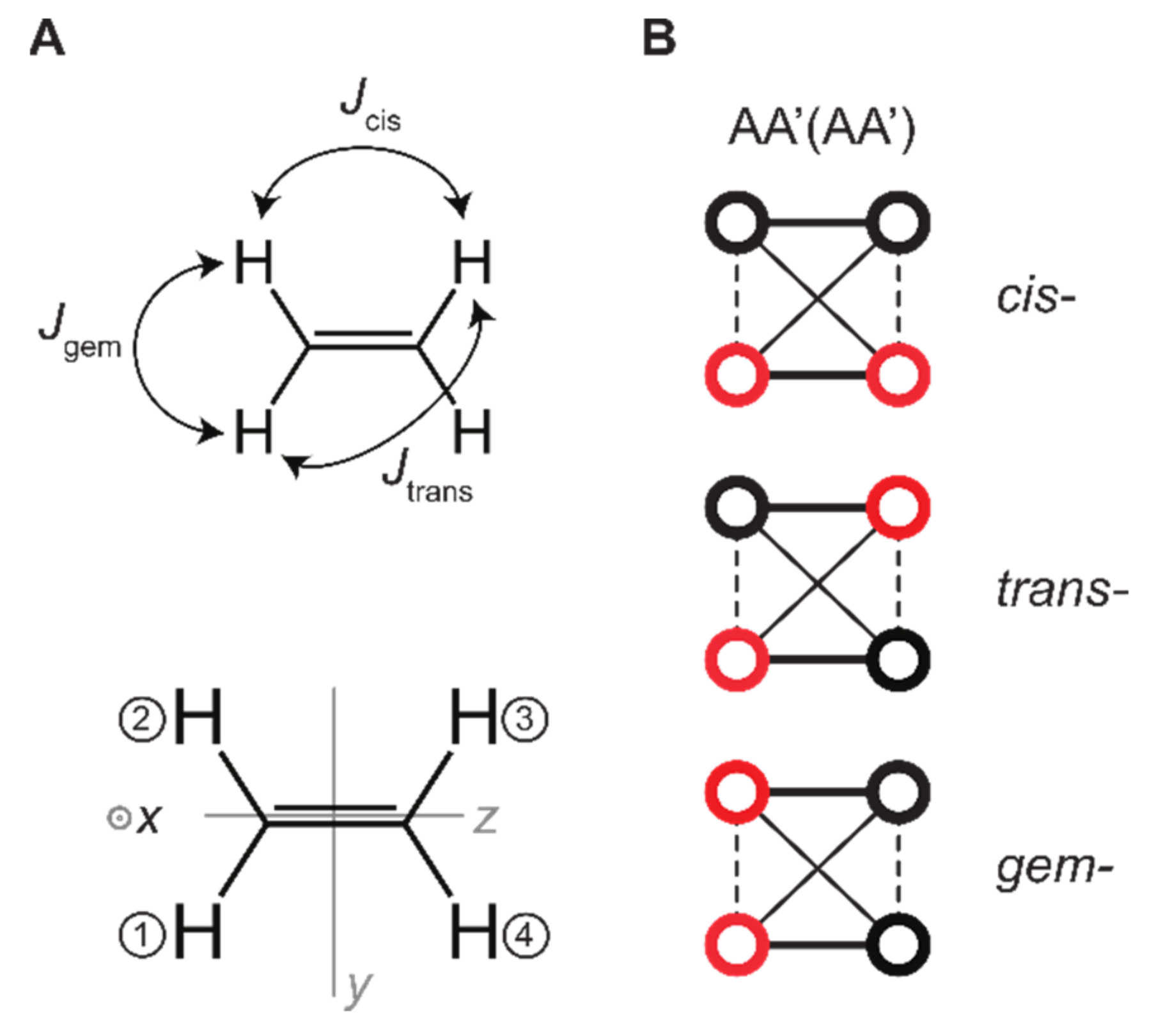
Unlike in H2 (with C2 symmetry), in ethylene, it seems possible to transfer singlet state and ZZ spin order into magnetization:
with Am and An being one of four protons. Using the ethylene J-coupling constants to set the basis of spin states (Appendix A and ref [7], = 1.07 Hz, = 11.47 Hz, and = 17.78 Hz [42]), the following values were obtained for different hydrogenation sites: and . According to Figure 5, here cis corresponds to protons 1 and 4 (or 2 and 3), trans to 1 and 3 (or 2 and 4), and gem to 1 and 2 (or 3 and 4). Note that using other simulations or isotope-labeling other (but similar) values were obtained and can be used [43]: = 2.23–2.39 Hz, = 11.62–11.66 Hz and = 18.99–19.03 Hz; [44]: = 2.5 Hz, = 11.6 Hz and = 19.1 Hz. However, because the constants were of the same order, we did not compare the spin order transfer for different J-coupling values.
It may come as a surprise that the efficient transfer of singlet spin order to polarization is feasible in a highly symmetric system like ethylene. So far, this transfer was demonstrated only in aligned media (nematic liquid crystals), where dipole–dipole spin–spin interactions remain, and the Hamiltonian of the system resembles (Equation (25)). For such a Hamiltonian as discussed above, instead of pure , a mixture of and should be considered and spin order is observable in NMR.
Normally, the transition between two states with spin 0 that are represented by and symmetries cannot be observed by NMR. However, the theory applied here still allows the transfer of spin order to polarization. Let us take a closer look and determine why this is possible.
After hydrogenation with pH2, six states with B-symmetry and two singlets of symmetry is populated under each hydrogenation scenario (Table 1). The average polarization that we considered in Equation (28) depends on the populations of all symmetry states and is not straightforward to analyze. However, let us instead consider the state with the maximum for the system value of spin and spin projection of 2; one of five states of symmetry. It means that if there is a way to transfer polarization from one of two spin states () to the state (both have the same symmetry, ethylene hyperpolarization will be revealed. Now the question of how to achieve this remains, and if any existing spin order transfer methods, e.g., spin-lock-induced crossing (SLIC) [45], adiabatic passage spin order conversion (APSOC) [29], or magnetic field cycling (MFC) [46], are suitable. This analysis goes beyond the scope of this paper and will be considered elsewhere.
3.3. PHIP-SAH and the Transfer of pH2 Spin Order to the Magnetization of X-Nuclei
3.3.1. PHIP-SAH
Polarization transfer from pH2 to X-nuclei also attracts significant attention in the context of hyperpolarized MRI applications. The lack of background signal and extended lifetime of polarization (compared to 1H) makes hyperpolarized MRI of X-nuclei highly interesting for biomedical applications, spearheaded by hyperpolarized MRI of xenon and 13C-pyruvate [19,47,48,49,50].
3.3.2. No Symmetry Constraints
We considered the transfer of and to X-nuclear polarization () without symmetry constraints (Table 2). In both cases, 100% polarization can be achieved: . This, for example, was predicted for the hydrogenation of perdeuterated 1-13C-vinyl-acetate-d6 [12]. More than 50% polarization was achieved on 13C in the system consisting of three nonequivalent spin-½ and six spin-1 nuclei (2H). The direct loss of polarization is due to S-T0 mixing of pH2–derived hydrogens at the catalyst and relaxation during hydrogenation [55].
3.3.3. Symmetry Constraints
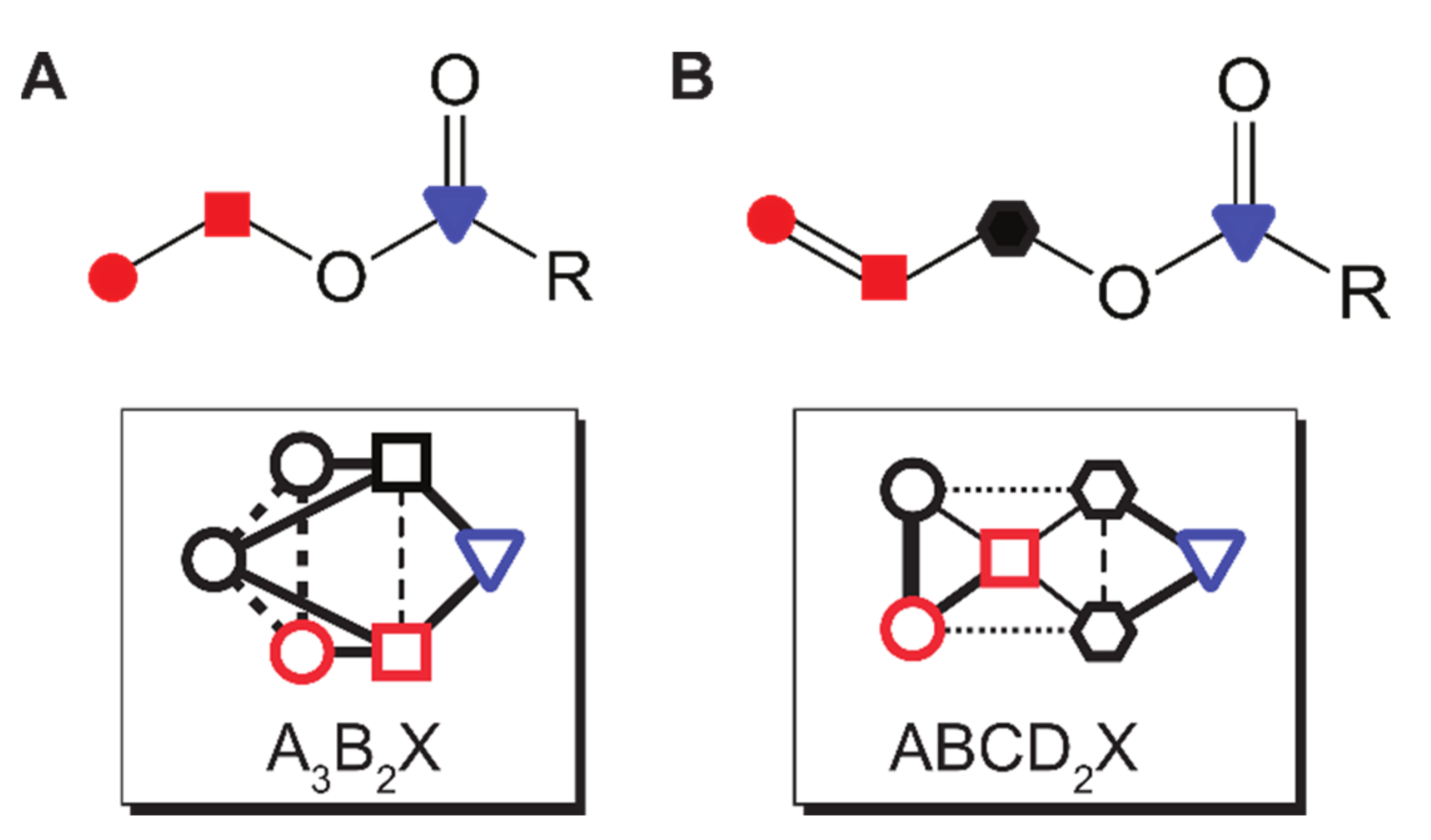
If some other (i.e., non-nascent pH2) protons possess any symmetry, 100% polarization transfer can still be achieved on X (theoretically). This situation is realized e.g., in 1-13C-allyl-pyruvate, an ABCD2X system.
If one of the pH2-nascent protons ends up in a symmetric site of the product, the maximum polarization that can be transferred to X is reduced to 75% for A2BX (3/4) and 66.(6)% for A3BX (2/3). Again, the number of the “other” protons and their symmetry does not play a role.
If both pH2 spins bind to two different symmetric spin sites, such as A3B2X, A3B2C2X, the maximum polarization that can be transferred to X is further reduced to 50% (). This situation is found, for example, in ethyl- and propyl pyruvate.
In the literature, 13C-polarization about 20% was reported on ethyl pyruvate. Here, pH2 was added to vinyl pyruvate at a high field and spin order was transferred to 13C using INEPT [56]. About 20–35% 13C-polarization was reported in a similar experiment, where pH2 was added at a low field and the detection took place at the high field after magnetic field variation [46,57]. However, one should remember that, as with the ALTADENA discussed above, the Hamiltonian of the system changes during the magnetic field variation.
The other interesting case, however exotic for PHIP, is the transfer of pH2 spin order in an A3X system if two of the A spins are in the singlet state ( for ). If they are in the “reduced” singlet state , however, the transfer is possible (1/3 for ). For example, as we discussed before, pH2-derived spin order after chemisorption is partially in the ZZ state.
3.3.4. Double Hydrogenation
Now we turn to the following question: Is it beneficial to add two pH2 molecules to one target? For example, 1-13C-ethynyl pyruvate is transformed into 1-13C-ethyl pyruvate, an A3B2X system, by double hydrogenation (addition of two pH2 molecules). Likewise, 1-13C-propargyl pyruvate becomes 1-13C propyl pyruvate, an A3B2C2X system, upon the addition of two pH2 (pH2 is added to A and B in both cases).
Let us assume that the hydrogenation reaction is so fast that only the spin state is populated, which can be written as
The theoretically maximum transfer from to X-nuclear polarization in A3B2X and A3B2C2X systems is , which is 2 times lower than for the single hydrogenation. Note that it is also system-specific, e.g., for the A2B2X system double hydrogenation results in , while 9/16 is the maximum predicted for single hydrogenation (Table 2).
In reality, however, there will be a finite time between the first and second hydrogenation, such that the system will start to evolve. As a result, the final state will be different than and the polarization of X nucleus will also be different.
The situation is similar for multiple hydrogenations in different positions, for example, in trivinyl orthoacetate [58]. If we simplify the product to two ethyl groups, the system becomes (A3B2)(A3B2)’X, where one pH2 is part of the A3B2 subsystem and the other is part of (A3B2)’ subsystem. In this case, polarization transfer amplitude was predicted, while for single hydrogenation (yielding an A3B2X system), it was .
We did not find instantaneous double hydrogenation beneficial for spin order transfer to X-nuclei, which may be different for slow and stepwise double hydrogenation.
3.4. Examples of Isotopic and Chemical Symmetry Breaking
3.4.1. Ethylene
Ethylene produced by adding pH2 to acetylene in an isotropic environment does not demonstrate any enhanced observable magnetization. The spin state of pH2 should be converted into the ZZ state instead: This was demonstrated after the hydrogenation and subsequent dissolution of ethylene in a liquid crystal [7]. However, one can imagine doing it in a different order. First, generate the ZZ-state of pH2-derived H2, which was estimated for a solid catalyst [32,33]. Second, hydrogenate acetylene using this H2.
In general, the pH2-derived protons are observed only when they are attached to chemically or magnetically inequivalent sites that for ethylene can be achieved in at least two ways:
- The two pairs of hydrogens in ethylene can be made magnetically nonequivalent by 13C labeling. In the case of a single-sided 13C labeling system, symmetry drops down to C2 and polarization transfer is possible to 1H or 13C nuclei. In addition, the chemical shifts of two gem pairs of protons are different.
- The other way to break the ethylene symmetry is a chemical reaction. So, polarized ethylene gas bubbled through a CCl4 solution of perfluoro(para-tolylsulfenyl) chloride (PTSC) yields an asymmetric PTSC/ethylene adduct [7]. As a result, a normal PASADENA spectrum can be obtained.
3.4.2. Fumarate and Maleate
Fumarate and maleate are two metabolites with symmetry-imposed spin order transfer restrictions; the solutions are also the same. The symmetry can be broken by 13C labeling [59,60] or as a result of a chemical reaction: “hyperpolarized” fumarate was converted by fumarase to asymmetric malate revealing itself in the PASADENA spectrum [61].
4. Discussion
We considered several cases of polarization transfer from pH2 to proton and X nuclei magnetization using the methods introduced in refs. [17,24,25] (see Supplementary Materials). The approach used here helps to provide some general answers to several nonintuitive questions. However, a few situations remain unclear and may indicate some limitations of the presented theory. Namely:
Q1. How do we estimate maximum polarization transfer from a state that is not diagonal in the basis of the system’s symmetry?
Q2. How do we estimate polarization transfer in systems that experience symmetry change during the polarization transfer, e.g., A2AB during magnetic field variation in the ALTADENA experiment? Is there a general solution for an N-spin system?
Discussion of Q1. This situation corresponds to the third case of -diagonalization (Equation (16)) as described in methods. For example, in an A2BX system, the basis consists of the functions where is one of S-T basis functions and , are spin up and down, and . On this basis, is not diagonal. Instead, projecting this state on the symmetry basis results in , meaning that we lose part of the initial spin order and potentially underestimate the level of polarization transfer.
Discussion of Q2. This problem was discussed in the context of ALTADENA, but it is also very important for magnetic field variation e.g., in PHIP-SAH. Let us consider a simple ABX system. At low fields, when the proton chemical shift difference can be neglected, the ABX system becomes equivalent to an A2X system meaning that for protons, an S-T basis is more appropriate at low fields. is the initial state of the system after pH2 addition (). Then, we increase the field slowly so that the system changes from A2X to ABX and the basis changes from S-T to Zeeman. This means that the symmetry basis of the system before and after (and during) the transformation is different. The theory presented here cannot be applied.
Although in three-spin systems, we can still reach 100% X nuclear polarization, we again could underestimate the efficiency of polarization transfer in more complex systems.
It seems as though the methodology used here for the static high magnetic field can be translated to low fields (and zero fields). However, the basis will be system-symmetry-specific and, in addition, will depend on the J-coupling network.
Assessing the validity of this approach is not straightforward. To date, however, experimental results have not contradicted the calculated results presented here.
5. Conclusions
The mathematical framework presented here allows for determining an upper limit for the polarization transfer from pH2 to X-nuclei or other protons with an emphasis on the effect of molecular spin symmetry. Solutions were presented for the most current experimental situations, although more complex cases remain unaddressed. This method may serve as a first check to estimate if and how much polarization transfer is possible in a given situation. Naturally, identifying and optimizing a dedicated transfer strategy is the next essential step, which is not addressed here.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/sym14030530/s1. All used Matlab scripts together with MOIN spin library [62] (.zip) are available online.
Author Contributions
A.N.P., conceptualization and software; D.A.B. and J.-B.H. visualization; A.N.P. and D.A.B., original draft; A.N.P., D.A.B., and I.V.K., investigation. All authors, review and editing and funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge funding from the German Federal Ministry of Education and Research (BMBF) within the framework of the e:Med research and funding concept (01ZX1915C), DFG (PR 1868/3-1, HO-4602/2-2, HO-4602/3, GRK2154-2019, EXC2167, FOR5042, SFB1479, TRR287), Kiel University and the Faculty of Medicine. MOIN CC was founded by a grant from the European Regional Development Fund (ERDF) and the Zukunftsprogramm Wirtschaft of Schleswig-Holstein (Project no. 122-09-053). DAB acknowledges support from the Alexander von Humboldt Foundation in the framework of the Sofja Kovalevskaja Award. I.V.K. acknowledges the Russian Ministry of Science and Higher Education (grant no. 075-15-2020-779) for financial support.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data and software obtained and used are available with the paper on mdpi.com.
Acknowledgments
We are grateful to Dmitry Budker for the discussion and editing of the manuscript. We are particularly thankful for the insightful comments from Warren S. Warren regarding large polarization obtained in multi-spin systems without symmetry constraints.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
Appendix A.1. A2 Two Spin-½ System, C2 Group
The number of states is 22 = 4.
The basis for two equivalent spins can be divided into two groups with a total spin of 1 (three states) and 0 (one state), also known as the singlet–triplet (S-T) basis. This can be derived formally by finding eigenfunctions and eigenvalues () of the cyclic permutation operator . The C2 permutation group of the A2 system consists of two permutations, {(),(21)}. In the matrix form, it is written in the Zeeman basis (, , , ):
Eigenvalues of are given in superscript to the corresponding wavefunctions:
We indicate spin states by the total spin and its projection as and/or by using the Zeeman basis, and .

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table A1.
Table of characters for A2 two spin-½ system (C2 group).
| E | C2 | |
|---|---|---|
| A | 1 | 1 |
| B | 1 | −1 |
| SpinRep = 3A + B | 4 | 2 |
Therefore, there are three symmetric states and one asymmetric state with respect to permutation (or rotation about 180 degrees, C2). It follows from both Equation (A1) and Table A1. Therefore, the basis for A2 two spin-½ system consists of two sets (S) with multiplicity of 3 and 1 (SpinRep = 3A + B):
To calculate characters for spin permutations (SpinRep) in Table A1, one can (i) write a matrix of permutation and (ii) calculate the trace. For an identity transformation, () = E, and for N spin-½, character is
Analogously (Equation (A1)) .
For any character of a representation which is a superposition of irreducible representations of the same group, the multiplicity of the irreducible representation is given by
Here “*” is a complex conjugate, and is a character of an irreducible representation . Equation (A5) is useful to decompose the SpinRep line into a sum of characters for irreducible representations. So, for the A2 system SpinRep = 3A + B.
Appendix A.2. A3 Three Spin-½ System, C3 Group
The number of states is 23 = 8.
The basis of three equivalent spins can be grouped into three groups with a total spin of 3/2 (four states), 1/2 (two states), and 1/2 (two states). Here, to distinguish groups, we also introduce eigenvalues for the cycling permutation operator , and its values are given as superscript to the corresponding wavefunctions.
The C3 permutation group G = {,, } of the A3 system consists of three permutations: The trivial identity permutation, permutation, or “+” rotation and rotation, with .
Table A2.
Table of characters for A3 three spin-½ system (C3 group). Here . Note the difference between three different “E” here. The characters for spin representations (SpinRep) are filled using Equation (A4) and the following discussions. Any permutation (or rotation) will leave only states and on the diagonal. This means that the sum of diagonal elements and corresponding character value is 2.
Table A2.
Table of characters for A3 three spin-½ system (C3 group). Here . Note the difference between three different “E” here. The characters for spin representations (SpinRep) are filled using Equation (A4) and the following discussions. Any permutation (or rotation) will leave only states and on the diagonal. This means that the sum of diagonal elements and corresponding character value is 2.
| E | |||
|---|---|---|---|
| A | 1 | 1 | 1 |
| E1 | 1 | ||
| E2 | 1 | ||
| SpinRep = 4A + 2E1 + 2E2 | 8 | 2 | 2 |
Summarizing, there are four symmetric (g) states and two pairs of rotationally symmetric states (Ei, ) states. It follows from both Equation (A6) and Table A2. Therefore, the basis for the A3 three spin-½ system consists of three sets with multiplicities 4, 2, and 2 (SpinRep = 4A + 2E1 + 2E2):
Appendix A.3. A4 Four Spin-½ System: C4 Group Example
The number of states is 24 = 16.
The basis of four spins can be grouped into four groups with total spin of 2 (five states), 1 (three groups, each consists of three states), and 0 (two groups, each with one state).
The C4 permutation group G = {, , , } of the A4 system consists of four permutations: Trivial identity permutation and three cyclic permutations that are equivalent to the rotation of a square by 90°, 180°, and 270° around the center axis perpendicular to its plane. Note that only 180° rotation can be represented as two consequent permutations .
Table A3.
Table of characters for A4 four spin-½ system (C4 group). The characters for SpinRep are filled using Equation (A4) and the following discussion. Any rotations will leave states and on diagonal. All other states change after an odd number of cyclic permutations. Hence, character for and is only 2. 180° rotation ( permutation) also does not change and states. Hence, the corresponding character is 2 + 2 = 4. This means that the sum of diagonal elements and corresponding character values are 4 for each rotation (permutation).
Table A3.
Table of characters for A4 four spin-½ system (C4 group). The characters for SpinRep are filled using Equation (A4) and the following discussion. Any rotations will leave states and on diagonal. All other states change after an odd number of cyclic permutations. Hence, character for and is only 2. 180° rotation ( permutation) also does not change and states. Hence, the corresponding character is 2 + 2 = 4. This means that the sum of diagonal elements and corresponding character values are 4 for each rotation (permutation).
| E | C4 | C2 = (C4)2 | (C4)3 | |
|---|---|---|---|---|
| A | +1 | +1 | +1 | +1 |
| B | +1 | −1 | +1 | −1 |
| E1 | +1 | +i | −1 | −i |
| E2 | +1 | −i | −1 | +i |
| SpinRep = 6A + 4B+3E1+3E2 | 16 | 2 | 4 | 2 |
Appendix A.4. AA’(AA’) Four Spin-½ System: D2 Group (Spin Symmetry of Ethylene)
The number of states is 24 = 16.
The basis of four spins can be grouped into four groups with total spin of 2 (five states), 1 (three groups, each consists of three states), and 0 (two groups, each with one state).
The D2 permutation group G = {,, , } of the AA’(AA’) system consists of four permutations: – Trivial identity permutation and three pairwise permutations that are equivalent to rotations of the rectangle by 180° around three orthogonal axes, which are orthogonal to the plane of the rectangle and (or) its edges. We do not present the corresponding basis for a general D2 group here, which is equivalent to D2h discussed below.
Table A4.
Table of characters for AA’(AA’) four spin-1/2 system (D2 group). The characters for SpinRep are filled using Equation A4 and the following discussion. Any rotations will leave states and on diagonal. In addition, states and do not change by the action of (21)(43) permutation. For two other rotations, one can also write the corresponding two states. It means that the sum of diagonal elements and corresponding character values are 4 for each rotation (permutation).
Table A4.
Table of characters for AA’(AA’) four spin-1/2 system (D2 group). The characters for SpinRep are filled using Equation A4 and the following discussion. Any rotations will leave states and on diagonal. In addition, states and do not change by the action of (21)(43) permutation. For two other rotations, one can also write the corresponding two states. It means that the sum of diagonal elements and corresponding character values are 4 for each rotation (permutation).
| E | C2(z) | C2(y) | C2(x) | |
|---|---|---|---|---|
| A | +1 | +1 | +1 | +1 |
| B1 | +1 | + | −1 | −1 |
| B2 | +1 | −1 | +1 | −1 |
| B3 | +1 | −1 | −1 | +1 |
| SpinRep = 7A + 3B1 + 3B2 + 3B3 | 16 | 4 | 4 | 4 |
Appendix A.5. AA’ (AA’) Four Spin-½ System, D2h Group (Molecular Symmetry of Ethylene)
The number of states is 24 = 16 [7].
The basis of four spins can be grouped into four groups with total spin of 2 (five states), 1 (three groups, each consists of three states), and 0 (two groups, each with one state).
The D2h group is a direct product of D2 and Ci groups. The D2 part consists of four spin permutations G(D2) = . The addition of the inversion operator of Ci results in four additional transformations : Inversion “I” and three mirror -planes: xy, xz, or yz (Figure A1 and Figure 4).
Figure A1.
The action of a mirror plane on an axial vector (spin or magnetic dipole). When an axial vector is perpendicular to the mirror plane, it does not change upon reflection. If, however, it is oriented along the mirror plane, it changes its orientation upon reflection [63].
Figure A1.
The action of a mirror plane on an axial vector (spin or magnetic dipole). When an axial vector is perpendicular to the mirror plane, it does not change upon reflection. If, however, it is oriented along the mirror plane, it changes its orientation upon reflection [63].

The eigenvectors for ethylene were found in [7] and are given in Equation (A8) without any changes.
It is not as trivial as before to fill the SpinRep line in the character Table A5 as in the previous cases; this needs some elaboration. However, the first four elements are identical to the one from Table A4 for group D2.
Let us consider the mirror plane (Figure 1A), which also can be referred to as . It does not change the position of atoms (a), and because spin is an axial vector (b) the spin states do not change under the action of . Hence, the corresponding character is 16 (number of spin states). We refer to this operator as parity in the main text, and it changes the sign of one coordinate axis (here x).
Now, let us consider two mirror planes and (Figure 1A), also referred to as . exchanges two neighbor protons (permutations (21)(43) or (41)(32)) and changes the sign of the spin projection (it is not convenient for NMR, but using this notation of the axis, we assume the projections of the spin states along the x-axis). So, when two pairs of protons are exchanged and their sign is inverted, then there are only four states that do not change under the action of this transformation: , , , and for the case of (21)(43) permutation with inversion. Four analogous states can be written for the other mirror transformation. Hence, the two corresponding characters are 4.
Finally, the inversion operator, , exchanges the protons as (31)(42) (a) and changes the sign of the spin projections, meaning that only four states , , , and will stay the same; hence, the corresponding character is 4.
Table A5.
Table of characters for AA’ (AA’) four spin-1/2 system (D2h group). This can be obtained as a direct product of Ci (the same as C2) and D2 character groups. See text for how to fill SpinRep line.
Table A5.
Table of characters for AA’ (AA’) four spin-1/2 system (D2h group). This can be obtained as a direct product of Ci (the same as C2) and D2 character groups. See text for how to fill SpinRep line.
| E | i | |||||||
|---|---|---|---|---|---|---|---|---|
| Ag | +1 | +1 | +1 | +1 | +1 | +1 | +1 | +1 |
| B1g | +1 | −1 | −1 | +1 | +1 | −1 | −1 | |
| B2g | +1 | −1 | +1 | −1 | +1 | −1 | +1 | −1 |
| B3g | +1 | −1 | −1 | +1 | +1 | −1 | −1 | +1 |
| Au | +1 | +1 | +1 | +1 | −1 | −1 | −1 | −1 |
| B1u | +1 | +1 | −1 | −1 | −1 | −1 | +1 | +1 |
| B2u | +1 | −1 | +1 | −1 | −1 | +1 | −1 | +1 |
| B3u | +1 | −1 | −1 | +1 | −1 | +1 | +1 | −1 |
| SpinRep = 7Ag + 3B1u + 3B1u + 3B3g | 16 | 4 | 4 | 4 | 4 | 4 | 4 | 16 |
There are seven symmetrical (Ag symmetry) states and three states for each of the three B symmetries (B1u, B2u, and B3g). This follows from both Equation (A8) and Table A5. Therefore, the basis for the AA’(AA’) four spin-1/2 system of D2h symmetry consists of four sets with multiplicities of 7, 3, 3, and 3 (Spins rep. = 7Ag + 3B1u + 3E2g + 3E3u):
Note that in the case of C4 symmetry considered earlier, six states belong to the A(g) group.
Appendix B
Table A6.
Maximum expected polarization transfer coefficient for systems without symmetry constraints and pH2-derived spin order transfer (states or ) to the longitudinal polarization of all protons of the same molecule (average polarization).
Table A6.
Maximum expected polarization transfer coefficient for systems without symmetry constraints and pH2-derived spin order transfer (states or ) to the longitudinal polarization of all protons of the same molecule (average polarization).
| Type of the System | Number of Spins | ||
|---|---|---|---|
| AB | 2 | ½ | 1 |
| ABC | 3 | ½ | 2/3 |
| ABCD | 4 | 3/8 | 5/8 |
| ABCDE | 5 | 3/8 | 11/20 |
| ABCDEF | 6 | 5/16 | ½ |
| ABCDEFG | 7 | 5/16 | 0.4821 |
| ABCDEFGH | 8 | 0.2734 | 0.4336 |
| ABCDEFGHI | 9 | 0.2734 | 0.4323 |
| ABCDEFGHIJ | 10 | 0.2461 | 0.3906 |
| ABCDEFGHIJK | 11 | 0.2461 | 0.3835 |
| ABCDEFGHIJKL | 12 | 0.2256 | 0.3597 |
Table A7.
Maximum expected polarization transfer coefficient for systems with symmetry constraints and pH2-derived spin order transfer (state ) to the longitudinal polarization of all protons of the same molecule (average polarization).
Table A7.
Maximum expected polarization transfer coefficient for systems with symmetry constraints and pH2-derived spin order transfer (state ) to the longitudinal polarization of all protons of the same molecule (average polarization).
| Type of the System | Number of Spins | |
|---|---|---|
| A2B | 3 | 1/3 |
| A2BC | 4 | 0.3125 |
| A3B | 4 | 0.25 |
| A2B2 | 4 | 0.1875 |
| A3BC | 5 | 0.175 |
| A2B2C | 5 | 0.175 |
| A3B2 | 5 | 0.1167 |
| A3BCD | 6 | 0.2014 |
| A3B2C | 6 | 0.1424 |
| A3B3 | 6 | 0.1204 |
Appendix C
Here, we exemplify that more than 200% polarization per molecule is predicted in the absence of symmetry constraints for . We also investigate analytically asymptotic behavior for .
First, we will write down the eigenvalues of the target state (Equation (8)) for a different number of spins. Because the Zeeman basis is the eigenbasis of , the eigenvalues of are:
where is the binomial coefficient and is an integer number spanning from 0 to .
Eigenvalues for N = 2, 3, and 4 are:
Second, we write down eigenvalues for the initial singlet spin order (Equation (1)):
This operator is diagonal on the basis where the (S-T)-basis is used for A and B spins and the Zeeman basis is used for the rest of (N-2) spins, respectively. The general rule is as follows:
This follows directly from the definition of (Equation (1)) where one has times value and times value 0 on the diagonal; this corresponds to the singlet state of A and B spins. The corresponding eigenvalues for N = 2, 3, and 4 are as follows:
Now, using Equation (11) and eigenvalues of the target state (A13) and initial state (A16), one obtains the amplitude of the polarization transfer :
It follows that average polarization decreases as the number of spins increases, but polarization per molecule increases and can exceed two units of 100% spin-½ polarization. This is illustrated in Figure 3 and Figure 4.
For arbitrary , one can use the known sums of binomial coefficients to find that
and
where is the sum of last elements in . Note that is a quarter of all elements, and this will be important in the following evaluation. Then,
The explicit formula for calculating S is cumbersome:
with defined such that the sum over binomial coefficients from 0 to does not exceed a quarter of all states (elements): . To the best of our knowledge, this expression does not have a simple solution; however, it can be simply calculated numerically. In the following, we derive an asymptotic solution for when .
For the large N, the summation over binomial coefficients can be replaced by the integration over a normal distribution with a mean value of and variance (note that we use for variance instead of conventional to avoid confusion with spin operators):
Then, the value of S (Equations (A19) and (A21)) can be estimated as
where the value with . This was found by solving an equation for the integral over the normal distribution equal to a quarter: . This is equivalent to a summation over a quarter of all elements in Equation (A21). Note that . Thus, for large N values, Equation (A19) is approximated as
Now we can find the maximum expected average polarization per spin and per molecule as
This approximation (Equation (A23)) fits well the numerical simulations using Equations (A18) and (A19) and values for demonstrated in the main manuscript (Figure 4).
Figure A2.
Average polarization per molecule—in units of one-spin-1/2 polarization—that can be achieved theoretically by adding pH2 to a precursor producing a molecule with N spins without symmetry constraints. Numerically calculated values fit well to the approximation .
Figure A2.
Average polarization per molecule—in units of one-spin-1/2 polarization—that can be achieved theoretically by adding pH2 to a precursor producing a molecule with N spins without symmetry constraints. Numerically calculated values fit well to the approximation .

References
- Natterer, J.; Bargon, J. Parahydrogen Induced Polarization. Prog. Nucl. Magn. Reson. Spectrosc. 1997, 31, 293–315. [Google Scholar] [CrossRef]
- Green, R.A.; Adams, R.W.; Duckett, S.B.; Mewis, R.E.; Williamson, D.C.; Green, G.G.R. The Theory and Practice of Hyperpolarization in Magnetic Resonance Using Parahydrogen. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 67, 1–48. [Google Scholar] [CrossRef]
- Ellermann, F.; Pravdivtsev, A.; Hövener, J.-B. Open-Source, Partially 3D-Printed, High-Pressure (50-Bar) Liquid-Nitrogen-Cooled Parahydrogen Generator. Magn. Reson. 2021, 2, 49–62. [Google Scholar] [CrossRef]
- Hövener, J.-B.; Bär, S.; Leupold, J.; Jenne, K.; Leibfritz, D.; Hennig, J.; Duckett, S.B.; von Elverfeldt, D. A Continuous-Flow, High-Throughput, High-Pressure Parahydrogen Converter for Hyperpolarization in a Clinical Setting. NMR Biomed. 2013, 26, 124–131. [Google Scholar] [CrossRef]
- Feng, B.; Coffey, A.M.; Colon, R.D.; Chekmenev, E.Y.; Waddell, K.W. A Pulsed Injection Parahydrogen Generator and Techniques for Quantifying Enrichment. J. Magn. Reson. 2012, 214, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Knopp, G.; Kirch, K.; Beaud, P.; Mishima, K.; Spitzer, H.; Radi, P.; Tulej, M.; Gerber, T. Determination of the Ortho-/Para Deuterium Concentration Ratio with Femtosecond CARS. J. Raman Spectrosc. 2003, 34, 989–993. [Google Scholar] [CrossRef]
- Zhivonitko, V.V.; Kovtunov, K.V.; Chapovsky, P.L.; Koptyug, I.V. Nuclear Spin Isomers of Ethylene: Enrichment by Chemical Synthesis and Application for NMR Signal Enhancement. Angew. Chem. Int. Ed. 2013, 52, 13251–13255. [Google Scholar] [CrossRef]
- Kilaj, A.; Gao, H.; Rösch, D.; Rivero, U.; Küpper, J.; Willitsch, S. Observation of Different Reactivities of Para and Ortho- Water towards Trapped Diazenylium Ions. Nat. Commun. 2018, 9, 2096. [Google Scholar] [CrossRef]
- Bowers, C.R.; Weitekamp, D.P. Parahydrogen and Synthesis Allow Dramatically Enhanced Nuclear Alignment. J. Am. Chem. Soc. 1987, 109, 5541–5542. [Google Scholar] [CrossRef] [Green Version]
- Pravica, M.G.; Weitekamp, D.P. Net NMR Alignment by Adiabatic Transport of Parahydrogen Addition Products to High Magnetic Field. Chem. Phys. Lett. 1988, 145, 255–258. [Google Scholar] [CrossRef]
- Adams, R.W.; Aguilar, J.A.; Atkinson, K.D.; Cowley, M.J.; Elliott, P.I.P.; Duckett, S.B.; Green, G.G.R.; Khazal, I.G.; López-Serrano, J.; Williamson, D.C. Reversible Interactions with Para-Hydrogen Enhance NMR Sensitivity by Polarization Transfer. Science 2009, 323, 1708–1711. [Google Scholar] [CrossRef] [Green Version]
- Korchak, S.; Mamone, S.; Glöggler, S. Over 50 % 1H and 13C Polarization for Generating Hyperpolarized Metabolites—A Para-Hydrogen Approach. ChemistryOpen 2018, 7, 672–676. [Google Scholar] [CrossRef] [Green Version]
- Hövener, J.-B.; Schwaderlapp, N.; Borowiak, R.; Lickert, T.; Duckett, S.B.; Mewis, R.E.; Adams, R.W.; Burns, M.J.; Highton, L.A.R.; Green, G.G.R.; et al. Toward Biocompatible Nuclear Hyperpolarization Using Signal Amplification by Reversible Exchange: Quantitative in Situ Spectroscopy and High-Field Imaging. Anal. Chem. 2014, 86, 1767–1774. [Google Scholar] [CrossRef]
- Buckenmaier, K.; Scheffler, K.; Plaumann, M.; Fehling, P.; Bernarding, J.; Rudolph, M.; Back, C.; Koelle, D.; Kleiner, R.; Hövener, J.-B.; et al. Multiple Quantum Coherences Hyperpolarized at Ultra-Low Fields. ChemPhysChem 2019, 20, 2823–2829. [Google Scholar] [CrossRef]
- Glöggler, S.; Müller, R.; Colell, J.; Emondts, M.; Dabrowski, M.; Blümich, B.; Appelt, S. Para-Hydrogen Induced Polarization of Amino Acids, Peptides and Deuterium–Hydrogen Gas. Phys. Chem. Chem. Phys. 2011, 13, 13759–13764. [Google Scholar] [CrossRef]
- Barskiy, D.A.; Tayler, M.C.D.; Marco-Rius, I.; Kurhanewicz, J.; Vigneron, D.B.; Cikrikci, S.; Aydogdu, A.; Reh, M.; Pravdivtsev, A.N.; Hövener, J.-B.; et al. Zero-Field Nuclear Magnetic Resonance of Chemically Exchanging Systems. Nat. Commun. 2019, 10, 3002. [Google Scholar] [CrossRef] [Green Version]
- Levitt, M.H. Symmetry Constraints on Spin Dynamics: Application to Hyperpolarized NMR. J. Magn. Reson. 2016, 262, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, K.L.; Pravdivtsev, A.N.; Yurkovskaya, A.V.; Vieth, H.-M.; Kaptein, R. The Role of Level Anti-Crossings in Nuclear Spin Hyperpolarization. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 81, 1–36. [Google Scholar] [CrossRef]
- Bhattacharya, P.; Harris, K.; Lin, A.P.; Mansson, M.; Norton, V.A.; Perman, W.H.; Weitekamp, D.P.; Ross, B.D. Ultra-Fast Three Dimensional Imaging of Hyperpolarized 13C in Vivo. Magn. Reson. Mater. Phy. 2005, 18, 245–256. [Google Scholar] [CrossRef]
- Schmidt, A.B.; Berner, S.; Braig, M.; Zimmermann, M.; Hennig, J.; von Elverfeldt, D.; Hövener, J.-B. In Vivo 13C-MRI Using SAMBADENA. PLoS ONE 2018, 13, e0200141. [Google Scholar] [CrossRef]
- Svyatova, A.; Skovpin, I.V.; Chukanov, N.V.; Kovtunov, K.V.; Chekmenev, E.Y.; Pravdivtsev, A.N.; Hövener, J.-B.; Koptyug, I.V. 15N MRI of SLIC-SABRE Hyperpolarized 15N-Labelled Pyridine and Nicotinamide. Chem. Eur. J. 2019, 25, 8465–8470. [Google Scholar] [CrossRef]
- Cavallari, E.; Carrera, C.; Aime, S.; Reineri, F. Metabolic Studies of Tumor Cells Using [1-13C] Pyruvate Hyperpolarized by Means of PHIP-Side Arm Hydrogenation. ChemPhysChem 2019, 20, 318–325. [Google Scholar] [CrossRef]
- Sellies, L.; Aspers, R.; Feiters, M.C.; Rutjes, F.; Tessari, M. Para-Hydrogen Hyperpolarization Allows Direct NMR Detection of α-Amino Acids in Complex (Bio)Mixtures. Angew. Chem. Int. Ed. 2021, 60, 26954–26959. [Google Scholar] [CrossRef]
- Nielsen, N.C.; Sorensen, O.W. Conditional Bounds on Polarization Transfer. J. Magn. Reson. A 1995, 114, 24–31. [Google Scholar] [CrossRef]
- Nielsen, N.C.; Schulte-Herbrüggen, T.; Sørensen, O.W. Bounds on Spin Dynamics Tightened by Permutation Symmetry Application to Coherence Transfer in I2S and I3S Spin Systems. Mol. Phys. 1995, 85, 1205–1216. [Google Scholar] [CrossRef]
- Sengstschmid, H.; Freeman, R.; Barkemeyer, J.; Bargon, J. A New Excitation Sequence to Observe the PASADENA Effect. J. Magn. Reson. A 1996, 120, 249–257. [Google Scholar] [CrossRef]
- Kiryutin, A.S.; Ivanov, K.L.; Yurkovskaya, A.V.; Vieth, H.-M.; Lukzen, N.N. Manipulating Spin Hyper-Polarization by Means of Adiabatic Switching of a Spin-Locking RF-Field. Phys. Chem. Chem. Phys. 2013, 15, 14248–14255. [Google Scholar] [CrossRef] [Green Version]
- Pravdivtsev, A.N.; Yurkovskaya, A.V.; Petrov, P.A.; Vieth, H.-M. Coherent Evolution of Singlet Spin States in PHOTO-PHIP and M2S Experiments. Phys. Chem. Chem. Phys. 2017, 19, 25961–25969. [Google Scholar] [CrossRef] [Green Version]
- Pravdivtsev, A.N.; Kiryutin, A.S.; Yurkovskaya, A.V.; Vieth, H.-M.; Ivanov, K.L. Robust Conversion of Singlet Spin Order in Coupled Spin-1/2 Pairs by Adiabatically Ramped RF-Fields. J. Magn. Reson. 2016, 273, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Pravdivtsev, A.N.; Sönnichsen, F.; Hövener, J.-B. OnlyParahydrogen SpectrosopY (OPSY) Pulse Sequences—One Does Not Fit All. J. Magn. Reson. 2018, 297, 86–95. [Google Scholar] [CrossRef]
- Aguilar, J.A.; Adams, R.W.; Duckett, S.B.; Green, G.G.R.; Kandiah, R. Selective Detection of Hyperpolarized NMR Signals Derived from Para-Hydrogen Using the Only Para-Hydrogen SpectroscopY (OPSY) Approach. J. Magn. Reson. 2011, 208, 49–57. [Google Scholar] [CrossRef]
- Ratajczyk, T.; Gutmann, T.; Dillenberger, S.; Abdulhussaein, S.; Frydel, J.; Breitzke, H.; Bommerich, U.; Trantzschel, T.; Bernarding, J.; Magusin, P.C.M.M.; et al. Time Domain Para Hydrogen Induced Polarization. Solid State Nucl. Magn. Reson. 2012, 43–44, 14–21. [Google Scholar] [CrossRef]
- Buntkowsky, G.; Gutmann, T.; Petrova, M.V.; Ivanov, K.L.; Bommerich, U.; Plaumann, M.; Bernarding, J. Dipolar Induced Para-Hydrogen-Induced Polarization. Solid State Nucl. Magn. Reson. 2014, 63–64, 20–29. [Google Scholar] [CrossRef]
- Nasibulov, E.A.; Pravdivtsev, A.N.; Yurkovskaya, A.V.; Lukzen, N.N.; Vieth, H.-M.; Ivanov, K.L. Analysis of Nutation Patterns in Fourier-Transform NMR of Non-Thermally Polarized Multispin Systems. Z. Phys. Chem. 2013, 227, 929–953. [Google Scholar] [CrossRef] [Green Version]
- Barskiy, D.A.; Salnikov, O.G.; Shchepin, R.V.; Feldman, M.A.; Coffey, A.M.; Kovtunov, K.V.; Koptyug, I.V.; Chekmenev, E.Y. NMR SLIC Sensing of Hydrogenation Reactions Using Parahydrogen in Low Magnetic Fields. J. Phys. Chem. C 2016, 120, 29098–29106. [Google Scholar] [CrossRef]
- Vinogradov, E.; Grant, A.K. Hyperpolarized Long-Lived States in Solution NMR: Three-Spin Case Study in Low Field. J. Magn. Reson. 2008, 194, 46–57. [Google Scholar] [CrossRef]
- Franzoni, M.B.; Buljubasich, L.; Spiess, H.W.; Münnemann, K. Long-Lived 1H Singlet Spin States Originating from Para-Hydrogen in Cs-Symmetric Molecules Stored for Minutes in High Magnetic Fields. J. Am. Chem. Soc. 2012, 134, 10393–10396. [Google Scholar] [CrossRef]
- Stevanato, G.; Roy, S.S.; Hill-Cousins, J.; Kuprov, I.; Brown, L.J.; Brown, R.C.D.; Pileio, G.; Levitt, M.H. Long-Lived Nuclear Spin States Far from Magnetic Equivalence. Phys. Chem. Chem. Phys. 2015, 17, 5913–5922. [Google Scholar] [CrossRef] [Green Version]
- Grohmann, T.; Leibscher, M. Nuclear Spin Selective Alignment of Ethylene and Analogues. J. Chem. Phys. 2011, 134, 204316. [Google Scholar] [CrossRef]
- Chapovsky, P.L.; Hermans, L.J.F. Nuclear Spin Conversion in Polyatomic Molecules. Annu. Rev. Phys. Chem. 1999, 50, 315–345. [Google Scholar] [CrossRef] [Green Version]
- Chapovsky, P.L.; Zhivonitko, V.V.; Koptyug, I.V. Conversion of Nuclear Spin Isomers of Ethylene. J. Phys. Chem. A 2013, 117, 9673–9683. [Google Scholar] [CrossRef]
- San Fabián, J.; Casanueva, J.; Díez, E.; Esteban, A.L. Spin–Spin Coupling Constants in Ethylene: Equilibrium Values. Chem. Phys. Lett. 2002, 361, 159–168. [Google Scholar] [CrossRef]
- Kaski, J.; Lantto, P.; Vaara, J.; Jokisaari, J. Experimental and Theoretical Ab Initio Study of the 13C−13C Spin−Spin Coupling and 1H and 13C Shielding Tensors in Ethane, Ethene, and Ethyne. J. Am. Chem. Soc. 1998, 120, 3993–4005. [Google Scholar] [CrossRef]
- Carrington, A.; McLachlan, A.D. Introduction to Magnetic Resonance with Applications to Chemistry and Chemical Physics; Harper & Row: New York, NY, USA, 1967. [Google Scholar]
- DeVience, S.J.; Walsworth, R.L.; Rosen, M.S. Preparation of Nuclear Spin Singlet States Using Spin-Lock Induced Crossing. Phys. Rev. Lett. 2013, 111, 173002. [Google Scholar] [CrossRef]
- Cavallari, E.; Carrera, C.; Boi, T.; Aime, S.; Reineri, F. Effects of Magnetic Field Cycle on the Polarization Transfer from Parahydrogen to Heteronuclei through Long-Range J-Couplings. J. Phys. Chem. B 2015, 119, 10035–10041. [Google Scholar] [CrossRef]
- Schmidt, A.B.; Berner, S.; Schimpf, W.; Müller, C.; Lickert, T.; Schwaderlapp, N.; Knecht, S.; Skinner, J.G.; Dost, A.; Rovedo, P.; et al. Liquid-State Carbon-13 Hyperpolarization Generated in an MRI System for Fast Imaging. Nat. Commun. 2017, 8, ncomms14535. [Google Scholar] [CrossRef]
- Cavallari, E.; Carrera, C.; Sorge, M.; Bonne, G.; Muchir, A.; Aime, S.; Reineri, F. The 13 C Hyperpolarized Pyruvate Generated by ParaHydrogen Detects the Response of the Heart to Altered Metabolism in Real Time. Sci. Rep. 2018, 8, 8366. [Google Scholar] [CrossRef]
- Kovtunov, K.V.; Pokochueva, E.; Salnikov, O.; Cousin, S.; Kurzbach, D.; Vuichoud, B.; Jannin, S.; Chekmenev, E.; Goodson, B.; Barskiy, D.; et al. Hyperpolarized NMR: D-DNP, PHIP, and SABRE. Chem. Asian J. 2018, 13, 1857–1871. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.T.; Zheng, G.; Talbot, C.L.; Tong, X.; D’Adam, T.; Parnell, S.R.; Veer, M.D.; Jenkin, G.; Polglase, G.R.; Hooper, S.B.; et al. Hyperpolarised Gas Filling Station for Medical Imaging Using Polarised 129Xe and 3He. Magn. Reson. Imag. 2021. [Google Scholar] [CrossRef]
- Reineri, F.; Viale, A.; Ellena, S.; Boi, T.; Daniele, V.; Gobetto, R.; Aime, S. Use of Labile Precursors for the Generation of Hyperpolarized Molecules from Hydrogenation with Parahydrogen and Aqueous-Phase Extraction. Angew. Chem. Int. Ed. 2011, 50, 7350–7353. [Google Scholar] [CrossRef]
- Chukanov, N.V.; Salnikov, O.G.; Shchepin, R.V.; Kovtunov, K.V.; Koptyug, I.V.; Chekmenev, E.Y. Synthesis of Unsaturated Precursors for Parahydrogen-Induced Polarization and Molecular Imaging of 1-13C-Acetates and 1-13C-Pyruvates via Side Arm Hydrogenation. ACS Omega 2018, 3, 6673–6682. [Google Scholar] [CrossRef] [Green Version]
- Nelson, S.J.; Kurhanewicz, J.; Vigneron, D.B.; Larson, P.E.Z.; Harzstark, A.L.; Ferrone, M.; van Criekinge, M.; Chang, J.W.; Bok, R.; Park, I.; et al. Metabolic Imaging of Patients with Prostate Cancer Using Hyperpolarized [1-13C]Pyruvate. Sci. Transl. Med. 2013, 5, 198ra108. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C.H.; Lau, J.Y.C.; Chen, A.P.; Geraghty, B.J.; Perks, W.J.; Roifman, I.; Wright, G.A.; Connelly, K.A. Hyperpolarized 13C Metabolic MRI of the Human Heart. Circ. Res. 2016, 119, 1177–1182. [Google Scholar]
- Berner, S.; Schmidt, A.B.; Zimmermann, M.; Pravdivtsev, A.N.; Glöggler, S.; Hennig, J.; von Elverfeldt, D.; Hövener, J.-B. SAMBADENA Hyperpolarization of 13C-Succinate in an MRI: Singlet-Triplet Mixing Causes Polarization Loss. ChemistryOpen 2019, 8, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.B.; Brahms, A.; Ellermann, F.; Knecht, S.; Berner, S.; Hennig, J.; von Elverfeldt, D.; Herges, R.; Hövener, J.-B.; Pravdivtsev, A. Selective Excitation of Hydrogen Doubles the Yield and Improves the Robustness of Parahydrogen-Induced Polarization of Low-γ Nuclei. Phys. Chem. Chem. Phys. 2021, 23, 26645–26652. [Google Scholar] [CrossRef]
- Olsson, L.E.; Chai, C.-M.; Axelsson, O.; Karlsson, M.; Golman, K.; Petersson, J.S. MR Coronary Angiography in Pigs with Intraarterial Injections of a Hyperpolarized 13C Substance. Magn. Reson. Med. 2006, 55, 731–737. [Google Scholar] [CrossRef]
- Pravdivtsev, A.; Brahms, A.; Kienitz, S.; Sönnichsen, F.D.; Hövener, J.-B.; Herges, R. Catalytic Hydrogenation of Trivinyl Orthoacetate: Mechanisms Elucidated by Parahydrogen Induced Polarization. ChemPhysChem 2021, 22, 370–377. [Google Scholar] [CrossRef]
- Pravdivtsev, A.N.; Yurkovskaya, A.V.; Lukzen, N.N.; Ivanov, K.L.; Vieth, H.-M. Highly Efficient Polarization of Spin-1/2 Insensitive NMR Nuclei by Adiabatic Passage through Level Anticrossings. J. Phys. Chem. Lett. 2014, 5, 3421–3426. [Google Scholar] [CrossRef]
- Knecht, S.; Blanchard, J.W.; Barskiy, D.; Cavallari, E.; Dagys, L.; Dyke, E.V.; Tsukanov, M.; Bliemel, B.; Münnemann, K.; Aime, S.; et al. Rapid Hyperpolarization and Purification of the Metabolite Fumarate in Aqueous Solution. Proc. Natl. Acad. Sci. 2021, 118. [Google Scholar] [CrossRef]
- Eills, J.; Cavallari, E.; Kircher, R.; Matteo, G.D.; Carrera, C.; Dagys, L.; Levitt, M.H.; Ivanov, K.L.; Aime, S.; Reineri, F.; et al. Singlet-Contrast Magnetic Resonance Imaging: Unlocking Hyperpolarization with Metabolism. Angew. Chem. Int. Ed. 2021, 60, 6791–6798. [Google Scholar] [CrossRef]
- Pravdivtsev, A.N.; Hövener, J.-B. Simulating Non-Linear Chemical and Physical (CAP) Dynamics of Signal Amplification By Reversible Exchange (SABRE). Chem. Eur. J. 2019, 25, 7659–7668. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J.; Bourée, F. Symmetry and Magnetic Structures. EPJ Web Conf. 2012, 22, 00010. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Schematic view of hydrogenative ((A), left) and non-hydrogenative ((B), right) PHIP for a 3-spin-½ system with asymmetric couplings (top) and a 4-spin-½ system with symmetric couplings (bottom). Here, we focus on hydrogenative PHIP in symmetric and asymmetric systems (A). The case of 4-spin-½ SABRE ((B), bottom) was considered by Levitt in a seminal paper [17]. Note that different lines represent different strengths of nuclear spin–spin interactions. pH2 is represented by red circles, other spins or other reagents by blue squares, and J-couplings by thin, thick, and dashed lines.
Figure 1.
Schematic view of hydrogenative ((A), left) and non-hydrogenative ((B), right) PHIP for a 3-spin-½ system with asymmetric couplings (top) and a 4-spin-½ system with symmetric couplings (bottom). Here, we focus on hydrogenative PHIP in symmetric and asymmetric systems (A). The case of 4-spin-½ SABRE ((B), bottom) was considered by Levitt in a seminal paper [17]. Note that different lines represent different strengths of nuclear spin–spin interactions. pH2 is represented by red circles, other spins or other reagents by blue squares, and J-couplings by thin, thick, and dashed lines.

Figure 2.
Spin topologies considered for simulating the effect of symmetry on the transformation of pH2-derived spin order (red) into observable polarization. Red symbols indicate the pH2-nascent spins, different lines indicate J-coupling constants, and circles, squares, hexagons are spins of the same type. Note that different lines represent different strengths of nuclear spin–spin interactions.
Figure 2.
Spin topologies considered for simulating the effect of symmetry on the transformation of pH2-derived spin order (red) into observable polarization. Red symbols indicate the pH2-nascent spins, different lines indicate J-coupling constants, and circles, squares, hexagons are spins of the same type. Note that different lines represent different strengths of nuclear spin–spin interactions.

Figure 3.
Average polarization per spin achieved theoretically by adding pH2 to a precursor yielding a molecule with 2–10 spins with (black) and without (orange, blue) symmetry constraints (SC). In general, higher polarization can be achieved if there are no constraints (compare black with orange and blue) and if the initial density matrix is (blue) rather than (orange, compare blue and orange in A). We assumed pH2 to be added in positions A and B. The reported values are given in Table A6 and Table A7 (Appendix B).
Figure 3.
Average polarization per spin achieved theoretically by adding pH2 to a precursor yielding a molecule with 2–10 spins with (black) and without (orange, blue) symmetry constraints (SC). In general, higher polarization can be achieved if there are no constraints (compare black with orange and blue) and if the initial density matrix is (blue) rather than (orange, compare blue and orange in A). We assumed pH2 to be added in positions A and B. The reported values are given in Table A6 and Table A7 (Appendix B).

Figure 4.
Average polarization per molecule—in units of one-spin-1/2 polarization—that can be achieved theoretically by adding pH2 to a precursor yielding a molecule with 2–10 spins with (black,) and without symmetry constraints (SC) for(blue) and(orange). If the polarization of all spins in one molecule is summed up, up to ~ 4 was obtained for large spin systems (blue). The reported values can be obtained from the average values given in Table A6 and Table A7 (Appendix B) and calculations for N = 2, 3, and 4 exemplified in Appendix C.
Figure 4.
Average polarization per molecule—in units of one-spin-1/2 polarization—that can be achieved theoretically by adding pH2 to a precursor yielding a molecule with 2–10 spins with (black,) and without symmetry constraints (SC) for(blue) and(orange). If the polarization of all spins in one molecule is summed up, up to ~ 4 was obtained for large spin systems (blue). The reported values can be obtained from the average values given in Table A6 and Table A7 (Appendix B) and calculations for N = 2, 3, and 4 exemplified in Appendix C.

Figure 5.
Interactions and symmetry axis of ethylene. (A) Ethylene structure and nuclear spin–spin couplings (J-couplings, top), the numbering of the atomic positions, and the cartesian axis x, y, z. (B) Graphs corresponding to the spin system AA’(AA’) where pH2 was added at cis-, trans-, or geminal positions (red circles). Different lines correspond to different values of spin–spin interactions.
Figure 5.
Interactions and symmetry axis of ethylene. (A) Ethylene structure and nuclear spin–spin couplings (J-couplings, top), the numbering of the atomic positions, and the cartesian axis x, y, z. (B) Graphs corresponding to the spin system AA’(AA’) where pH2 was added at cis-, trans-, or geminal positions (red circles). Different lines correspond to different values of spin–spin interactions.

Figure 6.
Molecular structures (top) and spin topologies (bottom) of ethyl (A) and allyl (B) esters of carboxylic acids-products of PHIP-SAH. Different lines (bottom) represent different spin–spin interaction values.
Figure 6.
Molecular structures (top) and spin topologies (bottom) of ethyl (A) and allyl (B) esters of carboxylic acids-products of PHIP-SAH. Different lines (bottom) represent different spin–spin interaction values.

Table 1.
Relative populations of spin symmetries in ethylene after addition of pH2 in germinal (gem), cis, and trans position. Note that there are also coherences between and states, and between the two respective populated B-symmetries (e.g., and in case of pos. = gem).
Table 1.
Relative populations of spin symmetries in ethylene after addition of pH2 in germinal (gem), cis, and trans position. Note that there are also coherences between and states, and between the two respective populated B-symmetries (e.g., and in case of pos. = gem).
| Pos. | ||||||
|---|---|---|---|---|---|---|
| gem | 0 | 0.2409 | 0.009 | 0 | 1/8 | 1/8 |
| cis | 0 | 0.1075 | 0.1425 | 1/8 | 0 | 1/8 |
| trans | 0 | 0.0266 | 0.2234 | 1/8 | 1/8 | 0 |
Table 2.
Polarization transfer from pH2 () to an X nucleus (). One hydrogen of pH2 is in A, the other is in B position.
Table 2.
Polarization transfer from pH2 () to an X nucleus (). One hydrogen of pH2 is in A, the other is in B position.
| Type of the System | Examples of Molecules, R = Acetate, Pyruvate | |
|---|---|---|
| ABX, ABCX, ABCDX, ABCDEX,… ABCD2X | 1 | 1-13C-vinyl-R |
| 1-13C-allyl-R | ||
| AA’BB’X | 1 | 1-13C-ethylene |
| A2BX | ¾ | |
| A2BC2X | ¾ | |
| A3BX | 2/3 | |
| A2B2X | 9/16 | |
| A3B2X | ½ | 1-13C-ethyl-R |
| A3B2C2X | ½ | 1-13C-propyl-R |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pravdivtsev, A.N.; Barskiy, D.A.; Hövener, J.-B.; Koptyug, I.V. Symmetry Constraints on Spin Order Transfer in Parahydrogen-Induced Polarization (PHIP). Symmetry 2022, 14, 530. https://doi.org/10.3390/sym14030530
AMA Style
Pravdivtsev AN, Barskiy DA, Hövener J-B, Koptyug IV. Symmetry Constraints on Spin Order Transfer in Parahydrogen-Induced Polarization (PHIP). Symmetry. 2022; 14(3):530. https://doi.org/10.3390/sym14030530
Chicago/Turabian StylePravdivtsev, Andrey N., Danila A. Barskiy, Jan-Bernd Hövener, and Igor V. Koptyug. 2022. "Symmetry Constraints on Spin Order Transfer in Parahydrogen-Induced Polarization (PHIP)" Symmetry 14, no. 3: 530. https://doi.org/10.3390/sym14030530
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.