2. Cyclization of ortho-Hydroxystyrene Derivatives
Activated 2-vinylphenols have been demonstrated to be very useful starting materials for building the chromane nucleus through an oxa-Michael–Michael cascade reaction. Since 1999 [
7], many chiral metal complexes have catalyzed this cascade reaction, and with organocatalysts being introduced only later. In particular, acid-based bifunctional catalysts have been revealed to be very efficient [
8].
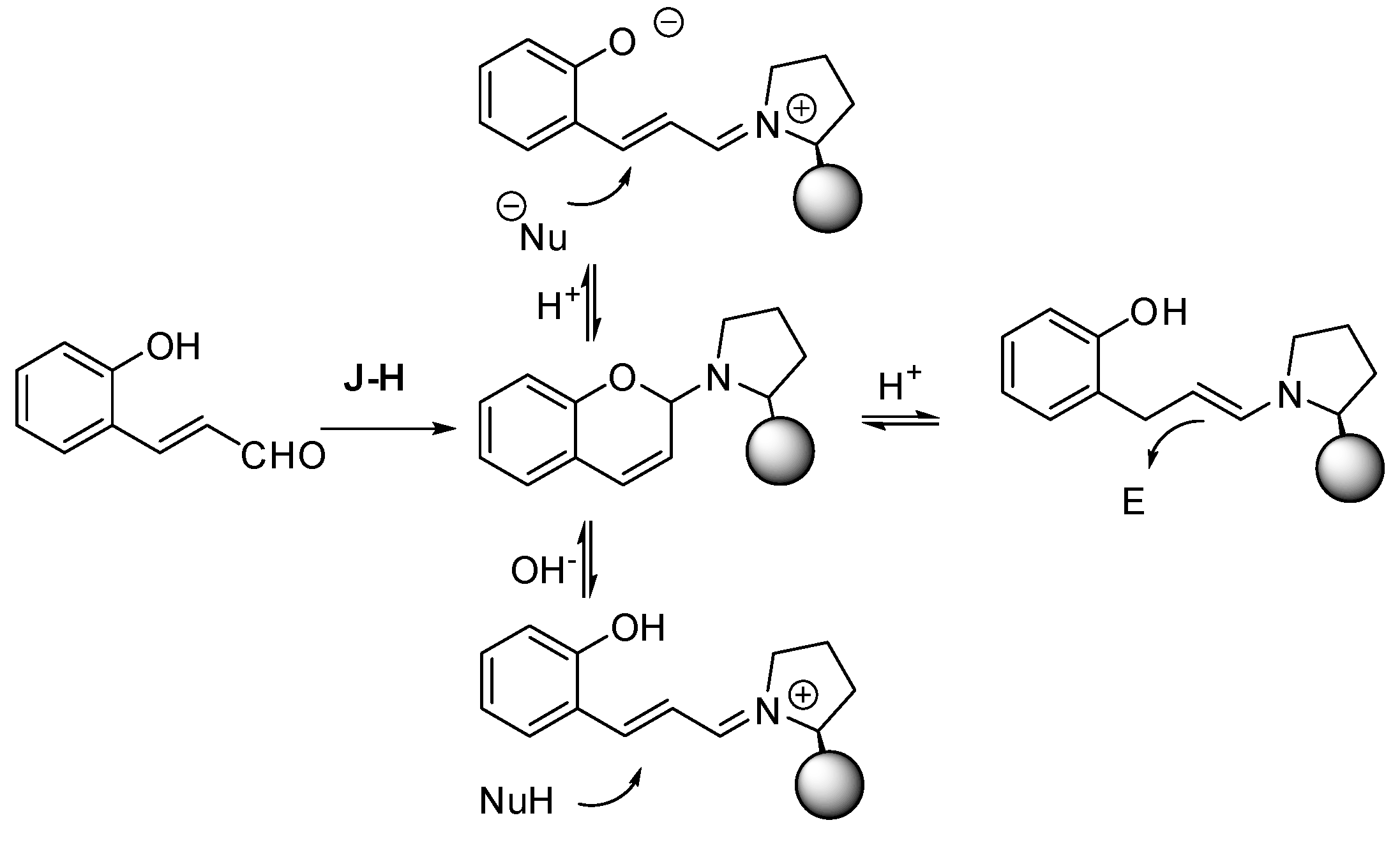
In the time range covered by this review, some interesting papers have appeared in the literature. For example, An and co-workers proposed a heterogeneous catalyst with an achiral acid site (silanols of mesoporous silica) and a chiral basic site (an immobilized chiral amine) [
9].
As depicted in
Scheme 2, the authors envisaged a mechanism similar to homogenous catalysis. By this hypothesis, the surface silanols activate the 2-nitrovinyl phenol, while the immobilized amine forms an iminium ion intermediate with the aldehyde. The hydroxy framework attacks the iminium ion intermediate. Then, C-1 of the nitro-olefin closes the six-membered ring by attacking the enamine derived from the first step. Moreover, the pore wall provides a steric shield, enhancing the enantioselectivity, as demonstrated by the higher enantioselectivity obtained with larger groups adjacent to the nitrogen atom in the catalyst. This reaction was also extended to the Michael–hemiacetalization reaction followed by oxidation, with good results (
Scheme 2, eq 3). Finally, the catalysts were recovered and could be reused up to five times without significantly affecting the yield and enantioselectivity. In
Scheme 2, eq 1, the (3
S,4
S)-stereochemistry is depicted as reported by authors in their schemes, but in the
Supplementary Material, they affirmed that the depicted compound had a (3
R,4
R)-stereochemistry. The stereochemistry of compounds of eqs 2 and 3 were, conversely, correctly assigned as (4
R) and (3
R,4
S) respectively. However, how the stereochemistry was obtained was not reported in either the text or the
Supplementary Material; perhaps this was done by comparison of data with previous works. It should be noted that the oxa-Michael-Henry reaction between two molecules of nitrostyrene was not observed.
Xia, Xu and co-workers performed an enantioselective, organocatalytic oxa-Michael-nitro-Michael reaction, but in this case, too, 2-hydroxynitrostyrenes reacted only with β-nitroolefins different from themselves [
10]. The reaction afforded chiral chromane derivatives bearing three contiguous stereogenic centers (
Scheme 3). The all-
S stereochemistry was assigned by X-ray analysis.
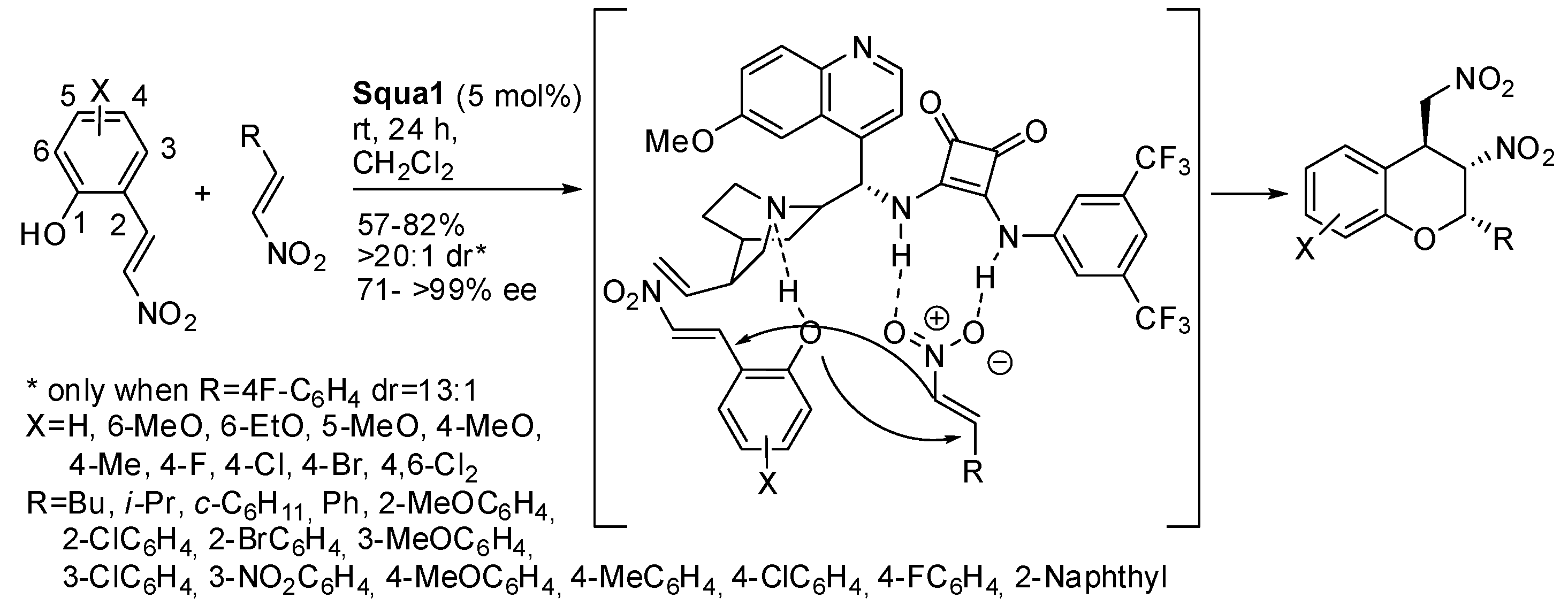
Subsequently, Andrés, Pedrosa and co-workers extended the reaction to polymer-supported squaramides [
11]. Initially, they tested some 4-vinylphenyl-substituted squaramides and found that
Squa2 (see
Scheme 1, 5 mol%, rt, 12–72 h in dichloromethane) gave the best results (65–88% yields, 63:27 to >99:1 dr, 70– > 99% ee, 13 examples). The absolute configuration was once more (2
S,3
S,4
S) and was established by X-ray analysis. The configuration of the minor isomer was instead established as (2
S,3
R,4
S) by comparison of the
1H-NMR coupling constants of the two diastereomers. Then
Squa2 was co-polymerized with styrene and divinylbenzene and the experiments were repeated (65–86% yields, 58:42 to >99:1 dr, 56–74% ee). The polymer-supported catalyst was also recovered and recycled five times without affecting yields and selectivity. Finally, the enantiomers were obtained from a squaramide derived from (1
R,2
R)-1,2-cyclohexanediamine instead of
Squa2 derived from α-amino acid. The authors attributed the opposite stereochemistry to a different assembly of the ternary complex formed by catalyst, nitroalkene and 2-hydroxynitrostyrene in the two cases.
Another domino Michael/hemiacetalization (see also
Scheme 2, eq 3) was carried out between aliphatic aldehydes and (
E)-2-(2-nitrovinyl)phenols under modularly designed organocatalysts (
Scheme 4) [
12].
Cis-3,4-disubstituted chroman-2-ones were obtained after oxidation of the hemiacetal intermediate or chromanes after dehydroxylation. The reaction was scaled up to 0.5 mmol scale, without significant modification of yield and selectivity. The absolute stereochemistry was determined by the optical rotation of known compounds.
(
E)-2-(2-Nitrovinyl)phenols were also allowed to react with azlactones in high excess (twelve-fold) (
Scheme 5) [
13]. The reaction was also performed at a gram scale (84% yield (1.06 g) with 92% ee and >19:1 dr). The absolute configuration was determined by X-ray analysis. It is noteworthy that these compounds have the (3
R,4
R) stereochemistry, that is, the 4-position has the opposite configuration to all those reported in the above schemes.
The reaction of
ortho-hydroxystyrene and azlactone catalyzed by chiral BINOL-phosphoric acid with and without chiral guanidine as the co-catalyst was studied with the density functional theory method [
14]. Both reactions were found to proceed through three steps and not by a classical [4+2] Diels-Alder reaction: C–C then C–O bond formation and, finally, azlactone ring-opening.
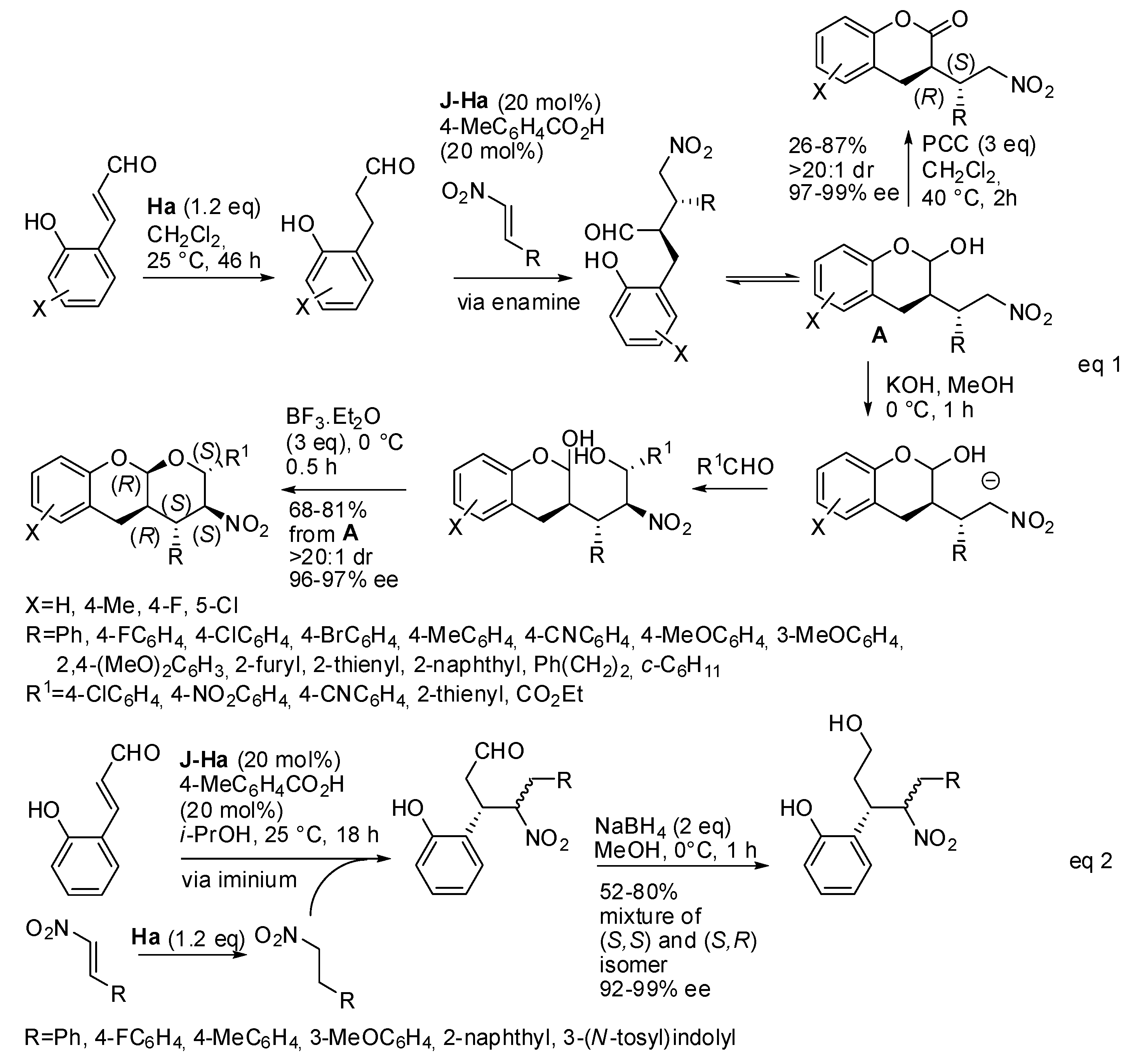
The enantioselective Michael-acetalization-Henry reaction of 2-hydroxynitrostyrene and 5-oxohexanal allowed the synthesis of hexahydro-6H-benzo[c]chromenones with four stereogenic centers under Jørgensen-Hayashi catalyst (
J-H) followed by oxidation with pyridinium chlorochromate (PCC) (
Scheme 6) [
15]. It was noted that longer reaction times were required with electron-donating groups on the phenyl ring. Only one of the four stereogenic centers was obtained in both configurations; thus, a mixture of diastereomers was recovered with predominance of the
cis isomer. Moreover, the reduction of these products followed by Nef-cyclization with ZnBr
2 afforded an aflatoxin skeleton.
Chiral 2-arylbenzopyranones (flavanones) can be obtained by the oxa-Michael reaction of 2′-hydroxychalcone or 1-(2-hydroxyaryl) α,β-unsaturated aliphatic ketones. In 2017, Ranganath and co-workers introduced heterogeneous catalysts prepared from magnetite and cobalt ferrite nanoparticles with a binaphthol phosphate as the chiral ligand [
16]. The reaction presented two interesting features: higher enantioselectivity and the opposite configuration when the chiral ferrite nanoparticles were alone or in cobalt ferrite nanoparticles (
Scheme 7). The authors attributed these behaviors to the confinement effect inside the cobalt ferrite nanoparticles and to the π-interactions of aromatic groups and cobalt ferrite. No reaction was observed with 1-(2-hydroxyaryl) α,β-unsaturated aliphatic ketones. The catalyst can be recovered by external magnet and reused five times without a decrease in yield and selectivity.
Although it did not produce enantioenriched products, Wang, Bu and co-workers performed a highly diasteroselective triple cascade reaction (Michael addition/condensation/Friedel-Crafts) of 3-indolyl-substituted oxindoles with
ortho-hydroxychalcones (
Scheme 8) [
16,
17]. The reaction yields were influenced by the bulkiness of the N-protecting group and the little methyl group was found to be the best. The structure of products was confirmed by X-ray analysis. The bromo-substituted products were then successfully submitted to Suzuki coupling.
o-Hydroxycinnamaldehydes are another valuable substrate for the asymmetric synthesis of chromane derivatives. This substrate is particularly suitable for iminium catalysis (
Scheme 9); in fact, the Jørgensen-Hayashi catalyst easily forms a relatively stable cyclic aminal. The reaction medium shifts the aminal towards a zwitterionic intermediate, under basic conditions or towards the iminium ion in acidic conditions. Then both open-chain intermediates can be attacked by nucleophiles from the less hindered face. Alternatively, the acidic conditions can form an enamine that can attack electrophiles.
An example of acidic conditions is the reaction with
trans-β-nitrostyrenes. This reaction was already envisaged as a formal cycloaddition [
18], but, recently, the reaction was further investigated and a diversity-oriented synthesis made it possible to prepare some different compounds (
Scheme 10) [
19].
The hydride donor diethyl 1,4-dihydro-2,6-dimethyl-3,5-pyridine dicarboxylate (Hantzsch ester,
H) can reduce either the 2-hydroxycinnamaldehyde to chroman-2-ol or the trans-β-nitrostyrene to nitroalkane. The first reaction (
Scheme 10, eq 1) occurs via enamine intermediate, and the subsequent oxidation with PCC or the basic treatment led to chroman-2-ones or to polycyclic
O,O-acetals with five adjacent stereocenters, respectively. In the second reaction (
Scheme 9, eq 2), which occurs via iminium ion intermediate, the reduction of the aldehyde led to open-chain compounds as a mixture of two diastereomers in different ratios, in which only the stereochemistry of the benzyl position is defined. In this reaction, it should be noted that the lower yields occurred with aliphatic trans-β-nitrostyrenes, because significant amounts from iminium ion pathway (eq 2) were recovered. The second reaction was carried out allowing reduction of the nitrostyrene before adding the
o-hydroxycinnamaldehydes. The absolute configuration was determined by X-ray analysis of one of the tricyclic
O,O-acetals and the other attributed by analogy.
In
Scheme 4, we reported the synthesis of 4-(nitromethyl) chroman-2-ols from
o-hydroxynitrostyrene and aldehydes. However, the same compounds were also obtained from
o-hydroxycinnamaldehydes and α-nitroketones, via iminium ion (
Scheme 11) [
20]. It should be noted that the acyl transfer avoided protection or transformation of the unstable chromanol. A
t-butyl group in the 6 position of cinnamaldehyde impeded the reaction for steric reason. Products were also successfully employed in further transformations. The absolute configuration was established by X-ray analysis.
4-Diphenylphosphinyl chroman-2-ols were obtained in moderate to good yields and in high stereoselectivities by addition of diphenylphosphine oxide to
o-hydroxycinnamaldehydes via zwitter ion pathway (
Scheme 12) [
21]. The much higher diasteroselectivity should be noted with respect to the same reaction of
o-hydroxycinnamaldehydes with nitromethane [
22] or malonates [
23]. Owing to the intrinsic instability of the products, they were characterized as acetates.
The stereochemistry was determined as (2R,4S) by X-ray analysis. The asymmetric (o-anisyl) phenylphosphine oxide was also tested, and a 1:1 mixture of separable diastereomers was obtained in 84% yield with 85% and 94% ee, but the absolute configuration of the chiral phosphorus was not identified. Some transformations of the products were also successfully carried out.
o-Hydroxycinnamaldehydes have also been employed as the partner in the cycloaddition to electron-deficient 1-aza-1,3-butadienes which was used both as the four-atom and the two-carbon unit in a highly regio- and stereo-controlled manner (
Scheme 13) [
23,
24]. The role of the Hantzsch ester was still as a hydride source in the iminium-catalyzed reduction of 2-hydroxycinnamaldehydes to chroma-2-ols (see
Scheme 10), the two-atoms partner in the cycloaddition with 1-aza-1,3-butadienes. Then, the aminal intermediate was cyclized by medium acidification. In the absence of the Hantzsch ester, 2-hydroxycinnamaldehydes worked as a four-atom partner instead. This second reaction was also carried out in 0.5-mmol scale, and the product was successfully used for further transformations. Finally, the structure was determined by X-ray analysis on a product of each reaction.
Tetrahydrofuran/tetrahydropyran-fused 3,4-dihydrocoumarin was prepared by a two-step-one-pot synthesis from 2-hydroxycinnamaldehydes and γ/δ-hydroxy enones activated by iminium ion catalysis and followed by PCC oxidation (
Scheme 14) [
25]. The absolute configuration of products was determined by X-ray analysis. The oxidation did not work with aliphatic γ-hydroxy enones, but the hemiacetal intermediate could be converted in high enantiopurity in acetals, submitted to Wittig-Michael reaction without affecting the ArCO moiety, or converted into an allyl derivative with allyl trimethylsilane and BF
3.Et
2O. Finally, the reduction with triethylsilane and BF
3.OEt
2 led to the exclusive reduction of the hemiacetal group at −78 °C, and also of the ArCO moiety at 0 °C.
Above (
Scheme 7), we have already reported a synthesis of chiral spiro-bridged compounds starting from hydroxychalcones. The same compounds can also be obtained from hydroxycinnamaldehydes, and this topic has been widely explored by the group of Y.-K. Liu. In the first paper, the target spiro-bridged compounds were obtained as a mixture of the diastereomers (3
R,6′
S,13′
S) and (3
S,6′
S,13′
S) with high enantiomeric excess (
Scheme 15, eq 1) [
26]. As in the reaction described in
Scheme 9, eq 2, the organocatalyst only controlled the stereochemistry of the 4-position of the chroman-2-ol intermediate. In the same paper, the authors also prepared spiro-fused heterocyclic starting from pre-formed chroman-2-ols and 3-indolyl-3-methoxyoxindoles (
Scheme 15, eq 2). Once more, a mixture of the diastereomers (5a
R,11
R,11a
R) and (5a
R,11
S,11a
R) with high enantiomeric excess was obtained. The absolute configurations of both spiro-bridged and spiro-fused products were determined by X-ray analysis. The synthetic utility of these compounds was also tested, and some transformations were successfully carried out.
Then the research group set up other syntheses of bridged compounds. One example was the asymmetric reaction of cyclic
N-sulfonyl ketimines (
Scheme 16) [
27]. Surprisingly, the conjugate addition to 2-hydroxy cinnamaldehydes gave an inseparable equilibrating mixture of isomers, but the oxidation with PCC only led to spiro-bridged benzo-fused aminals, while the dehydration of this mixture initially provided a mixture of regioisomers, but the spiro-bridged aminal slowly converted into the polycyclic aminal. The reaction can be scaled up to 1 mmol with similar results and extended to
N-protected 2-amino cinnamaldehydes. It should be noted that the reaction of the six-membered cyclic
N-sulfonyl ketimine afforded only the four-substituted chroman-2-ol which was isolated after oxidation. However, the bridged benzo-fused aminal was obtained by a one-pot acid-catalyzed reaction. To increase the significance of the reaction some chemical transformations of these compounds were successfully carried out.
Another organocatalytic enantioselective synthesis reported by Y.-K. Liu’s group allowed the preparation of a 6,6,5-bridged or a 6,6,6-bridged ring system from β-oxoesters and
o-hydroxycinnamaldehydes in the presence or in the absence of
m-chloroperbenzoic acid (
m-CPBA), respectively (
Scheme 17) [
28]. It should be noted that, after oxidation of the oxoester, the reduced
m-chlorobenzoic acid could act as the acidic additive to activate the Jørgensen-Hayashi catalyst. Actually, the authors found that the acid must be neutralized in order to obtain the products. 2-Furyl substituted oxoester afforded an inseparable 3:1 mixture of epimers. Also, the aliphatic oxoester gave a 2:1 mixture of epimers, but they are separable. The reaction was extended to
N-protected 2-amino cinnamaldehydes with worse yields. The structure depicted in
Scheme 17 was determined by X-ray analysis. Moreover, several transformations were successfully attempted on these products.
Then the reaction was extended to cyclic β-oxoaldehydes [
29]. A bifunctional tertiary amine-thiourea catalyst was employed to activate the oxoaldehyde toward the attack to the iminium ion from 2-hydroxycinnamaldehyde (
Scheme 18). Benzofused oxoaldehydes led to spiro-bridged hemiacetal products, while aliphatic cyclic oxoaldehydes gave cage-like polycyclic hemiacetal products. In the former reaction, an increase to a 1 mmol scale did not affect yields and selectivity. In the latter reaction, longer reaction times were required, owing to the poorer reactivity of aliphatic cyclic oxoaldehydes. Moreover, products underwent some transformations without affecting stereoselectivity. The authors unsuccessfully attempted recovering of the catalysts, but they reused the catalysts in situ. Thus, they charged 100 mol% catalysts and added 0.1 mmol of reactants, waited the complete consumption of reactants, added fresh reactants and repeated the procedure five times. Product was recovered with a slightly lower average yield and comparable stereochemistry, but the reaction was faster (in our opinion, this procedure did not demonstrate the recyclability of the catalyst, since the first 100 mol% loading ensured 20 mol% of fresh catalyst for every addition of reactants. They should have loaded only 20 mol% of catalyst). The absolute configuration of both spiro-bridged and cage-like products was determined by X-ray analysis.
A quite different
o-vinylphenol is the imine from salicylaldehyde. It can be deprotonated by a base leading to an azomethine ylide, which in turn can undergo 1,3-dipolar cycloaddition. Recently, Lin and co-workers applied this reaction to 2-hydoxybenzylidene indandiones [
30]. Most of the paper is devoted to the racemic reaction, but at the end they tried a chiral base similar to 1,4-diazabicyclo [2.2.2]octane to perform an asymmetric version.
Squa1 was found the best catalyst for this reaction (
Scheme 19).
The absolute configuration of products was established via X-ray analysis. Ortho-substituted salicylaldehyde derivatives were unreactive, perhaps for steric hindrance. A mechanism is reported for neither the racemic nor for the asymmetric reaction.
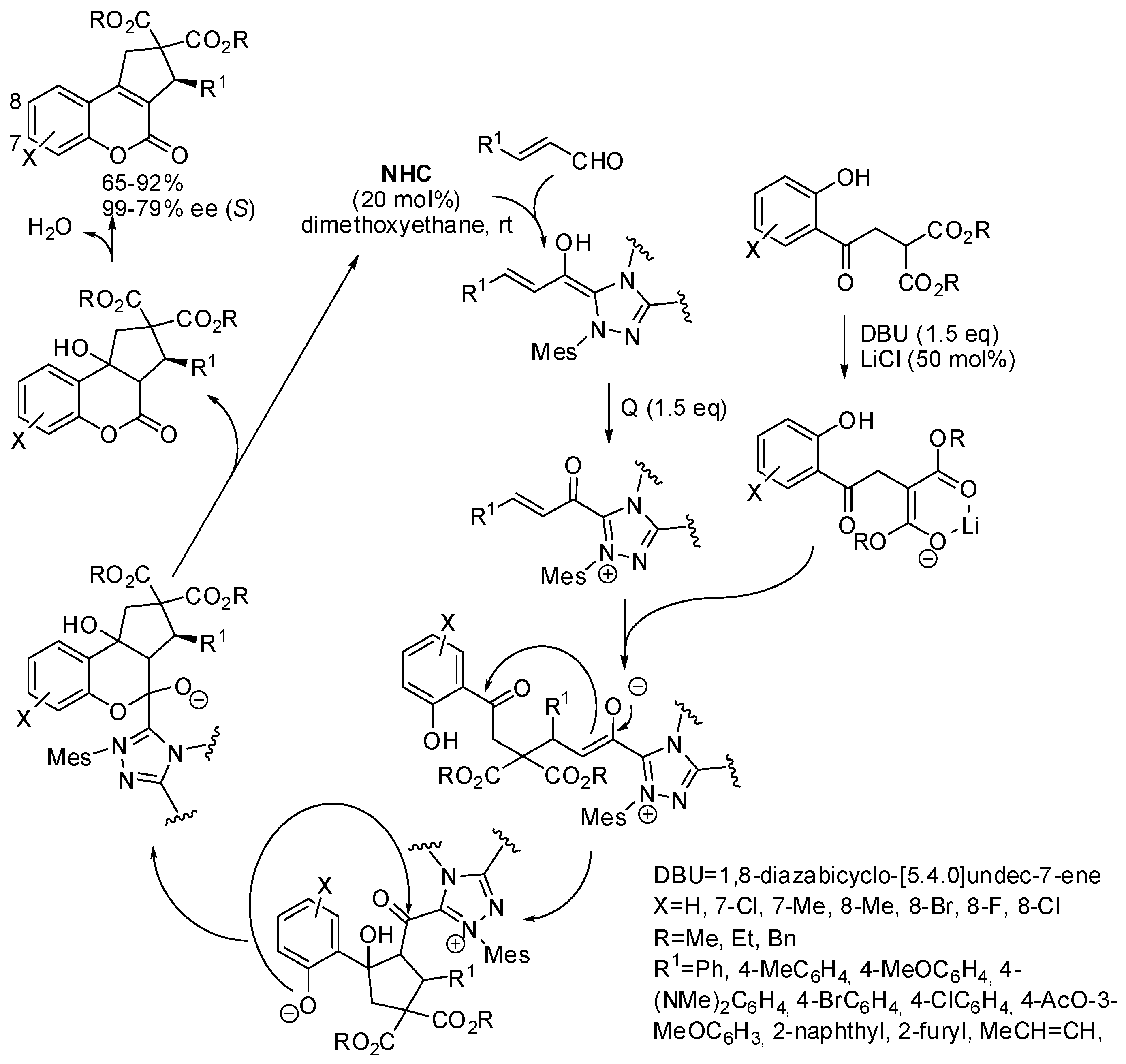
Enantioenriched cyclopenta[c]-fused chromenones have also been synthesized by a cascade reaction between α,β-unsaturated aldehydes with a fourfold reactive reagent such as a malonate bearing an
ortho-hydroxybenzoyl group catalyzed by an
N-heterocyclic carbene (
NHC,
Scheme 20) [
31]. It is worth noting the control of the reactivity of the three nucleophilic and the electrophilic sites of the substrate, which led to a very high chemoselectivity. However, when R
1 = Et or
ortho-substituted phenyl, the reaction did not work. The absolute configuration of compounds was determined by X-ray analysis, and the mechanism depicted in
Scheme 20 was supported by DFT calculations, which demonstrated that the δ-lactonization mechanism is more thermodynamically favored with respect to the β-lactonization.
3. Cycloaddition of ortho-Quinone Methides
Another method for preparing the benzopyran nucleus is the cycloaddition of
o-quinone methides. A recent review was devoted to this topic [
32]. Before describing the more recent papers describing reactions affording chiral chromane derivatives, the organocatalytic dearomative [4+2] cycloadditions of biomass-derived 2,5-dimethylfuran should be mentioned [
33]. This reaction provided two different types of chromanes, and one of these pathways used (-)-camphor sulfonic acid as the catalyst, but the authors reported excellent diasteroselectivities but not enantiomeric excesses.
Chromane [
4] arenes were obtained by the Diels-Alder reaction between the o-quinone methides of resorcin [
4] arene and styrenes [
34]. The authors identified their compounds as chiral, but actually, they were racemic mixtures. However, it should be noted that only two of the possible diastereoisomers (just the C4-symmetric) were formed in 44–88% yield and up to 95:5 dr.
o-Quinone methides can be obtained in situ from
ortho-alkylphenols bearing a leaving group in the benzylic position, by acid or basic catalysis, and then they can undergo [4+2] annulation with allenes. If the catalyst is a chiral acid or base, we can obtain enantioenriched product. In the time range covered by this review, some papers performed this reaction smartly. For example, the reaction with allenoates was reported. Fan and co-workers reported that aryl butadienoates can react with
o-quinone methides generated by fluorine deprotection of 2-silyloxybenzyl chlorides (
Scheme 21) [
35]. Albeit phenyl- and 4-methoxyphenyl-butadienoates gave the expected products, the best yields were obtained with
ortho-substituted aryl butadienoates. However, 2,6-dimethylphenylbutadienoate was unreactive, perhaps due to too-high steric requirements. The absolute configuration was determined by X-ray analysis. It should be noted that 6-arylmethylidenebenzo[
d][1,3]dioxol-5(6
H)-ones are stable, while their precursors are not.
Allene ketones were then allowed to react with
o-quinone methides (
Scheme 22) [
36]. An amino acid-derived bifunctional phosphine was used as the catalyst. Both enantiomers can be obtained by using two different catalysts. However, two different catalysts had to be used when Ar = 4-MeOC
6H
4 or other different Ar groups were present on the quinone methides. The reaction worked on both stable quinone methides and on in situ prepared ones. The (
S)-configuration was determined by X-ray analysis. The reaction was successfully scaled up to 1 mmol. The mechanism depicted in
Scheme 22 was suggested by the authors without experimental evidence or ab initio calculations.
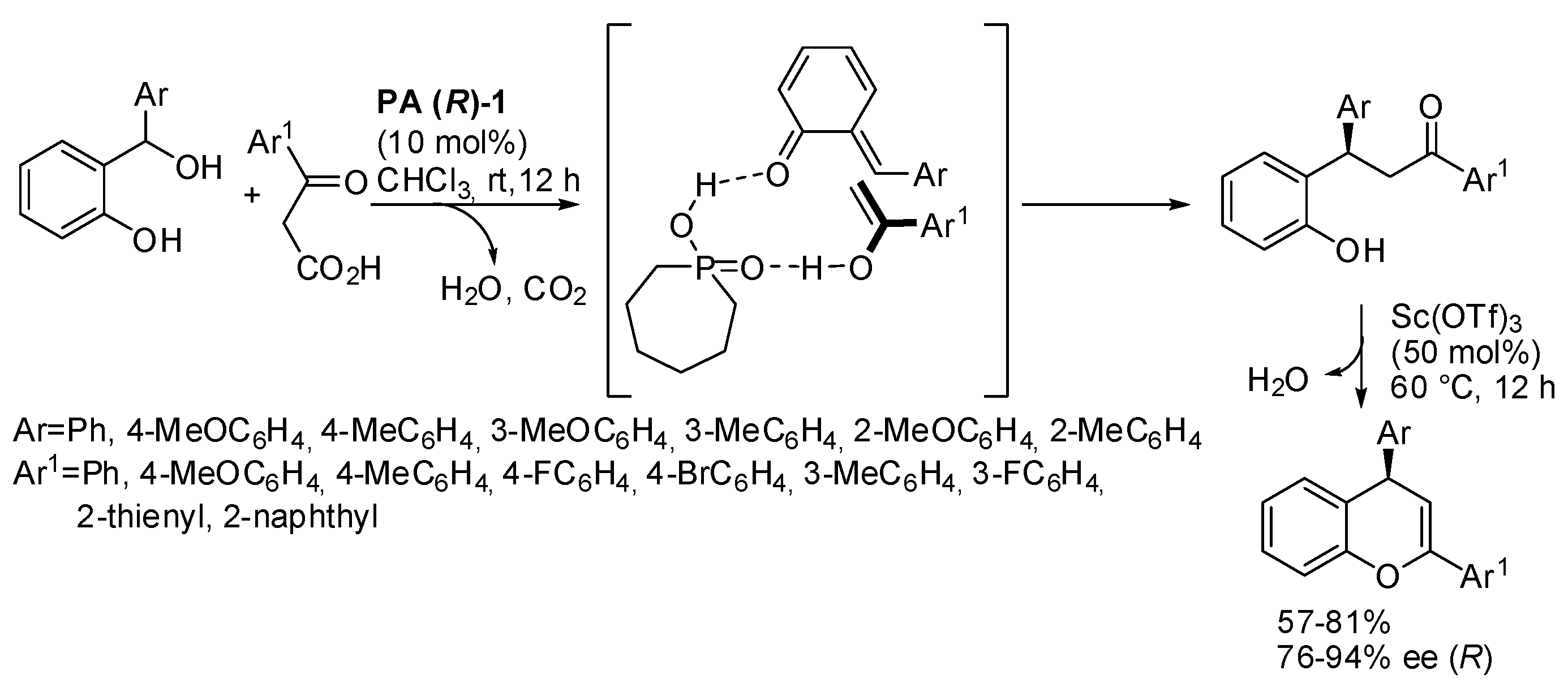
o-Quinone methides, generated in situ by dehydration of the corresponding
o-hydroxy benzhydryl alcohols, have been widely used in reaction with enolizable carbonyl compounds. The common features of these reactions are: (i) chiral phosphoric acid catalysis; (ii) significant amounts of enol formed in the keto-enol tautomerism; (iii) formation of two hydrogen bonds to give a three-membered
o-quinone methide-catalyst-enol intermediate; (iv) this intermediate is responsible of the enantioselectivity. For instance, 2,4-diaryl-1-benzopyrans were obtained from β-keto acids, followed by decarboxylation and dehydration (
Scheme 23) [
37]. The absolute configuration of products was determined by comparison of their optical values with the literature. The reaction was scaled up to gram scale with a 78% yield and 91% ee. The authors surmised that chiral phosphoric acid could also promote both dehydration of the
o-hydroxy benzhydryl alcohols and decarboxylation of the β-keto acids. Then scandium triflate promoted cyclization and dehydration of the lactol intermediate.
Although not organocatalyzed, the synthesis of enantioenriched 4
H-chromenes performed by the group of Schneider catalyzed by a chiral manganese (III) phosphate is worthy of mention here, because it is preparatory to a subsequent organocatalyzed reaction [
38]. The
o-hydroxy benzhydryl alcohol was formed in situ by aerobic oxidation from 2-alkyl-substituted phenols and then dehydrated to
o-quinone methides. Then they reacted with β-dicarbonyl compounds. The same research group prepared
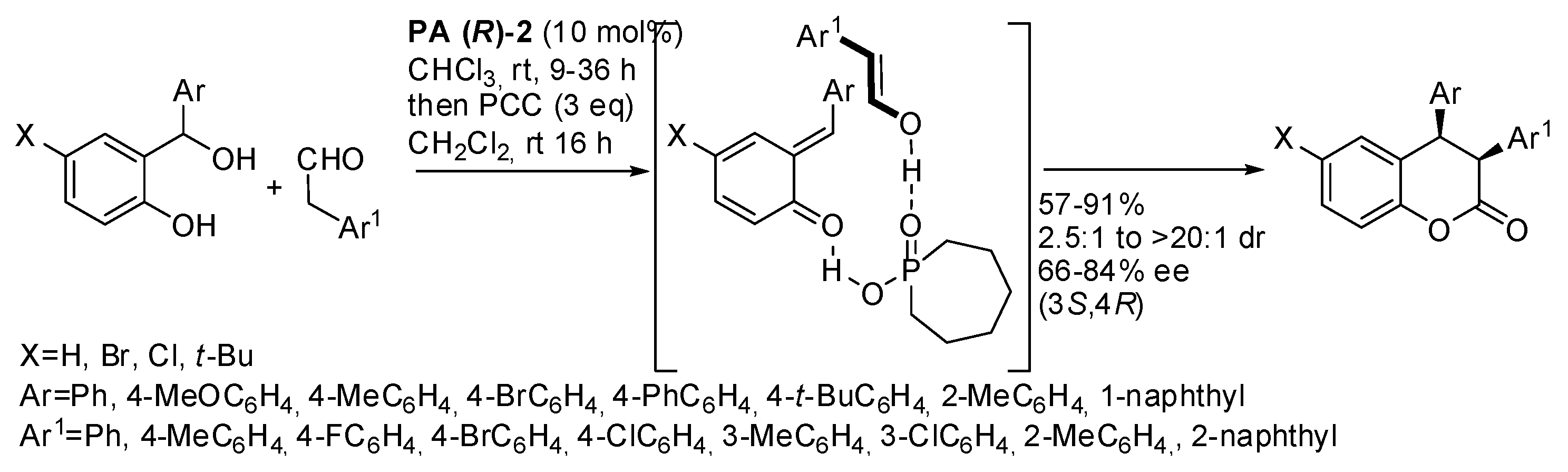
cis-3,4-diarylchromanols through reaction of
o-hydroxybenzhydryl alcohols and aryl acetaldehydes (
Scheme 24) [
39].
Ortho substitution in the aryl group of
o-hydroxybenzhydryl alcohol enhanced the enantioselectivity. The substituted aryl group of
o-hydroxybenzhydryl alcohol gave >20:1
cis/trans ratio, while generally the diasteroselectivity was only 3−4:1. The authors found that the acidic conditions could open the initially obtained lactol so that the
cis/trans ratio was dynamic, but they did not explain why the 4-methoxy group locked the ratio. Aliphatic aldehydes did not react, perhaps because the very low enol content in their keto-enol tautomerism. The lactol intermediate was also transformed into a variety of other useful compounds.
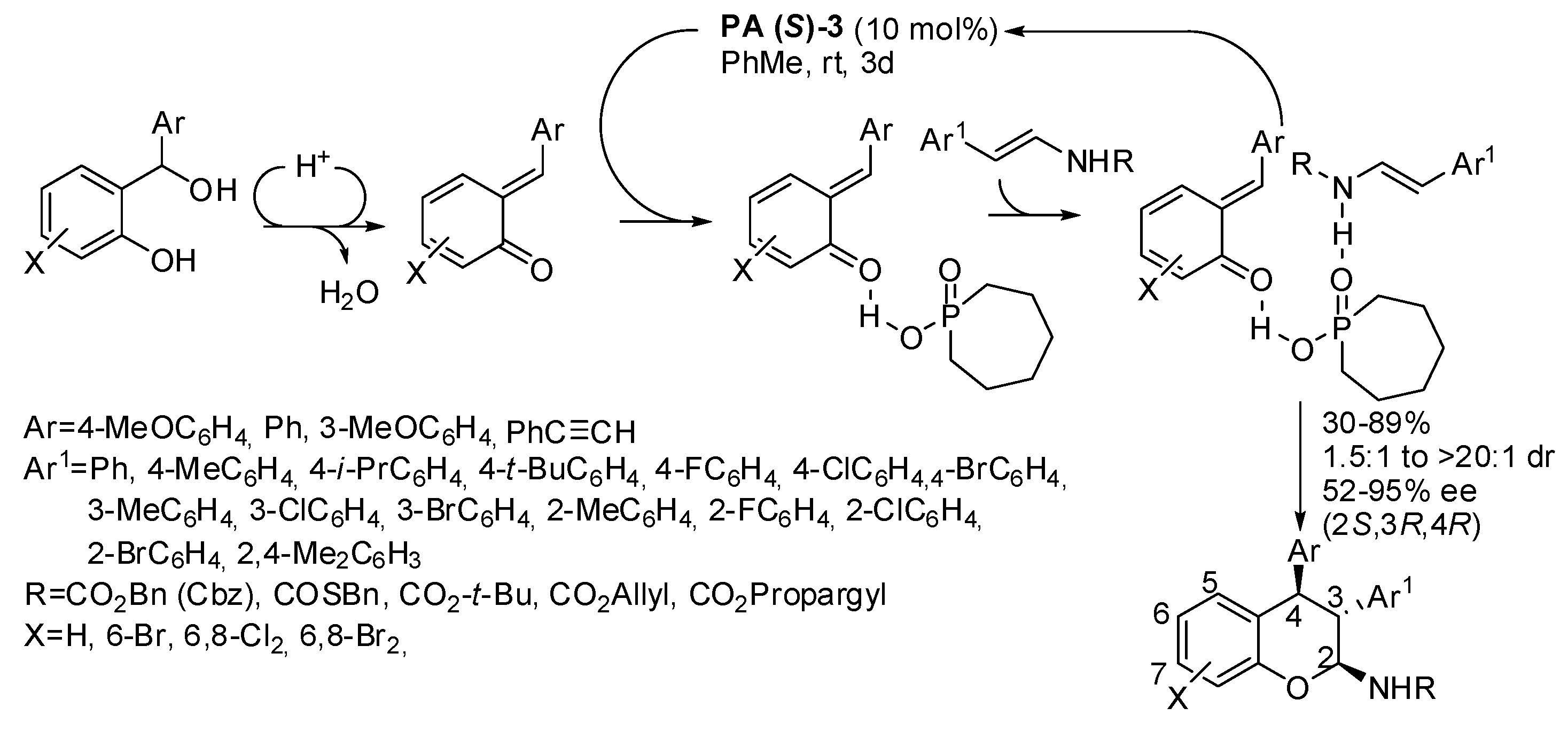
The organocatalytic asymmetric reaction of acyclic enecarbamates with
o-quinone methides led to 2,3,4-trisubstituted chromanes (
Scheme 25) [
40]. The reaction was sluggish when Ar = 4-MeOC
6H
4, X = 6,8-Cl
2, 6,8-Br
2, R = Cbz (only 30–33% yields after 3–6 days at 50 °C). The (2
S,3
R,4
R)-configuration was determined by the Mosher amide method.
Ukis and Schneider performed an intramolecular version of this reaction by involving suitable benzhydryl alcohols (
Scheme 26) [
41]. Simple benzyl alcohols with a double bond at a suitable distance to give stable five- or six-membered rings were unreactive. On the other hand, cinnamyl ether is peculiar to the reaction, because prenyl and phenylpropargylic ethers gave very low enantioselectivity, while other allyl- or vinyl ethers did not react at all. The reactions worked better with (
E)-cinnamyl ethers leading to
exo-trans compounds. Only when the 3′-position (see
Scheme 26) was occupied were the
endo-trans-isomers recovered. When the 3-position (see
Scheme 26) was occupied, the
endo-exo-selectivity was completely lost, very likely due to a fast equilibrium between
E- and
Z-configured
o-quinone methide. The reaction of the (
Z)-cinnamyl ethers led to the exclusive formation of the
endo-cis product, but with very low ee’s. The absolute configuration of products was determined by X-ray analysis. Finally, both (
E)- and (
Z)- products were allowed to react in a large-scale experiment (3.30 mmol): 913 mg (80% yield, 92:8 dr, 92% ee), and 530 mg, (51% yield, single diastereomer, 50% ee) were recovered respectively.
Other enolizable carbonyls were instead tested only with stable
o-quinone methides. For instance, chiral amidine derivative catalyzed tandem Michael addition-lactonization of carboxylic acids and stable
o-quinone methides (
Scheme 27) [
42]. Only diastereomers arising from the reaction of indole-3-acetic acid could be separated by column chromatography, while the other diastereomeric mixtures remained unseparated. The authors found that
N-protected indole gave better results, but only reported two examples. The absolute configuration of products was determined by X-ray analysis. The authors also proposed the mechanism depicted in
Scheme 27, but they did not provide evidence for it.
In other reactions, chiral squaramides with a tertiary amine framework were used as the catalyst. These catalysts can deprotonate the carbonyl compound generating an enolate and link the
o-quinone methides by hydrogen bonds. Thus, the two partners are kept together by ionic and hydrogen bond interactions. The authors often refer to the reaction as a formal asymmetric [4+2] cyclization, but all the mechanisms reported are stepwise (see
Scheme 28 and
Scheme 29 below). One instance was the reaction of enolizable homophthalic anhydrides or a succinic anhydride with
o-quinone methides (
Scheme 28) [
43]. The reaction was scaled up to 1 mmol with a slight decrease in yield while maintaining excellent stereoselectivity. The absolute configuration of products was determined by X-ray analysis.
Aryl β-keto acylpyrazoles and
o-quinone methides also reacted to give
trans-3,4-dihydrocoumarins (
Scheme 29) [
44]. Unfortunately, aliphatic β-keto acylpyrazoles were unreactive. The absolute configuration of products was determined by X-ray analysis. To extend the scope of the reaction, it was also attempted on an
o-quinone methide generated in situ, and it proceeded smoothly, but with a decrease of both yield and enantioselectivity.
o-Quinone methides can be also obtained from salicylaldehydes and they were allowed to react with unsaturated alcohols to give furano- and pyrano-chromanes (
Scheme 30) [
45]. The absolute configuration of products was determined by X-ray analysis. The reaction was also successfully tested with chiral unsaturated alcohols with retention of the configuration of the chiral alcohol. Kinetic resolution from racemic chiral unsaturated alcohols was also performed. Some experiments were carried out to elucidate the mechanism and the result seems to be consistent with a [4+2] addition, but the authors did not rule out a stepwise mechanism.
A particular
p-quinone methide was introduced by Li and co-workers as the substrate for the synthesis of chromane derivatives. It can be in equilibrium with
o-quinone methide structures, which can undergo [4+2] addition as above. However, its reactivity can also be explained in terms of 1,6-conjugated addition, and both of these different mechanisms can explain the reactivity. The isomerization energy was calculated by DFT and was found to be very low (7.0 kcal/mol) [
46], so that all papers generally explained the observed stereochemistry with both mechanisms.
One instance of this reaction made possible the preparation of dihydrocoumarins from azlactones (
Scheme 31) [
47]. The authors scaled their reaction up to gram scale and found that a lower loading of the catalyst (3 mol% vs. 5 mol%) was enough to obtain the product in higher yield 87% vs. 65%) but with little low enantiomeric excess (95% vs. 98%). The products were also submitted to some transformation in order to discover the potentiality of the reaction. Control experiments did not allow author discrimination between the two possible mechanisms. The absolute configuration of products was determined by X-ray analysis.
Another example from the same research group used 1-oxotetralin-2-carbaldehydes and
Squa1 as the catalyst to give spiro-dihydrocoumarins (
Scheme 32) [
48]. All
p-quinone methides gave high diasteroselectivity (>19:1), while the different tested 1-oxotetralin-2-carbaldehydes gave sometimes poor diasteroselectivity (1.5:1 to >19:1). The absolute configuration of products was determined by X-ray analysis. The reaction was scaled up to gram-scale, without significant modification of yield and selectivity. The products were also submitted to some transformation in order to discover the potentiality of the reaction.
Only the detert-butylation with a mixture of Tf2O and TfOH at 60 °C gave racemization, perhaps due to the higher required temperature. In this paper, the authors did not speculate about what mechanism was operative, although in the title the conjugate addition is presupposed.
Another substrate used with
o-hydroxyphenyl-substituted
p-quinone methides is enamides (
Scheme 33) [
46]. All reactions afforded products in high yields and selectivity, except that when X = H, X
1 = 4-MeO, the diastereomeric ratio was only 64:36. The absolute configuration of products was determined by X-ray analysis. A gram-scale reaction gave product in a 96% yield with 95:5 dr and 96% ee.
3-alkyl-2-vinylindoles were also used as reactants in this catalytic asymmetric [4+2] cyclization (
Scheme 34) [
49]. The absolute configuration of products was determined by X-ray analysis. The authors found that NH group of indole is very important, in fact,
N-methyl-protected indole was unreactive. This feature demonstrated that a hydrogen bond between the chiral phosphoric acid and the NH group of indole must be involved in the transition state. The other hydrogen bond could be formed with the OH group of the substrate in a fashion of a 1,6-conjugate addition, or with the OH group of the
o-quinone methide form in a fashion of a [4+2] cycloaddition. Both hydrogen bonds can explain the observed stereochemistry. The reaction was scaled up to gram scale, and product was recovered in 96% yield with >95:5 dr and 96% ee.
Finally, unsaturated isoxazolones were used as the partner in the reaction with
o-hydroxyphenyl substituted
p-quinone methides affording spiro-isoxazolonechromans [
50]. The paper mainly describes the classical reaction and authors surmised a domino oxa-Michael/1,6-addition reactions, rather than a [4+2] cycloaddition. Only at the end of the paper is an example of the asymmetric version reported with quinine as the chiral catalyst, but information about the absolute configuration of this enantioenriched product is furnished. However, 4-(3,5-di-
tert-butyl-4-hydroxyphenyl)-3′-methyl-2-(
p-tolyl)-5′H-spiro[chromane-3,4′-isoxazol]5′-one was recovered in 82% yield with unsatisfactory diasteroselectivity and enantioselectivity (21:79 dr, 11/48% ee).
5. Miscellaneous
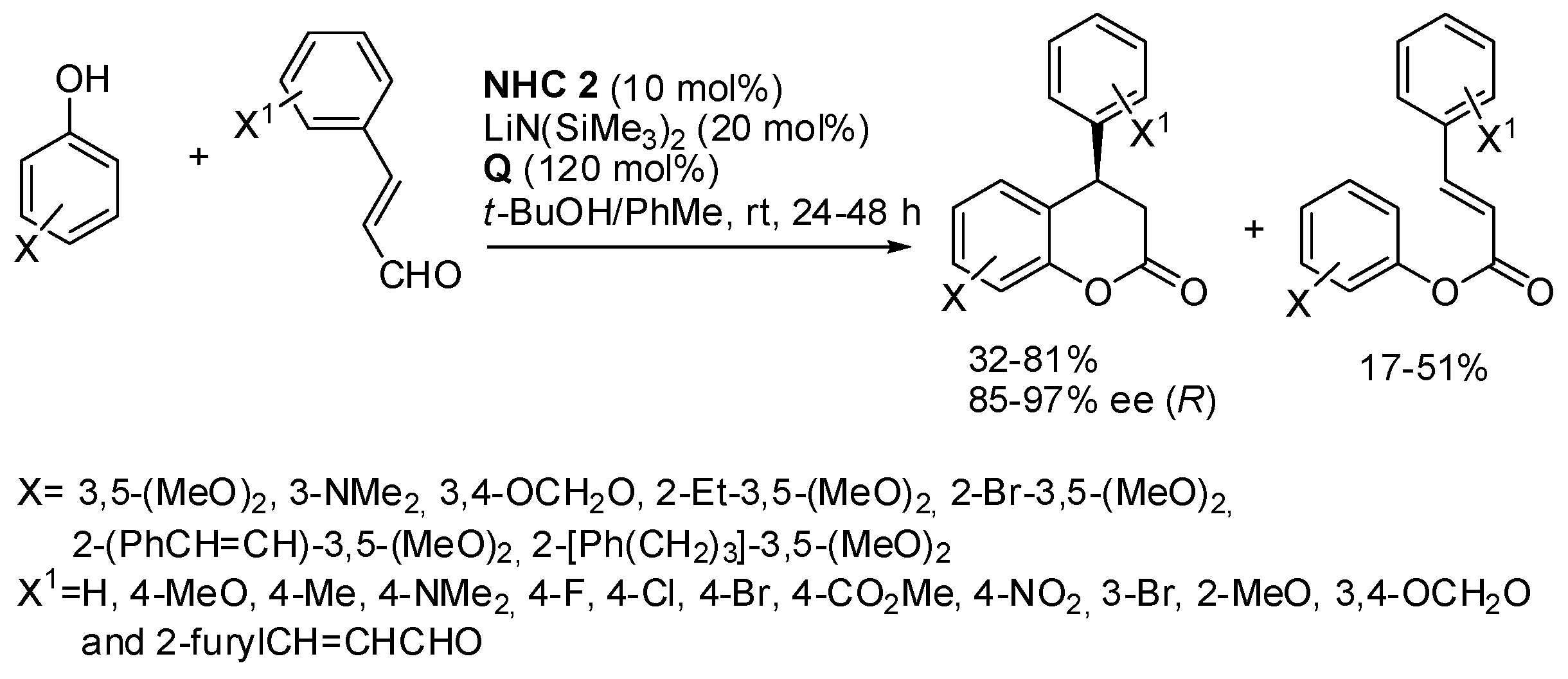
Other reactions have been used to obtain the benzopyran skeleton that could not be listed in the previous main sections. Therefore, they are discussed in the present section. One example is the enantioselective synthesis of 4-aryl-3,4-dihydrocoumarins by reaction of phenols with α,β-unsaturated arylaldehydes, catalyzed by NHC and with 3,3′,5,5′-tetra-
t-butyldiphenoquinone as the oxidant (
Scheme 38) [
54]. In contrast to the reaction described in
Scheme 20 [
29], simple electron-donating substituted phenols are reacted here. However, all reactions give a mixture of the desired products and open-chain byproducts. The absolute configuration of products was determined by X-ray analysis. α,β-Unsaturated aldehydes such as crotonaldehyde gave a sluggish reaction. Some enantioenriched products were submitted to transformation to key intermediates for the synthesis of natural products.
The group of Andrés reported the reaction of 2-hydroxy
N-Boc-α-amidosulfones with
trans-β-nitrostyrenes or amidosulfones by a cascade oxa-Michael-aza-Henry reaction and a cascade Mannich cyclization-tautomerization, respectively (
Scheme 39) [
55]. The authors found thioureas to be more efficient catalysts for the first reaction and squaramides for the second one. Moreover, in the second reaction, the enantiomeric compounds were also prepared by the use of
Squa5 as the catalyst (88–96% yield with 86–94% ee). The catalysts were also co-polymerized with styrene divinylbenzene and polymeric catalysts were tested with good results (91–96% yield with 82:18 to 99:1 dr and 86– >99% ee for polymeric thiourea; 78–84% yield, with 78–88% ee for polymeric squaramide). Polymeric catalysts were recovered and reused five times without loss of efficiency. The authors explained the difference in the catalytic activity of thioureas and squaramides in these reactions on the basis of the different distance of the two hydrogen atoms able to give hydrogen bonds, so that the nitro group and the acyl imine functionality were better accommodated by thiourea and squaramide moieties, respectively. A mechanism was also hypothesized in which 2-hydroxy
N-Boc imine was initially formed by the aqueous solution of K
2CO
3, and then the hydrogen bods were formed and the tertiary amines deprotonated the phenol framework.
Li, Liu and co-workers prepared a series of spiro-, dispiro-, fused, and bridged heterocycles by the reaction of quinones and 3-keto-oxindoles (
Scheme 40) [
56]. The absolute configuration of products was determined by X-ray analysis. Some control experiments enabled the understanding that the free hydroxy group of the catalyst played a key role in the catalytic process (the methoxy derivative is much more efficient) and that the two spiroxindoles arose both from the hemiacetal intermediate by dehydration or ketalization, respectively.
Disubstituted Meldrum’s acids can be used as an alternative to malonic esters. In particular, if the
ortho-hydroxyphenyl group is one of the substituents, an intramolecular cyclization/decarboxylation can occur (
Scheme 41) [
57]. Actually, these phenols were difficult to purify, and thus the corresponding pure silyl ethers were in situ desylilated. When the R group is a 4-methoxy substituted benzyl moiety, the reaction time was prolonged to 60 h in order to increase yield from 61 to 99%. The absolute configuration of products was determined by X-ray analysis. The reduction of the carbonyl group provided (
S)-3-benzylchromane without affecting enantiomeric excess. Moreover, chlorination instead of protonation was also performed. However, some amounts of protonated by-product were found (from 83:17 to 80:20), but the authors proved that chlorinated product did not arise from it. The absolute configuration was not determined for these compounds.
In the previous sections, salicylaldehydes have been already used as a starting material for the synthesis of chromane derivatives (see
Scheme 19 [
30] and
Scheme 30 [
45]). Here it is worth mentioning the reaction of salicylaldehydes with 3-
C-vinyl sugar nitro olefins prepared from glucofuranose diisopropylidine/dicyclohexylidine [
58]. Actually, this is not an organocatalyzed reaction, but it can be ascribed to the chiral pool reactions. In fact, the exclusive formation of the new chiral center in the C2 position of the chromane ring in the
S configuration arose from the chiral sugar and not from the catalyst, which is triethylamine. However, the reaction ended in very short times (8–15 min), affording 2-
C-spiro-glycosyl-3-nitrochromenes in 60–90% yields under neat conditions.
Finally, naphthopyran is another skeleton widely found in natural products. In recent years, its assembly has been made to lead to both racemic and enantioenriched products [
59,
60,
61,
62,
63], but the direct asymmetric assembly of tetrahydro-3
H-benzo[
f]chromen-3-one derivatives has been reported only very recently by the
N-heterocyclic carbene-catalyzed formal [3+3] annulation of bromoenal and β-tetralone and β-indanone (
Scheme 42) [
64]. However, 2-bromobut-2-enal and α-bromoenals containing heterocycles as well as α-tetralone did not give the expected products. The absolute configuration was assigned observing an opposite optical rotation of products with respect to that reported in the literature [
60]. The authors envisaged the possible mechanism depicted in
Scheme 42.
Finally, a DFT study of the mechanism of asymmetric intramolecular nucleophilic substitution of
ortho-allyloxy benzaldehydes catalyzed by
NHC should be mentioned [
65]. The following stages were found: (i) the combination of
NHC with the substrate from the Si face; (ii) the SN2′ reaction, which is the rate- and enantioselectivity-determining step giving the stereocenter in the favored
R configuration; and (iii) the release of the product.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










































