

Revealing the Gene Diversity and Candidate Gene Family for Adaption to Environment Depth in Leucosiid Crabs Comparing the Transcriptome Assembly

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and RNA Preparation
2.2. Illumina RNA-Seq Library Preparation, Sequencing and Assembly
2.3. PacBio cDNA Library Construction and Single-Molecule Real-Time (SMRT) Sequencing
2.4. Sequencing Data Treatment and Annotation
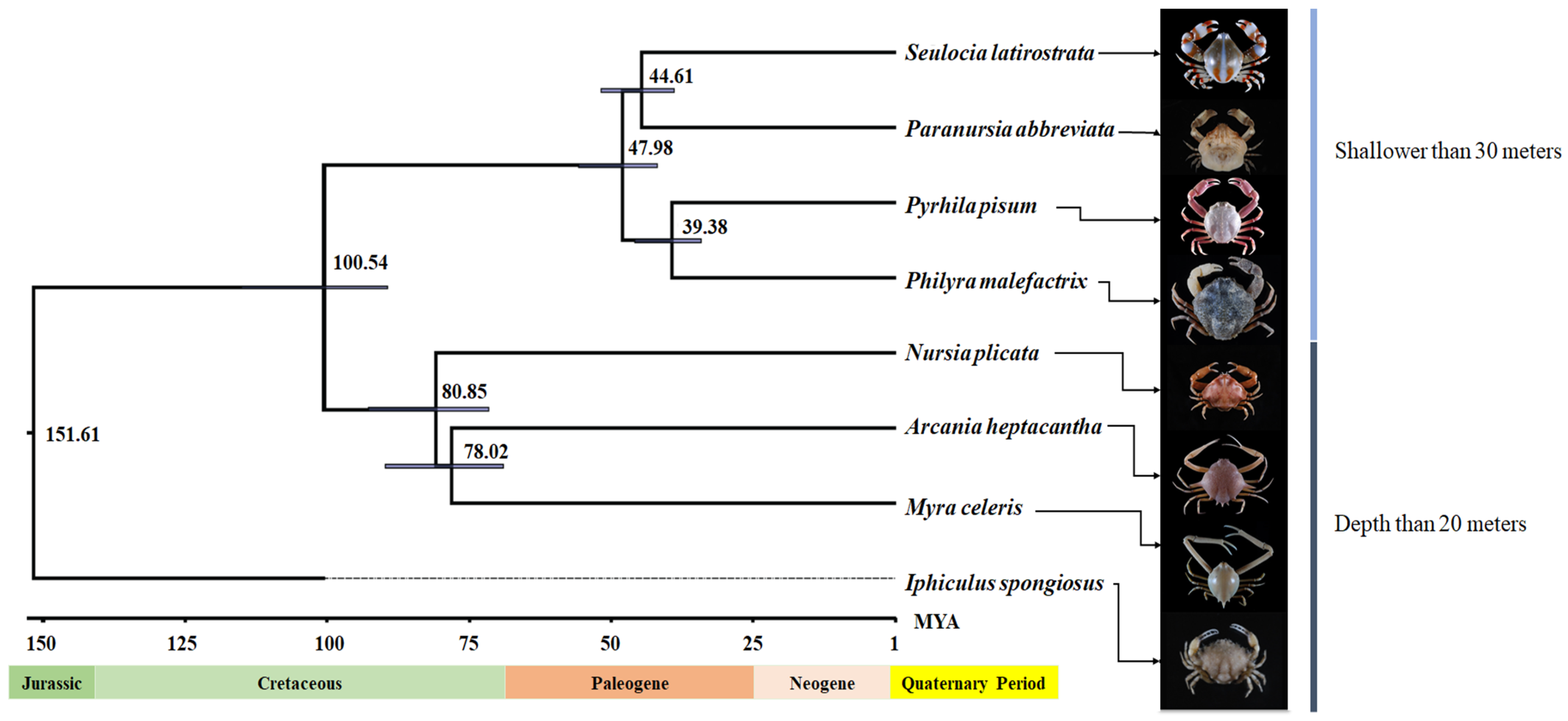
2.5. Analysis of Phylogenetic Cladogram and Divergence Time Estimation
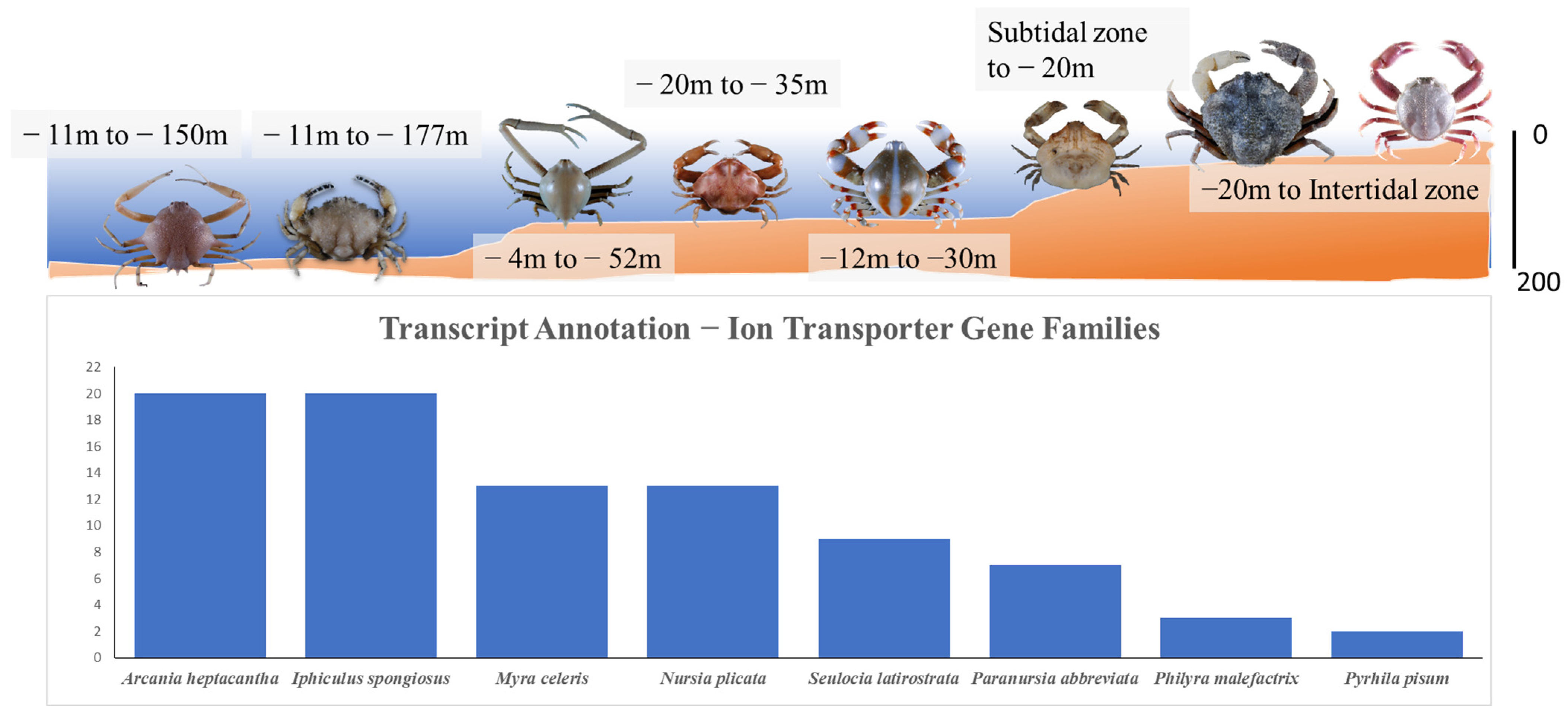
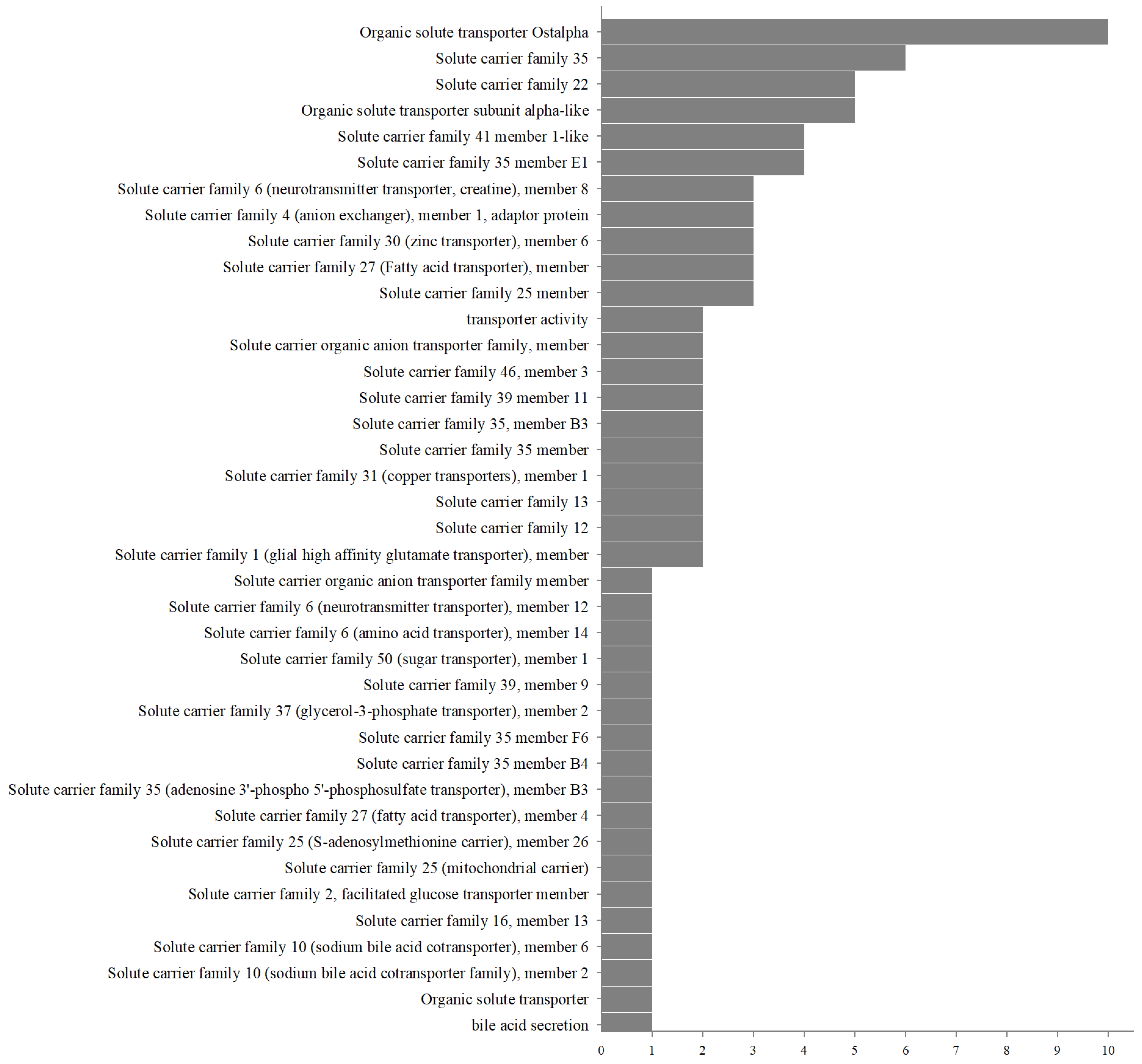
2.6. Analysis and Annotation of Ion Transporter Gene Families
3. Results and Discussion
3.1. The Results of the Eight Species’ De Novo Transcriptome
3.2. Phylogenetic Cladogram and Divergence Time Estimation
3.3. Ion Transporter Gene Families in Eight Leucosiid Species
3.4. Deposition of Samples and Transcript Data
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Noss, R.F.; Cooperrider, A. Saving Nature’s Legacy: Protecting and Restoring Biodiversity; Island Press: Washington, DC, USA, 1994. [Google Scholar]
- Sahney, S.; Benton, M.; Ferry, P.A. Links between global taxonomic diversity, ecological diversity and the expansion of vertebrates on land. Biol. Lett. 2010, 6, 544–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, T.J.; Shih, Y.J.; Shih, C.H.; Wang, J.Q.; Huang, L.M.; Tsai, S.C. Developing a model to select indicator species based on individual species’ contributions to biodiversity. Appl. Sci. 2022, 12, 6748. [Google Scholar] [CrossRef]
- Campbell, A.K. Save those molecules! Molecular biodiversity and life. J. Appl. Ecol. 2003, 40, 193–203. [Google Scholar] [CrossRef]
- Zhang, X.; Xue, Y.; Zhang, C.; Ren, Y.; Xu, B.; Chen, Y. Sampling intensity influences the estimation of functional diversity indices of fish communities. Ecol. Indic. 2021, 121, 107169. [Google Scholar] [CrossRef]
- Levin, B.A.; Komarova, A.S.; Rozanova, O.L.; Golubtsov, A.S. Unexpected diversity of feeding modes among chisel-mouthed ethiopian Labeobarbus (Cyprinidae). Water 2021, 13, 2345. [Google Scholar] [CrossRef]
- Jump, A.S.; Marchant, R.; Peñuelas, J. Environmental change and the option value of genetic diversity. Trends Plant Sci. 2009, 14, 51–58. [Google Scholar] [CrossRef]
- Avise, J.C. Molecular Markers, Natural History and Evolution; Chapman & Hall: New York, NY, USA, 1994; 511p. [Google Scholar]
- Avise, J.C. Mitochondrial DNA polymorphism and a connection between genetics and demography of relevance to conservation. Cons. Biol. 1995, 9, 686–690. [Google Scholar] [CrossRef]
- Hillis, D.M.; Moritz, C.; Mable, B.K. Molecular Systematics, 2nd ed.; Sinauer: Sunderland, MA, USA, 1996. [Google Scholar]
- Siepel, A. Phylogenomics of primates and their ancestral populations. Genome Res. 2009, 19, 1929–1941. [Google Scholar] [CrossRef] [Green Version]
- Kotov, A.A.; Van Damme, K. Species richness and diversity of aquatic ecosystems: Lessons from a special issue. Water 2022, 14, 2904. [Google Scholar] [CrossRef]
- Ng, P.K.L.; Shih, S.T.; Ho, P.H.; Wang, C.H. An updated annotated checklist of brachyuran crabs from Taiwan (Crustacea: Decapoda). J. Taiwan Mus. 2017, 70, 1–185. [Google Scholar] [CrossRef]
- Ng, P.K.L.; Guinot, D.; Davie, P.J.F. Systema Brachyurorum: Part I. An annotated checklist of extant brachyuran crabs of the world. Raffles Bull. Zool. 2008, 17, 1–286. [Google Scholar]
- Tsang, L.M.; Schubart, C.D.; Ahyong, S.T.; Lai, J.C.Y.; Au, E.Y.; Chan, T.Y.; Ng, P.K.L.; Chu, K.H. Evolutionary history of true crabs (Crustacea: Decapoda: Brachyura) and the origin of freshwater crabs. Mol. Biol. Evol. 2014, 31, 1173–1187. [Google Scholar] [CrossRef]
- Chu, T.J.; Shih, C.H.; Lu, Y.M.; Shih, Y.J.; Wang, J.Q.; Huang, L.M. Incorporating Species-Conditional Co-Occurrence When Selecting Indicator Species to Monitor Restoration after Mangrove Removal from the Siangshan Wetland, Taiwan. J. Mar. Sci. Eng. 2021, 9, 1044. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, J.; Wu, Q.; Xu, X.I.; Wang, P.; Wang, Z.F. Insights into the evolution of Brachyura (Crustacea: Decapoda) from mitochondrial sequences and gene order rearrangements. Int. J. Biol. Macromol. 2021, 170, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Ahyong, S.T.; Lai, J.C.Y.; Sharkey, D.; Colgan, D.J.; Ng, P.K.L. Phylogenetics of the brachyuran crabs (Crustacea: Decapoda): The status of Podotremata based on small subunit nuclear ribosomal RNA. Mol. Phylogenet. Evol. 2007, 45, 576–586. [Google Scholar] [CrossRef]
- Davie, P.J.; Guinot, D.; Ng, P.K.L. Phylogeny of Brachyura; Brill: Leiden, The Netherlands, 2015. [Google Scholar]
- Huang, Y.H.; Shih, H.T. Diversity in the Taiwanese swimming crabs (Crustacea: Brachyura: Portunidae) estimated through DNA barcodes, with descriptions of 14 new records. Zool. Stud. 2021, 60, 60. [Google Scholar] [CrossRef]
- Xu, X.Y.; Wang, Q.; Wu, Q.; Xu, J.Y.; Wang, J.; Wang, Z.F. The entire mitochondrial genome of Macrophthalmus abbreviatus reveals insights into the phylogeny and gene rearrangements of Brachyura. Biochem. Genet. 2021, 59, 617–636. [Google Scholar] [CrossRef] [PubMed]
- Grover, C.E.; Salmon, A.; Wendel, J.F. Targeted sequence capture as a powerful tool for evolutionary analysis. Am. J. Bot. 2012, 99, 312–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andermann, T.; Torres Jiménez, M.F.; Matos-Maraví, P.; Batista, R.; Blanco-Pastor, J.L.; Gustafsson, A.L.S.; Kistler, L.; Liberal, I.M.; Oxelman, B.; Bacon, C.D.; et al. A guide to carrying out a phylogenomic target sequence capture project. Front. Genet. 2020, 10, 1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, S.; Yu, X.; Chen, H.; Zhang, Y.; Wu, Q.; Tan, H.; Song, J.; Saqib, H.S.A.; Farhadi, A.; Ikhwanuddin, M.; et al. Full-Length Transcriptome Reconstruction Reveals the Genetic Mechanisms of Eyestalk Displacement and Its Potential Implications on the Interspecific Hybrid Crab (Scylla serrata × S. paramamosain). Biology 2022, 11, 1026. [Google Scholar] [CrossRef]
- Farrelly, C.A.; Greenaway, P. Morphology and ultrastructure of the gills of terrestrial crabs (Crustacea, Gecarcinidae and Grapsidae): Adaptations for air-breathing. Zoomorphology 1992, 112, 39–49. [Google Scholar] [CrossRef]
- Greenaway, P. Terrestrial adaptations in the anomura (Crustacea: Decapoda). Mem. Mus. Vic. 2003, 60, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Watson-Zink, V.M. Making the grade: Physiological adaptations to terrestrial environments in decapod crabs. Arthropod Struct. Dev. 2021, 64, 101089. [Google Scholar] [CrossRef] [PubMed]
- Jahn, M.P.; Cavagni, G.M.; Kaiser, D.; Kucharski, L.C. Osmotic effect of choline and glycine betaine on the gills and hepatopancreas of the Chasmagnathus granulata crab submitted to hyperosmotic stress. J. Exp. Mar. Biol. Ecol. 2006, 334, 1–9. [Google Scholar] [CrossRef]
- Lee, C.E. Ion transporter gene families as physiological targets of natural selection during salinity transitions in a copepod. Physiology 2021, 36, 335–349. [Google Scholar] [CrossRef]
- Pizzagalli, M.D.; Bensimon, A.; Superti-Furga, G. A guide to plasma membrane solute carrier proteins. FEBS J. 2021, 288, 2784–2835. [Google Scholar] [CrossRef]
- Shih, Y.J.; Ho, P.H.; Chan, T.Y. Leucosiid crabs (Crustacea: Decapoda: Brachyura) from Taiwan, with three new records. Zootaxa 2015, 4052, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Shih, Y.J.; Ho, P.H.; Jeng, M.S. Two new records of the leucosioid genus Pariphiculus Alcock, 1896 from Taiwan (Decapoda, Brachyura, Iphiculidae). Crustaceana 2017, 90, 1367–1371. [Google Scholar] [CrossRef]
- Shih, Y.J.; Ho, P.H.; Galil, B.S. Contributions to the knowledge of Leucosiidae VII. Liusius gen. nov.(Crustacea, Brachyura). Crustaceana 2020, 93, 1269–1276. [Google Scholar] [CrossRef]
- Miyake, K.; Baba, Y. De novo transcriptome assembly of the midgut glands of herbivorous land crabs, Chiromantes haematocheir, and identification of laccase genes involved in lignin degradation. J. Comp. Physiol. B 2022, 192, 247–261. [Google Scholar] [CrossRef]
- Andersen, Ø.; Johnsen, H.; Wittmann, A.C.; Harms, L.; Thesslund, T.; Berg, R.S.; Siikavuopio, S.; Mykles, D.L. De novo transcriptome assemblies of red king crab (Paralithodes camtschaticus) and snow crab (Chionoecetes opilio) molting gland and eyestalk ganglia-Temperature effects on expression of molting and growth regulatory genes in adult red king crab. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2022, 257, 110678. [Google Scholar] [CrossRef] [PubMed]
- Pfenninger, M.; Schönnenbeck, P.; Schell, T. ModEst: Accurate estimation of genome size from next generation sequencing data. Mol. Ecol. Resour. 2022, 22, 1454–1464. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. GPB 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calsbeek, R.; Thompson, J.N.; Richardson, J.E. Patterns of molecular evolution and diversification in a biodiversity hotspot: The California Floristic Province. Mol. Ecol. 2003, 12, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Karasawa, H.; Schweitzer, C.E.; Feldmann, R.M. Phylogenetic analysis and revised classification of podotrematous Brachyura (Decapoda) including extinct and extant families. J. Crustac. Biol. 2011, 31, 523–565. [Google Scholar] [CrossRef] [Green Version]
- Benson, J.; Stewart, B.; Close, P.; Lymbery, A. Evidence for multiple refugia and hotspots of genetic diversity for Westralunio carteri, a threatened freshwater mussel in south-western Australia. Aquat. Conserv. 2022, 32, 559–575. [Google Scholar] [CrossRef]
- Frusher, S.D.; Giddins, R.L.; Smith, T.J. Distribution and abundance of grapsid crabs (Grapsidae) in a mangrove estuary: Effects of sediment characteristics, salinity tolerances, and osmoregulatory ability. Estuaries 1994, 17, 647–654. [Google Scholar] [CrossRef]
- Yang, Z.; Zhu, L.; Zhao, X.; Cheng, Y. Effects of salinity stress on osmotic pressure, free amino acids, and immune-associated parameters of the juvenile Chinese mitten crab, Eriocheir sinensis. Aquac. 2022, 549, 737776. [Google Scholar] [CrossRef]
- Heiss, J.W.; Michael, H.A. Saltwater-freshwater mixing dynamics in a sandy beach aquifer over tidal, spring-neap, and seasonal cycles. Water Resour. Res. 2014, 50, 6747–6766. [Google Scholar] [CrossRef]
- Yancey, P.H. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 2005, 208, 2819–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Wiel, S.M.; Porteiro, B.; Belt, S.C.; Vogels, E.W.; Bolt, I.; Vermeulen, J.L.; de Waart, D.R.; Verheij, J.; Muncan, V.; Oude Elferink, R.P.J.; et al. Differential and organ-specific functions of organic solute transporter α and β in experimental cholestasis. JHEP Rep. 2022, 4, 100463. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Gaytán, M.F.; Gómez-Jiménez, S.; Gámez-Alejo, L.A.; Rosas-Rodríguez, J.A.; Figueroa-Soto, C.G.; Valenzuela-Soto, E.M. Effect of salinity on the synthesis and concentration of glycine betaine in osmoregulatory tissues from juvenile shrimps Litopenaeus vannamei. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2020, 240, 110628. [Google Scholar] [CrossRef]
- Taugbøl, A.; Solbakken, M.H.; Jakobsen, K.S.; Vøllestad, L.A. Salinity-induced transcriptome profiles in marine and freshwater threespine stickleback after an abrupt 6-hour exposure. Ecol. Evol. 2022, 12, e9395. [Google Scholar] [CrossRef] [PubMed]
- Mohindra, V.; Chowdhury, L.M.; Chauhan, N.; Paul, A.; Singh, R.K.; Kushwaha, B.; Maurya, R.K.; Lal, K.K.; Jena, J.K. Transcriptome Analysis Revealed Osmoregulation Related Regulatory Networks and Hub Genes in the Gills of Hilsa shad, Tenualosa ilisha, during the Migratory Osmotic Stress. Mar. Biotechnol. 2023, 25, 161–173. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| No. | Species | Method | Total Unigene Sequences | Total Length (bp) |
|---|---|---|---|---|
| 1 | Iphiculus spongiosus | PacBio SMRT sequencing | 73,809 | 56,483,844 |
| 2 | Arcania heptacantha | Next-Generation Sequencing | 98,279 | 95,471,088 |
| 3 | Myra celeris | Next-Generation Sequencing | 94,664 | 83,854,763 |
| 4 | Nursia plicata | Next-Generation Sequencing | 76,236 | 71,668,640 |
| 5 | Paranursia abbreviata | Next-Generation Sequencing | 85,700 | 52,623,429 |
| 6 | Philyra malefactrix | Next-Generation Sequencing | 65,617 | 48,006,211 |
| 7 | Pyrhila pisum | Next-Generation Sequencing | 73,709 | 53,931,646 |
| 8 | Seulocia latirostrata | Next-Generation Sequencing | 69,054 | 67,046,175 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shih, Y.-J.; Yang, Y.-M.; Luo, S.-T.; Liu, J.-Y. Revealing the Gene Diversity and Candidate Gene Family for Adaption to Environment Depth in Leucosiid Crabs Comparing the Transcriptome Assembly. Water 2023, 15, 1246. https://doi.org/10.3390/w15061246
Shih Y-J, Yang Y-M, Luo S-T, Liu J-Y. Revealing the Gene Diversity and Candidate Gene Family for Adaption to Environment Depth in Leucosiid Crabs Comparing the Transcriptome Assembly. Water. 2023; 15(6):1246. https://doi.org/10.3390/w15061246
Chicago/Turabian StyleShih, Yi-Jia, Yin-Ming Yang, Si-Te Luo, and Jia-Ying Liu. 2023. "Revealing the Gene Diversity and Candidate Gene Family for Adaption to Environment Depth in Leucosiid Crabs Comparing the Transcriptome Assembly" Water 15, no. 6: 1246. https://doi.org/10.3390/w15061246