
Properties Assessment by Quantum Mechanical Calculations for Azulenes Substituted with Thiophen– or Furan–Vinyl–Pyridine

Abstract
:1. Introduction
2. Computational Details
3. Results
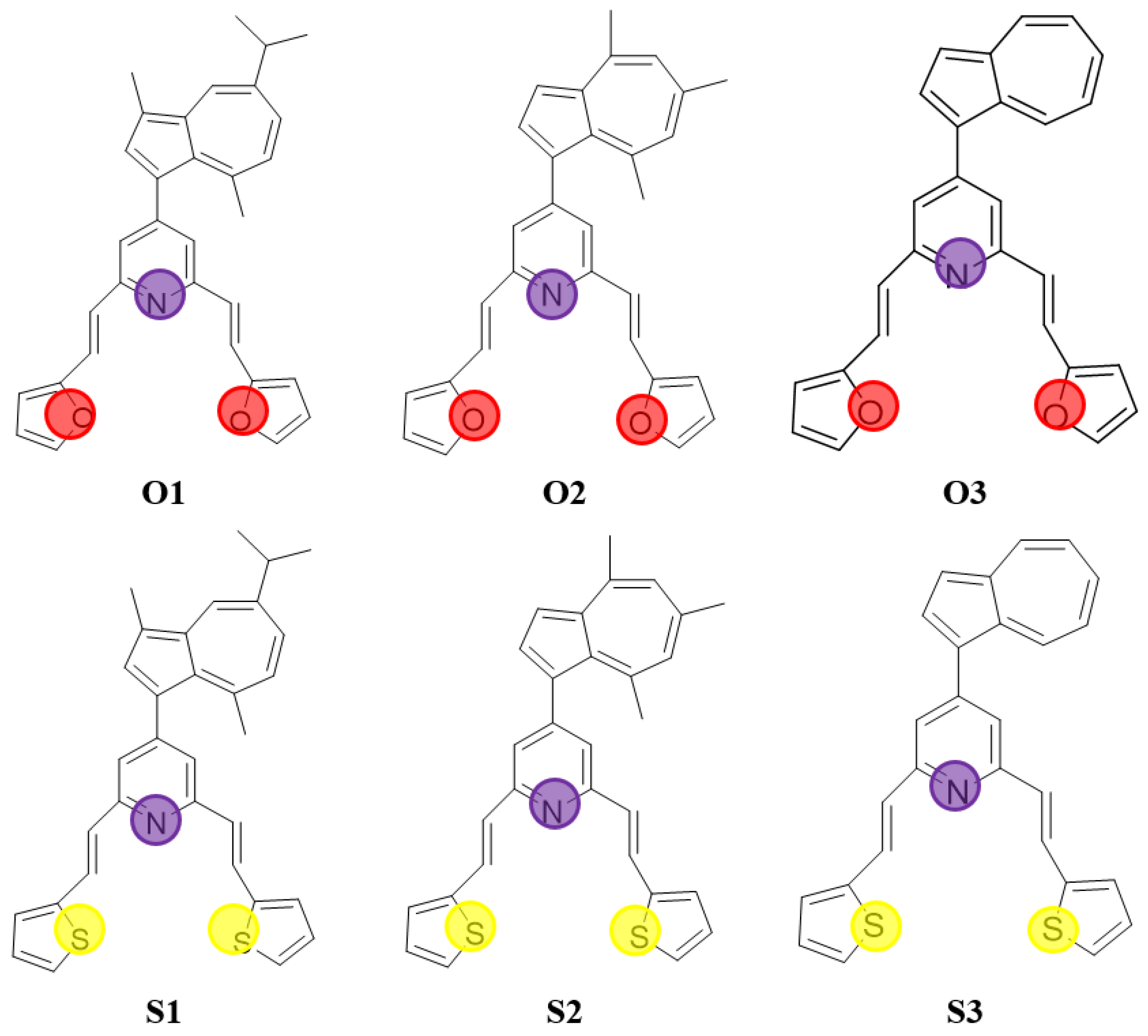
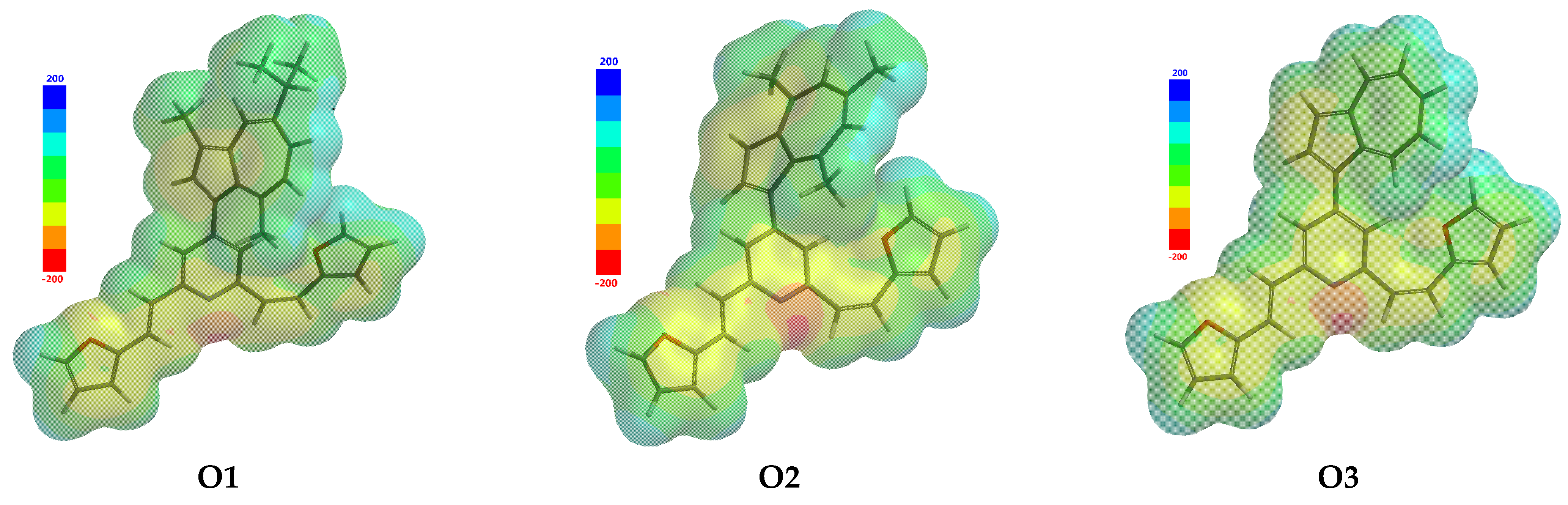
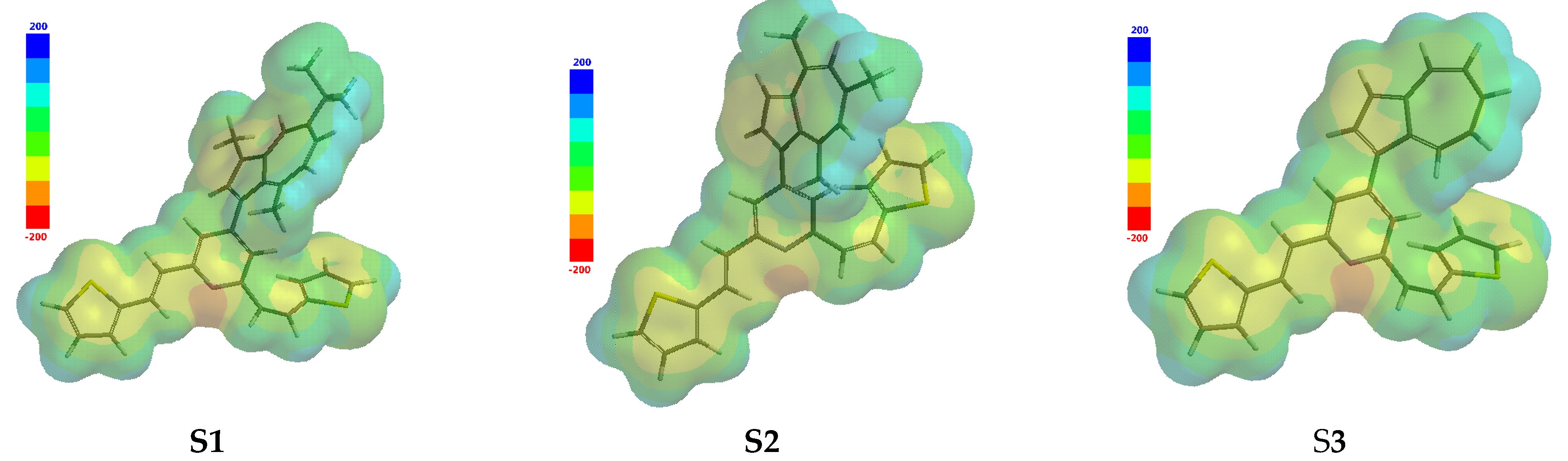
3.1. Molecular and QSAR Properties Computations
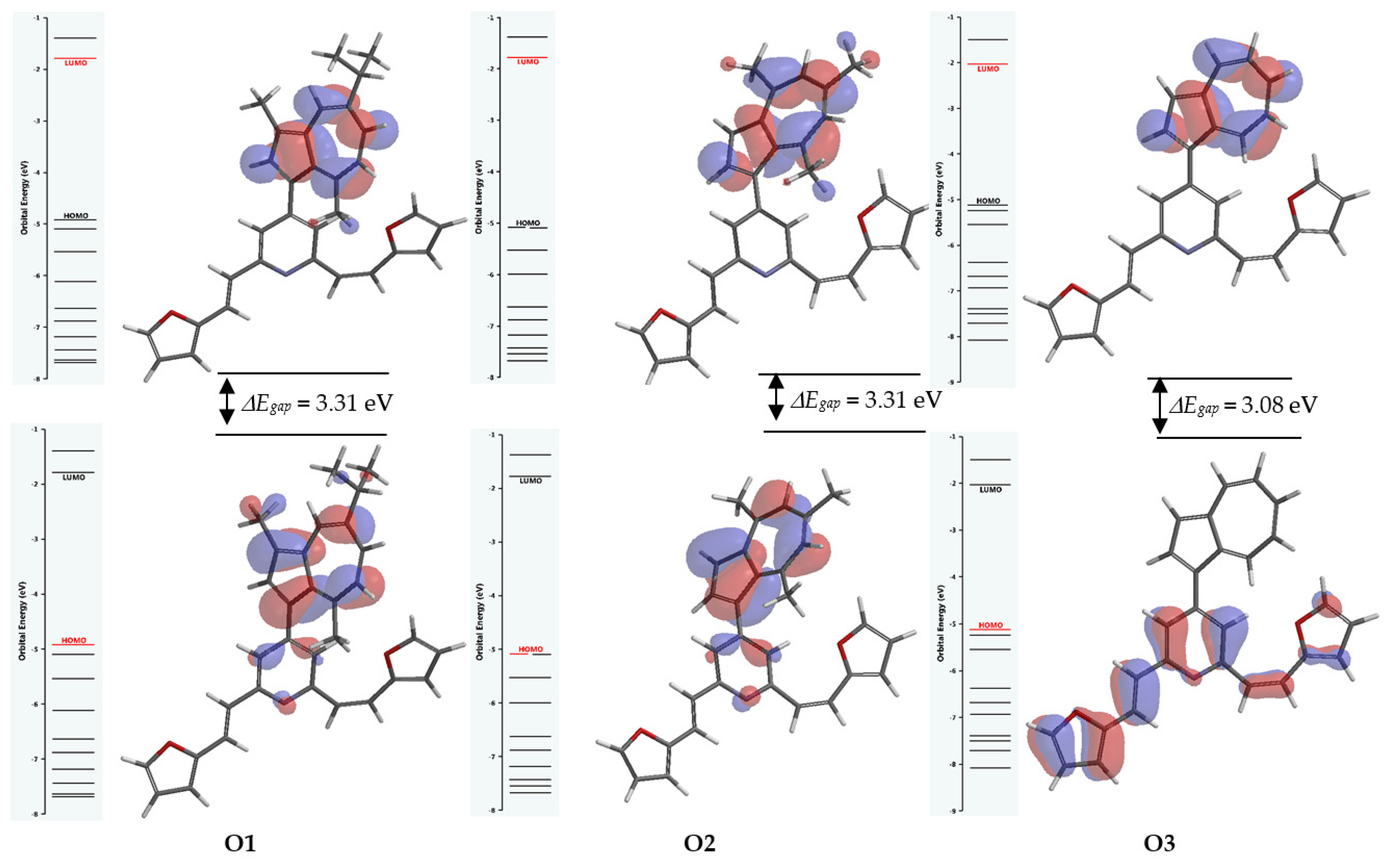
3.2. Correlations between DFT-Calculated Frontier Molecular Orbital’s Energies and Experimental Data
3.3. Correlation between DFT-Computed Molecular and QSAR Properties and Ionization Potential or Electron Affinity
3.4. Correlation of Quantum Chemical Reactivity Parameters
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ungureanu, E.-M.; Popescu, M.; Tatu, G.-L.; Birzan, L.; Isopescu, R.; Stanciu, G.; Buica, G.-O. Electrochemical Comparison on New (Z)-5-(Azulen-1-Ylmethylene)-2-Thioxo-Thiazolidin-4-Ones. Symmetry 2021, 13, 588. [Google Scholar] [CrossRef]
- Oprisanu, A.; Matica, O.-T.; Ungureanu, E.M.; Cristea, M.; Birzan, L. Electrochemical study of azulene-pyridine compounds and film formation. UPB Sci. Bull. Series B 2022, in press.
- Oprisanu, A.; Lazar, I.G.; Pop, M.D.; Ungureanu, E.M.; Isopescu, R.; Birzan, L. Polyazulenes Based Materials for Heavy Metal Ions Detection. Rev. Chim. 2017, 68, 2215–2218. [Google Scholar] [CrossRef]
- Pop, M.D.; Anastasoaie, V.; Apostoiu Popescu, M.; Oprisanu, A.; Ungureanu, E.-M.; Cristea, M. New Azulene Modified Electrodes for Heavy Metal Sensing. Rev. Chim. 2017, 68, 2172–2175. [Google Scholar] [CrossRef]
- Lazar, I.-G.; Diacu, E.; Ungureanu, E.-M.; Buica, G.-O.; Birzan, L.; Arnold, G.-L. Modified Electrodes Based on 2,6-Bis((E)-2-(Thiophen-2-yl)Vinyl)-4-(4,6,8-Trimethylazulen-1-yl)Pyridine For Heavy Metals Sensing. UPB Sci. Bull. Ser. B. 2017, 79, 23–36. [Google Scholar]
- Engwa, G.A.; Ferdinand, P.U.; Nwalo, F.N.; Unachukwu, M.N. Mechanism and Health Effects of Heavy Metal Toxicity in Humans. In Poisoning in the Modern World, New Tricks for an Old Dog? Karcioglu, O., Arslan, B., Eds.; IntechOpen: London, UK, 2019; ISBN 978-1-83880-786-3. [Google Scholar]
- Posadas, Y.; López-Guerrero, V.E.; Segovia, J.; Perez-Cruz, C.; Quintanar, L. Dissecting the copper bioinorganic chemistry of the functional and pathological roles of the prion protein: Relevance in Alzheimer’s disease and cancer. Curr. Opin. Chem. Biol. 2022, 66, 102098. [Google Scholar] [CrossRef]
- Gupta, R.; Kumar, P. Integrative analysis of OIP5-AS1/miR-129-5p/CREBBP axis as a potential therapeutic candidate in the pathogenesis of metal toxicity-induced Alzheimer’s disease. Gene Rep. 2022, 26, 101442. [Google Scholar] [CrossRef]
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal Toxicity Links to Alzheimer’s Disease and Neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef]
- Azevedo, J.A.; Azevedo, R.A. Heavy metals and oxidative stress: Where do we go from here? Commun. Biometry Crop. Sci. 2006, 1, 135–138. [Google Scholar]
- Pratt, J.M. Metalloenzymes as Molecular Switches: The Role of Conformation Changes in Controlling Activity. J. Inorg. Biochem. 1986, 28, 145–153. [Google Scholar] [CrossRef]
- Huang, J.; Huang, S.; Zhao, Y.; Feng, B.; Jiang, K.; Sun, S.; Ke, C.; Kymakis, E.; Zhuang, X. Azulene-Based Molecules, Polymers, and Frameworks for Optoelectronic and Energy Applications. Small Methods 2020, 4, 2000628. [Google Scholar] [CrossRef]
- Razus, A.C.; Nica, S.; Cristian, L.; Raicopol, M.; Birzan, L.; Dragu, A.E. Synthesis and physico-chemical properties of highly conjugated azo-aromatic systems. 4-(azulen-1-yl)-pyridines with mono- and bis azo-aromatic moieties at C3-position of azulene. Dye. Pigm. 2011, 91, 55–61. [Google Scholar] [CrossRef]
- Xin, H.; Li, J.; Lu, R.Q.; Gao, X.; Swager, T.M. Azulene–Pyridine-Fused Heteroaromatics. J. Am. Chem. Soc. 2020, 142, 13598–13605. [Google Scholar] [CrossRef] [PubMed]
- Murfin, L.C.; Lewis, S.E. Azulene-A Bright Core for Sensing and Imaging. Molecules 2021, 26, 353. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Gao, X. Application of azulene in constructing organic optoelectronic materials: New tricks for an old dog. ChemPlusChem 2017, 82, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Amir, E.; Murai, M.; Amir, R.J.; Cowart, J.S.; Chabinyc, M.L.; Hawker, C.J. Conjugated oligomers incorporating azulene building blocks–seven-vs. five-membered ring connectivity. Chem. Sci. 2014, 5, 4483–4489. [Google Scholar] [CrossRef]
- Algohary, A.M.; Hassan, M.M.; El-Hashash, M.A.; Rizk, S.A.; Elamin, M.B.; Ahmed, A.H. Novel colorimetric chemosensors containing pyridine moiety for detection of some cations in water and crops samples: Design, synthesis, and evaluation. J. Saudi Chem. Soc. 2021, 25, 101386. [Google Scholar] [CrossRef]
- Razus, A.C.; Birzan, L.; Pavel, C.; Lehadus, O.; Corbu, A.; Chiraleu, F.; Enache, C. Azulene--substituted pyridines and pyridinium salts. Synthesis and structure. 1. Azulene-substituted pyridines. J. Heterocycl. Chem. 2007, 44, 245–250. [Google Scholar] [CrossRef]
- Popa-Tudor, I.; Gugoasa, L.A.; Stefan-Van Staden, R.-I. Electrochemical Sensor for the Assay of Zinc Ions in Whole Blood Samples. UPB Sci. Bull. Ser. B 2019, 81, 103–108. [Google Scholar]
- Brotea, A.G.; Matica, O.-T.; Musina, C.; Ungureanu, E.-M. Polyazulene-Based Materials based on 4-(azulen-1-yl)-2,6-bis((E)-2-(thiophen-2-yl)vinyl)pyridine for Heavy Metals Ions Detection. UPB Sci Bull. 2021, in press.
- Enache, O.-I.; Matica, O.-T.; Bujduveanu, M.-R.; Ungureanu, E.-M.; Cristea, M. Electrochemical proprieties of 2,6-bis((E)-2-(furan-2-yl)vinyl)-4-(azulen-1-yl)pyridine. UPB Sci Bull. 2021, in press.
- Zarrouk, A.; Zarrok, H.; Salghi, R.; Hammouti, B.; Al-Deyab, S.S.; Touzani, R.; Hadda, T.B. A theoretical investigation on the corrosion inhibition of copper by quinoxaline derivatives in nitric acid solution. Int. J. Electrochem. Sci. 2012, 7, 6353–6364. [Google Scholar]
- Korichi, H.; Zouchoune, F.; Zendaoui, S.M.; Zouchoune, B.; Saillard, J.Y. The coordination chemistry of azulene: A comprehensive DFT investigation. Organometallics 2010, 29, 1693–1706. [Google Scholar] [CrossRef]
- Verdal, N.; Rivera, S.A.; Hudson, B.S. Inelastic neutron scattering and periodic DFT studies of crystalline aromatic materials: Azulene. A test of ab initio methods. Chem. Phys. Lett. 2007, 437, 38–44. [Google Scholar] [CrossRef]
- Vasile, A.-A.; Ungureanu, E.-M.; Stanciu, G.; Cristea, M.; Stefaniu, A. Evaluation of (Z)-5-(Azulen-1-ylmethylene)-2-thioxothiazolidin-4-ones Properties Using Quantum Mechanical Calculations. Symmetry 2021, 13, 1462. [Google Scholar] [CrossRef]
- Boulkroune, M.; Ignatovich, L.; Muravenko, V.; Spura, J.; Chibani, A.; Jouikov, V. Correlation of the HOMO–LUMO gap in furyl and thienyl nitrones and nitroethenes with their electrochemical redox potentials. Chem. Heterocycl. Compd. 2014, 49, 1579–1588. [Google Scholar] [CrossRef]
- Brovelli, F.; Rivas, B.L.; Bernède, J.C.; del Valle, M.A.; Díaz, F.R.; Berredjem, Y. Electrochemical and optical studies of 1,4-diaminoanthraquinone for solar cell applications. Polym. Bull. 2007, 58, 521–527. [Google Scholar] [CrossRef]
- Wu, T.Y.; Tsao, M.H.; Chen, F.L.; Su, S.G.; Chang, C.W.; Wang, H.P.; Lin, Y.C.; Sun, I.W. Synthesis and Characterization of Three Organic Dyes with Various Donors and Rhodanine Ring Acceptor for Use in Dye-Sensitized Solar Cells. J. Iran. Chem. Soc. 2010, 7, 707–720. [Google Scholar] [CrossRef]
- Méndez-Hernández, D.D.; Tarakeshwar, P.; Gust, D.; Moore, T.A.; Moore, A.L.; Mujica, V. Simple and accurate correlation of experimental redox potentials and DFT-calculated HOMO/LUMO energies of polycyclic aromatic hydrocarbons. J. Mol. Model. 2013, 19, 2845–2848. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Süß, D.; Sukuba, I.; Schauperl, M.; Probst, M.; Maihom, T.; Kaiser, A. Performance of DFT functionals for properties of small molecules containing beryllium, tungsten and hydrogen. Nucl. Mater. Energy. 2020, 22, 100731. [Google Scholar] [CrossRef]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989; Volume 16. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hehre, W.J. A Guide to Molecular Mechanics and Quantum Chemical Calculations; Wavefunction, Inc.: Irvine, CA, USA, 2003. [Google Scholar]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibsom, T.D.; Windus, T.L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Neese, F. Prediction of molecular properties and molecular spectroscopy with density functional theory: From fundamental theory to exchange-coupling. Coord. Chem. Rev. 2009, 253, 526–563. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and hardness: Application to inorganic chemistry. Inorg. Chem. 1988, 27, 734–740. [Google Scholar] [CrossRef]
- Yankova, R.; Genieva, S.; Halachev, N.; Dimitrova, G. Molecular structure, vibrational spectra, MEP, HOMO-LUMO and NBO analysis of Hf(SeO3)(SeO4)(H2O)4. J. Mol. Struct. 2016, 1106, 82–88. [Google Scholar] [CrossRef]
- Pearson, R.G. Recent Advances in the Concept of Hard and Soft Acids and Bases. J. Chem. Educ. 1987, 64, 561–567. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Wang, H.; Wang, L.; Liu, A. DFT study of new bipyrazole derivatives and their potential activity as corrosion inhibitors. J. Mol. Model. 2007, 13, 147–153. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpaly, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Vennila, P.; Govindaraju, M.; Venkatesh, G.; Kamal, C. Molecular structure, vibrational spectral assignments (FT-IR and FT-RAMAN), NMR, NBO, HOMO-LUMO and NLO properties of O-methoxybenzaldehyde based on DFT calculations. J. Mol. Struct. 2016, 1111, 151–156. [Google Scholar] [CrossRef]
- Alam, M.J.; Ahmad, S. Anharmonic vibrational studies of L-aspartic acid using HF and DFT calculations. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 96, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Demir, P.; Akman, F. Molecular structure, spectroscopic characterization, HOMO and LUMO analysis of PU and PCL grafted onto PEMA-co-PHEMA with DFT quantum chemical calculations. J. Mol. Struct. 2017, 1134, 404–415. [Google Scholar] [CrossRef]
- Ungureanu, E.M.; Buica, G.O.; Razus, A.C.; Birzan, L.; Weisz, R.; Bujduveanu, M.R. Electrochemical study on 4-(azulen-1-yl)-2, 6-bis (2-furyl)-and 4-(azulen-1-yl)-2, 6-bis (2-thienyl)-pyridines. Rev. Chim. 2012, 63, 27–33. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | O1 C32H29NO2 | O2 C30H25NO2 | O3 C27H19NO2 | |||
|---|---|---|---|---|---|---|
| B3LYP | ωB97XD | B3LYP | ωB97XD | B3LYP | ωB97XD | |
| M a (g⋅mol−1) | 459.59 | 431.54 | 389.45 | |||
| E b (au) | −1441.98 | −1441.51 | −1363.36 | −1362.9 | −1245.42 | −1244.99 |
| Eaq c (au) | −1441.99 | −1441.52 | −1363.37 | −1362.91 | −1245.43 | −1245 |
| Esolv d (kJ⋅mol−1) | −23.72 | −21.35 | −27.97 | −25.92 | −32.7 | −30.16 |
| μ e (D) | 3.10 | 3.27 | 3.43 | 3.67 | 2.91 | 3.11 |
| EHOMOf (eV) | −4.92 | −6.74 | −5.08 | −6.93 | −5.12 | −7.03 |
| ELUMO g (eV) | −1.79 | −0.13 | −1.77 | −0.13 | −2.04 | −0.36 |
| S h (Å2) | 520.42 | 514.08 | 481.21 | 474.27 | 429.84 | 423.09 |
| V i (Å3) | 508.94 | 506.68 | 472.01 | 469.83 | 418.75 | 416.98 |
| PSA j (Å2) | 18.27 | 16.79 | 18.31 | 16.80 | 18.6 | 17.93 |
| OI k | 1.69 | 1.67 | 1.64 | 1.62 | 1.59 | 1.57 |
| α l (10−30⋅m3) | 81.92 | 80.91 | 78.88 | 77.88 | 74.61 | 73.62 |
| Epot m (kJ⋅mol−1) | −166.66 | −167.73 | −167.67 | −168.84 | −164.84 | −163.65 |
| S1 C32H29NS2 | S2 C30H25NS2 | S3 C27H19NS2 | ||||
| B3LYP | ωB97XD | B3LYP | ωB97XD | B3LYP | ωB97XD | |
| M a (g⋅mol−1) | 491.72 | 463.67 | 421.59 | |||
| E b (au) | −2087.94 | −2087.47 | −2009.31 | −2008.86 | −1891.37 | −1890.95 |
| Eaq c (au) | −2087.95 | −2087.48 | −2009.32 | −2008.88 | −1891.38 | −1890.96 |
| Esolv d (kJ⋅mol−1) | −32.25 | −32.25 | −36.99 | −37.13 | −39.55 | −38.56 |
| μ e (D) | 3.11 | 3.22 | 3.33 | 3.55 | 2.79 | 2.82 |
| EHOMOf (eV) | −4.98 | −6.72 | −5.13 | −6.91 | −5.27 | −7.07 |
| ELUMO g (eV) | −1.84 | −0.14 | −1.83 | −0.14 | −2.06 | −0.36 |
| S h (Å2) | 535.76 | 532.51 | 496.12 | 491.29 | 444.51 | 442.36 |
| V i (Å3) | 526.71 | 524.23 | 489.67 | 487.32 | 436.52 | 434.71 |
| PSA j (Å2) | 6.297 | 6.187 | 6.34 | 6.21 | 6.31 | 6.25 |
| OI k | 1.70 | 1.69 | 1.65 | 1.64 | 1.60 | 1.59 |
| α l (10−30⋅m3) | 83.36 | 82.34 | 80.32 | 79.31 | 76.03 | 75.05 |
| Epot m (kJ⋅mol−1) | −160.04 | −152.77 | −165.18 | −154.99 | −159.77 | −160.26 |
| Parameter | O1 C32H29NO2 | O2 C30H25NO2 | O3 C27H19NO2 | |||
|---|---|---|---|---|---|---|
| B3LYP | ωB97XD | B3LYP | ωB97XD | B3LYP | ωB97XD | |
| I m = −EHOMO (eV) | 4.92 | 6.74 | 5.08 | 6.93 | 5.12 | 7.03 |
| A n = −ELUMO (eV) | 1.79 | 0.13 | 1.77 | 0.13 | 2.04 | 0.36 |
| ΔEgap o = I − A (eV) | 3.13 | 6.61 | 3.31 | 6.8 | 3.08 | 6.67 |
| χ p = (I + A)/2 (eV) | 3.36 | 3.44 | 3.43 | 3.53 | 3.58 | 3.70 |
| η q = (I − A)/2 (eV) | 1.57 | 3.31 | 1.66 | 3.40 | 1.54 | 3.34 |
| σ r = 1/η (eV−1) | 0.64 | 0.30 | 0.60 | 0.29 | 0.65 | 0.30 |
| ω s = μ2/2η (D2⋅ eV−1) | 3.07 | 1.62 | 3.55 | 1.98 | 2.75 | 1.45 |
| S1 C32H29NS2 | S2 C30H25NS2 | S3 C27H19NS2 | ||||
| B3LYP | ωB97XD | B3LYP | ωB97XD | B3LYP | ωB97XD | |
| I m = −EHOMO (eV) | 4.98 | 6.72 | 5.13 | 6.91 | 5.27 | 7.07 |
| A n = −ELUMO (eV) | 1.84 | 0.14 | 1.83 | 0.14 | 2.06 | 0.36 |
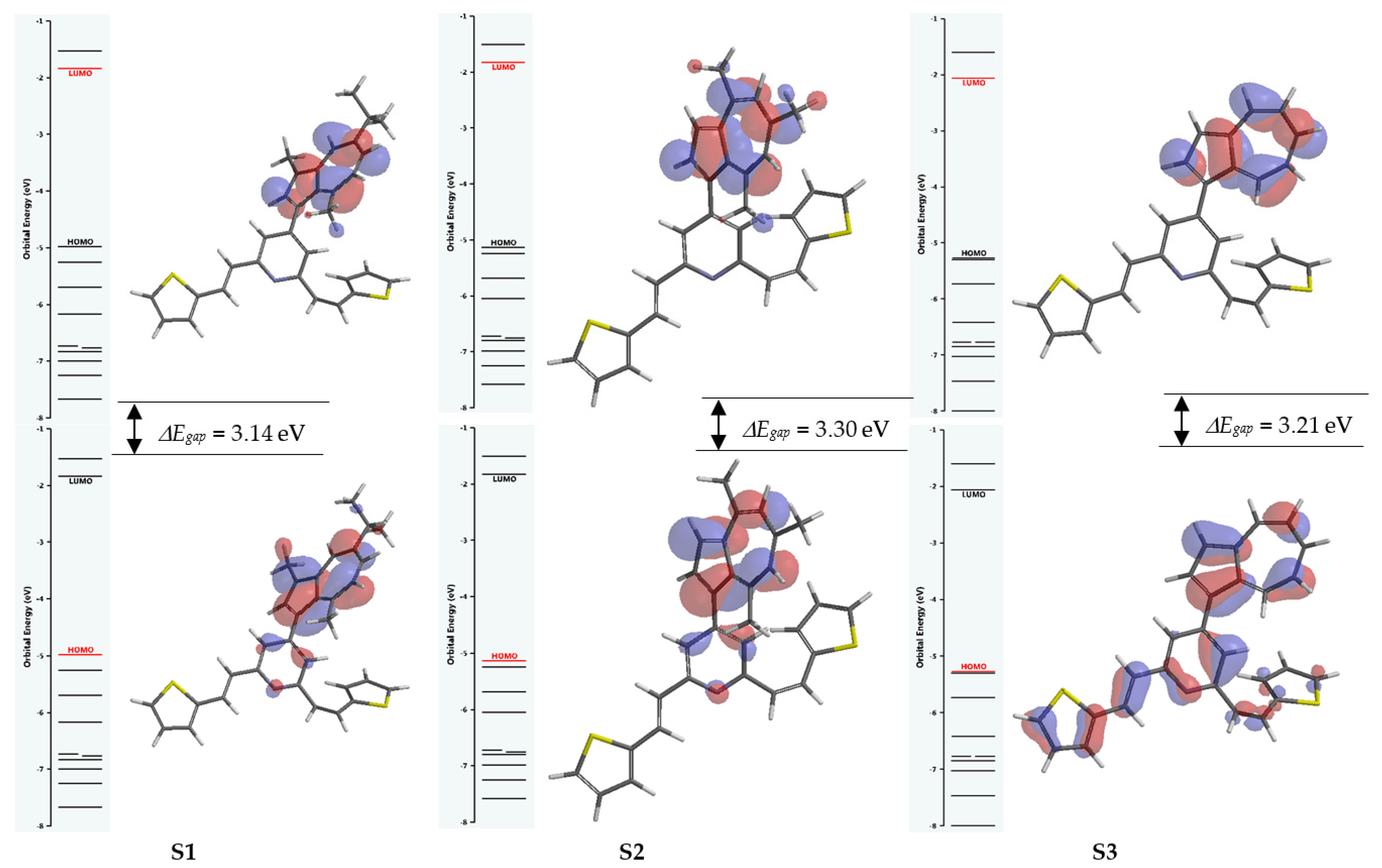
| ΔEgap o = I − A (eV) | 3.14 | 6.58 | 3.30 | 6.77 | 3.21 | 6.71 |
| χ p = (I + A)/2 (eV) | 3.41 | 3.43 | 3.48 | 3.53 | 3.67 | 3.72 |
| η q = (I − A)/2 (eV) | 1.57 | 3.29 | 1.65 | 3.39 | 1.61 | 3.36 |
| σ r = 1/η (eV−1) | 0.64 | 0.30 | 0.61 | 0.30 | 0.62 | 0.30 |
| ω s = μ2/2η (D2⋅ eV−1) | 3.08 | 1.58 | 3.36 | 1.86 | 2.42 | 1.19 |
| Property | Ligand | ||
|---|---|---|---|
| O1 | O2 | O3 | |
| Ea (V) | 0.318 | 0.487 | 0.553 |
| Ec (V) | −2.071 | −2.084 | −1.854 |
| Reference | [4] | [2] | [22] |
| S1 | S2 | S3 | |
| Ea (V) | 0.338 | 0.470 | 0.567 |
| Ec (V) | −2.065 | −2.090 | −1.858 |
| Reference | [3] | [5] | [21] |
| Correlation | B3LYP | ωB97XD | ||||
|---|---|---|---|---|---|---|
| a | b | R2 | a | b | R2 | |
| For O1–O3 | ||||||
| Ea vs. I | −5.298 | 1.141 | 0.993 | −5.211 | 0.821 | 0.995 |
| Ec vs. A | −3.606 | 0.859 | 0.9997 | −2.204 | 0.972 | 0.998 |
| For S1–S3 | ||||||
| Ea vs. I | 4.500 | 1.259 | 0.995 | 6.202 | 1.523 | 0.999 |
| Ec vs. A | 3.949 | 1.017 | 0.996 | 2.199 | 0.991 | 0.990 |
| Correlation * | B3LYP | ωB97XD | ||||
|---|---|---|---|---|---|---|
| a | b | R2 | a | b | R2 | |
| For O1–O3 | ||||||
| E vs. I | −5595.9 | 842.39 | 0.812 | −5786.5 | 642.99 | 0.917 |
| E vs. A | −2441.8 | 584.77 | 0.791 | −1491.1 | 683.54 | 0.842 |
| Eaq vs. I | −5595.9 | 842.39 | 0.812 | −5786.5 | 642.99 | 0.917 |
| Eaq vs. A | −2441.8 | 584.77 | 0.791 | −2441.8 | 584.77 | 0.791 |
| Esolv vs. I | 171.76 | −39.66 | 0.870 | 178.07 | −29.55 | 0.980 |
| Esolv vs. A | 19.12 | −25.31 | 0.720 | −19.95 | −28.37 | 0.730 |
| μ vs. I | nlc ** | nlc ** | ||||
| μ vs. A | nlc ** | nlc ** | ||||
| For S1–S3 | ||||||
| E vs. I | −5595.9 | 842.39 | 0.812 | −5786.5 | 642.99 | 0.917 |
| E vs. A | −3306.9 | 686.25 | 0.813 | −2148.2 | 714.61 | 0.842 |
| Eaq vs. I | −5461.9 | 676.00 | 0.982 | −5838.5 | 556.91 | 0.973 |
| Eaq vs. A | −3306.9 | 686.25 | 0.813 | −2148.2 | 714.64 | 0.842 |
| Esolv vs. I | 93.18 | −25.25 | 0.977 | 90.05 | −18.27 | 0.936 |
| Esolv vs. A | nlc ** | nlc ** | ||||
| μ vs. I | nlc ** | nlc ** | ||||
| μ vs. A | 6.784 | −1.941 | 0.863 | 3.745 | −2.568 | 0.796 |
| Correlation * | B3LYP | ωB97XD | ||||
|---|---|---|---|---|---|---|
| a | b | R2 | a | b | R2 | |
| For O1–O3 | ||||||
| S vs. I | 2460.50 | −393.52 | 0.841 | 2541.00 | −300.07 | 0.939 |
| S vs. A | 968.30 | −263.11 | 0.760 | 534.35 | −309.07 | 0.809 |
| V vs. I | 2422.40 | −388.05 | 0.802 | 2494.20 | −294.16 | 0.924 |
| V vs. A | 964.01 | −266.49 | 0.782 | 528.54 | −309.89 | 0.833 |
| PSA vs. I | nlc ** | nlc ** | ||||
| PSA vs. A | 16.19 | 1.18 | 0.969 | 16.15 | 4.94 | 0.999 |
| OI vs. I | 3.89 | −0.45 | 0.893 | 3.93 | −0.33 | 0.969 |
| OI vs. A | 2.16 | −0.28 | 0.690 | 1.69 | −0.33 | 0.750 |
| α vs. I | 237.41 | −31.54 | 0.826 | 242.59 | −23.93 | 0.927 |
| α vs. A | 118.60 | −21.50 | 0.776 | 82.66 | −25.11 | 0.829 |
| Epot vs. I | nlc ** | nlc ** | ||||
| Epot vs. A | −183.43 | 9.13 | 0.916 | −170.90 | 20.15 | 0.959 |
| For S1–S3 | ||||||
| S vs. I | 2102.20 | −314.06 | 0.991 | 2257.30 | −256.31 | 0.990 |
| S vs. A | 1086.40 | −311.13 | 0.781 | 556.15 | −316.09 | 0.791 |
| V vs. I | 2074.80 | −310.24 | 0.985 | 2233.80 | −253.87 | 0.978 |
| V vs. A | 1081.30 | −312.58 | 0.804 | 551.00 | −323.02 | 0.832 |
| PSA vs. I | nlc ** | 4.99 | 0.1782 | 0.959 | ||
| PSA vs. A | nlc ** | 6.17 | 0.2341 | 0.870 | ||
| OI vs. I | 3.42 | −0.345 | 0.999 | 3.61 | −0.285 | 0.998 |
| OI vs. A | 2.28 | −0.3254 | 0.716 | 1.71 | −0.3409 | 0.750 |
| α vs. I | 209.18 | −25.216 | 0.987 | 221.57 | −20.678 | 0.979 |
| α vs. A | 128.29 | −25.334 | 0.799 | 84.50 | −26.25 | 0.829 |
| Epot vs. I | nlc ** | −10.42 | −21.099 | 0.923 | ||
| Epot vs. A | nlc ** | −149.82 | −29.00 | 0.917 | ||
| Correlation * | B3LYP | ωB97XD | ||||
|---|---|---|---|---|---|---|
| a | b | R2 | a | b | R2 | |
| For O1–O3 | ||||||
| χ vs. I | nlc ** | nlc ** | ||||
| χ vs. A | 2.0664 | 0.743 | 0.942 | 3.3624 | 0.9239 | 0.870 |
| η vs. I | 4.4667 | −0.5714 | 1.000 | 5.1432 | −0.2604 | 0.624 |
| η vs. A | nlc ** | nlc ** | ||||
| σ vs. I | 0.4941 | 0.2232 | 0.999 | 0.14 | 0.023 | 0.617 |
| σ vs. A | nlc ** | nlc ** | ||||
| ω vs. I | 21.971 | −3.7393 | 0.954 | 14.382 | −1.8404 | 0.999 |
| ω vs. A | nlc ** | nlc ** | ||||
| For S1–S3 | ||||||
| χ vs. I | nlc ** | nlc ** | ||||
| χ vs. A | 1.6338 | 0.9867 | 0.948 | 3.3264 | 1.0795 | 0.893 |
| η vs. I | nlc ** | nlc ** | ||||
| η vs. A | nlc ** | nlc ** | ||||
| σ vs. I | nlc ** | nlc ** | ||||
| σ vs. A | nlc ** | nlc ** | ||||
| ω vs. I | 19.407 | −3.2091 | 0.940 | 14.783 | −1.9129 | 0.981 |
| ω vs. A | 9.6175 | −3.4881 | 0.892 | 2.0581 | −2.4249 | 0.823 |
| Correlated Parameters | a | b | R2 | DFT Method |
|---|---|---|---|---|
| Oxygen compounds | ||||
| E vs. I | −5786.5 | 642.99 | 0.917 | ωB97XD |
| Eaq vs. I | −5786.5 | 642.99 | 0.917 | ωB97XD |
| Esolv vs. I | 178.07 | −29.55 | 0.980 | ωB97XD |
| S vs. I | 2541.00 | −300.07 | 0.939 | ωB97XD |
| V vs. I | 2494.20 | −294.16 | 0.924 | ωB97XD |
| PSA vs. A | 16.15 | 4.94 | 0.999 | ωB97XD |
| OI vs. I | 3.93 | −0.33 | 0.969 | ωB97XD |
| α vs. I | 242.59 | −23.93 | 0.927 | ωB97XD |
| Epot vs. A | −170.90 | 20.15 | 0.959 | ωB97XD |
| Sulfur compounds | ||||
| E vs. I | −5461.8 | 676.00 | 0.982 | B3LYP |
| Eaq vs. I | −5461.9 | 676.00 | 0.982 | B3LYP |
| Esolv vs. I | 93.18 | −25.25 | 0.977 | B3LYP |
| S vs. I | 2102.20 | −314.06 | 0.991 | B3LYP |
| V vs. I | 2233.80 | −253.87 | 0.985 | B3LYP |
| PSA vs. I | 4.99 | 0.1782 | 0.959 | ωB97XD |
| OI vs. I | 3.42 | −0.345 | 0.999 | B3LYP |
| α vs. I | 209.18 | −25.216 | 0.987 | B3LYP |
| Epot vs. I | −10.42 | −21.099 | 0.923 | ωB97XD |
| Correlated Parameters | a | b | R2 | DFT Method |
|---|---|---|---|---|
| Oxygen compounds | ||||
| χ vs. A | 2.0664 | 0.743 | 0.942 | B3LYP |
| η vs. I | 4.4667 | −0.5714 | 1.000 | B3LYP |
| σ vs. I | 0.4941 | 0.2232 | 0.999 | B3LYP |
| ω vs. I | 21.971 | −3.7393 | 0.954 | B3LYP |
| ω vs. I | 14.382 | −1.8404 | 0.999 | ωB97XD |
| Sulfur compounds | ||||
| χ vs. A | 1.6338 | 0.9867 | 0.948 | B3LYP |
| ω vs. I | 19.407 | −3.2091 | 0.940 | B3LYP |
| ω vs. I | 14.783 | −1.9129 | 0.981 | ωB97XD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciocirlan, O.; Ungureanu, E.-M.; Vasile, A.-A.; Stefaniu, A. Properties Assessment by Quantum Mechanical Calculations for Azulenes Substituted with Thiophen– or Furan–Vinyl–Pyridine. Symmetry 2022, 14, 354. https://doi.org/10.3390/sym14020354
Ciocirlan O, Ungureanu E-M, Vasile A-A, Stefaniu A. Properties Assessment by Quantum Mechanical Calculations for Azulenes Substituted with Thiophen– or Furan–Vinyl–Pyridine. Symmetry. 2022; 14(2):354. https://doi.org/10.3390/sym14020354
Chicago/Turabian StyleCiocirlan, Oana, Eleonora-Mihaela Ungureanu, Alina-Alexandra Vasile (Corbei), and Amalia Stefaniu. 2022. "Properties Assessment by Quantum Mechanical Calculations for Azulenes Substituted with Thiophen– or Furan–Vinyl–Pyridine" Symmetry 14, no. 2: 354. https://doi.org/10.3390/sym14020354