3.1. Result of the Ball Milling: Size Parameters
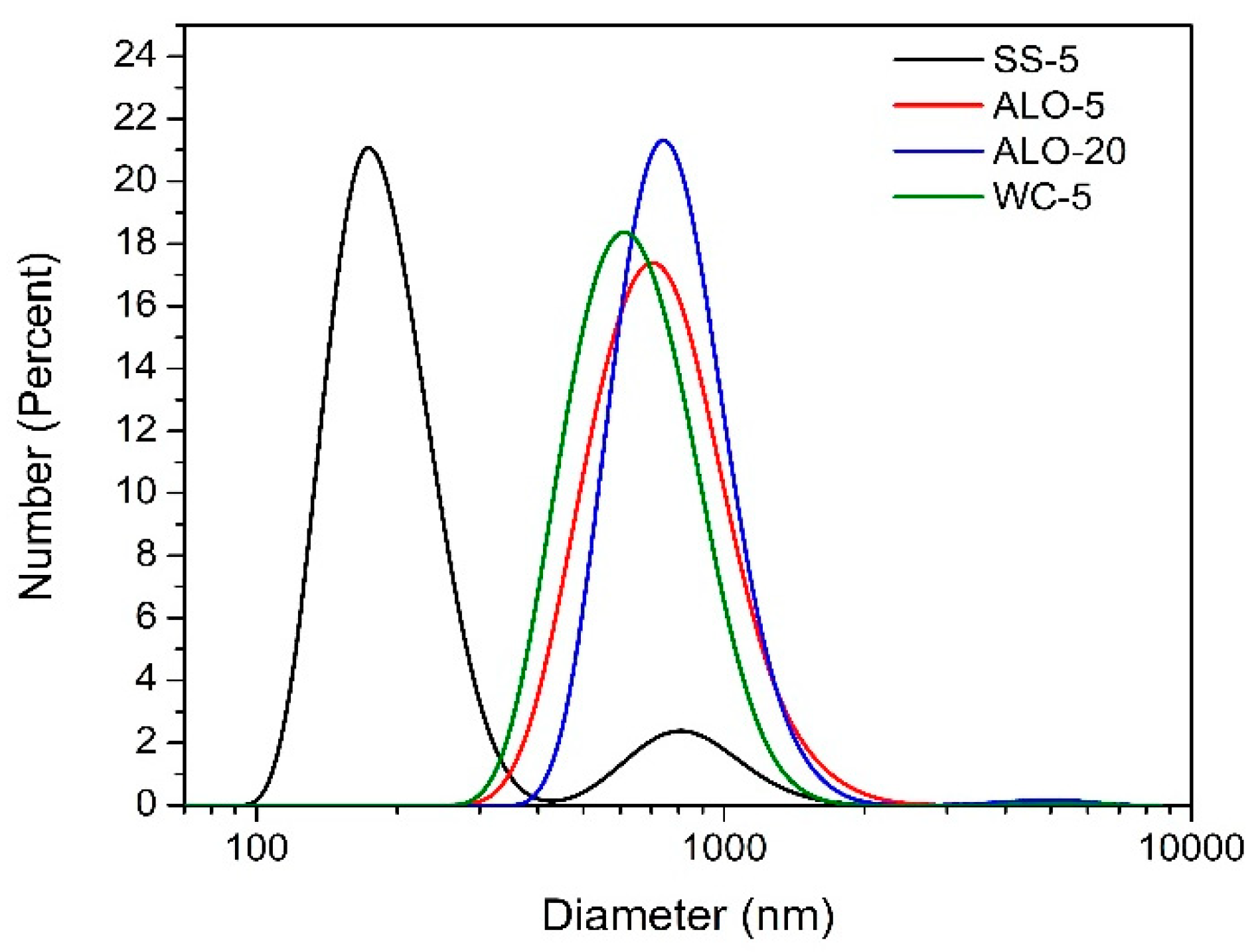
Figure 1 shows the as-received particle diameter distributions of the ground samples determined by the DLS method. All of them have a broad distribution in the range of a few hundred nanometers while the smallest particle diameter can be achieved by the ball-milling in stainless steel vial. Ball-milling in alumina or tungsten carbide vial resulted in similar particle diameter distributions which did not change considerably if a milling time longer than 5 h was chosen.
Since the samples darkened as a result of the grinding process, it was checked whether the greyish color was related to sample contamination originating from the ball or the vial. EDS results showed that no characteristic impurity element at
x > 0.1 at% concentration was present in the LN powders ground in either the tungsten carbide or the stainless steel vials, while aluminum could be detected when alumina vial was used in the milling process. This also means that the large fraction corresponding to smaller particle sizes observed in sample SS-5 milled in the stainless steel vial (see
Figure 1) cannot be ascribed to the presence of an impurity but has to be considered as LN, too.
Neither the occurrence of alumina contamination nor the absence of any stainless steel residue can be explained on the basis of the hardness properties of the materials involved in the ball milling process. Taking into account the order of the Vickers hardness values of the relevant materials (stainless steel: 200–240, LN: ~630 [
24], tungsten carbide: 1200–1700, alumina: 1400–1900), one might expect that stainless steel would cause the largest impurity level. However, alumina was the only vial material resulting in sample contamination, although it was the hardest material used. Al contamination was also confirmed by the XRD measurements as indicated by the presence of α-Al
2O
3 in the XRD pattern. This phenomenon was also proved by Heitjans and co-workers [
22]. Although the alumina impurity level was sufficiently high for both XRD and EDS detection (at least 2 vol% which was the detection limit of the XRD method), it did not appear as a discernible fragment in the size distribution curves of the samples ALO-5 and ALO-20 (see
Figure 1).
Although the alumina contamination cannot be explained on the basis of the hardness of the unmodified materials, it is understood by taking into account the mechanochemical reactions taking place in the system. As it will be shown in the forthcoming sections, lithium oxide is released from LN during the milling process that can react with the amphoteric alumina, resulting in the modification of its structure and facilitating its degradation. Various mixed oxide compounds of aluminum and lithium are known (such as LiAlO
2, LiAl
5O
8, Li
3AlO
4; see Ref. [
25]). In particular, the synthesis of LiAlO
2 is well described by using either sol-based [
26] or solid phase reaction-based [
27] routes. Although the quantitative yield of the solid-phase reaction is given for temperatures higher than 370 °C, a surface-limited reaction can be assumed for the milling conditions applied in the present work. The hardness data of LiAlO
2 is not known to the authors, but the Vickers hardness of other alkali aluminates (Na or K) is reported to be quite small (< 100, see [
28]). Hence, the sites where the released Li
2O reacts with the Al
2O
3 ball/vials can be assumed to serve as degradation initiation spots during the milling process.
While the particle size of a few hundred nm observed by light scattering did not diminish further for milling times longer than 5 h, a Williamson-Hall type evaluation of the X-ray diffractograms resulted in mean grain sizes decreasing from at about 63 nm to 37 nm for milling times increasing from 5 to 20 h (see
Table 2). This reflects the fact that the diffraction-based size analysis provides the diameter of the coherently scattering grains which differs from the much larger particle diameter visible by light scattering or imaging methods. This is direct evidence that the particles formed during high-energy milling consist of a multitude of small grains. The grain diameters found in the present work are in good agreement with those reported before [
13,
15,
20,
21,
22,
23] for ball-milled LN crystals.
3.2. Structure of the Samples: XRD, Raman and Optical Reflectance Measurements
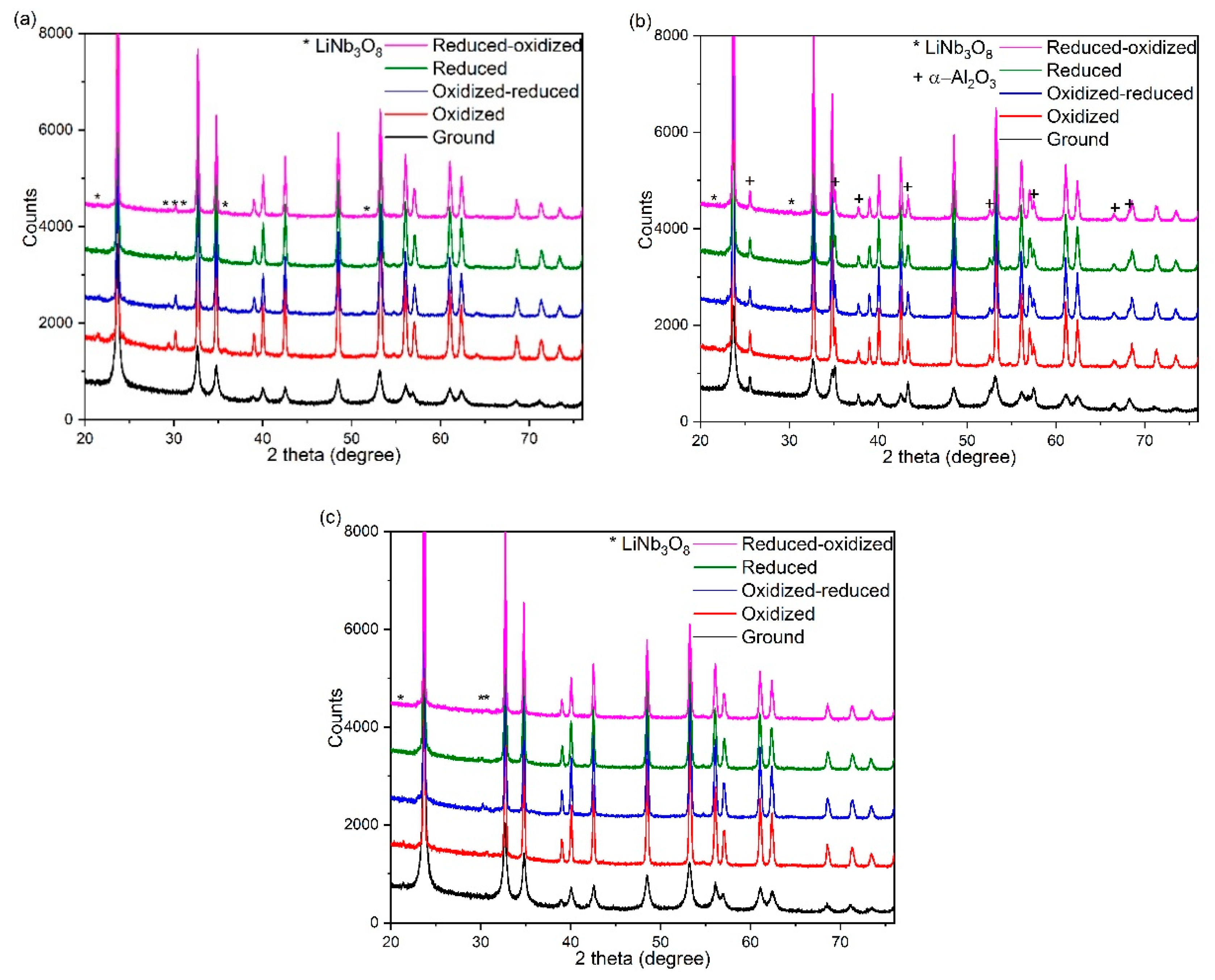
Figure 2 shows the XRD patterns of the ground materials compared with those oxidized and/or reduced at 800 °C for 3 h. The broad peaks of the XRD patterns of the as-ground samples indicate small grain sizes as determined numerically using the Williamson-Hall method (see
Table 2). Heat-treatment processes resulted in narrower diffraction lines due to increased grain sizes. The reflections of a LiNb
3O
8 (lithium triniobate) phase appeared in the diffraction patterns of all annealed samples (best seen for oxidized samples, especially for those ground in the stainless steel vial, see
Figure 2a). The formation of the LiNb
3O
8 phase taking place as a result of the combined ball-milling and annealing procedure can be described as
where lithium oxide is a volatile byproduct.
The fact that the lithium triniobate can only be identified in the heat-treated samples indicates that the structural rearrangement of the residual Nb containing oxides is not completed in the as-ground samples. However, the annealing process provides the activation energy required for crystallization of the new phase with sufficiently large crystallites to yield strong enough reflections in the diffraction patterns.
In the XRD pattern of the sample ball-milled in alumina vial the reflections of α-Al
2O
3 can be clearly identified due to abrasion of the vial and balls during the milling process already before the heat-treatments (see
Figure 2b). The fact that alumina was present in a crystalline form already in the as-ground samples indicates that the milling destroyed the ball/vial material and the majority of this impurity does not arise as a result of the side reaction that weakened the alumina structure. The lack of further crystalline compounds of aluminum in the annealed samples shows that the amount of possibly reacted alumina was insignificant as compared to alumina that entered the ground mixture by the mechanical effect of milling.
Figure 3 shows the Raman spectra of the as-ground and heat-treated samples ball-milled in stainless steel and tungsten carbide vials. The Raman intensities of the as-ground samples are weak, the bands are broad, not showing all characteristic features of LiNbO
3 crystals. The heat-treatment process resulted in line narrowing and increased intensity of the bands corresponding to the pure LiNbO
3 phase. In addition, in the oxidized samples some weak bands appeared at 59, 79 and 96 cm
−1 corresponding to the LiNb
3O
8 phase (
Figure 3a) [
29]. This confirms the XRD results, where the presence of the LiNb
3O
8 phase predicted by Equation (1) was best seen for oxidized samples, especially for those ground in stainless steel vial.
Equation (1) suggests that at least one new component without any niobium content has to appear during the milling process. Lithium oxide, Li
2O, may be present as the primary byproduct and can be transformed in air to another lithium compound (LiOH·
xH
2O, Li
2CO
3·
xH
2O) by water and/or CO
2 uptake. Indeed, the water suspensions of all ball-milled LN powders were found to be alkaline, regardless of the chemical state of the Li-rich segregate, which is an unambiguous confirmation of the decomposition of LN via Li
2O separation during the milling process. The as-ground LN particles were structurally disordered in the decomposed region but recrystallized upon annealing, hence both the Raman and XRD lines of LiNb
3O
8 could manifest themselves. The CO
2 uptake of Li
2O produced during ball-milling can also be observed, viz. in the Raman spectrum of the LN powder ground in tungsten carbide vial shown in
Figure 3b. The bands at about 190 and 1090 cm
−1 present in the as-ground samples are attributed to Li
2CO
3 generated from Li
2O (
Figure 3b) [
30]. Heat-treatments at 800 °C either in air or in vacuum resulted in the loss of CO
2 evidenced by the disappearance of those bands from the Raman spectra. The presence of α-Al
2O
3 contamination in the powder ball-milled in alumina vial was observed in the XRD diffractogram; however, it could not be detected by Raman spectroscopy as the corresponding bands at about 383 and 420 cm
−1 overlap with the larger bands of LN [
31].
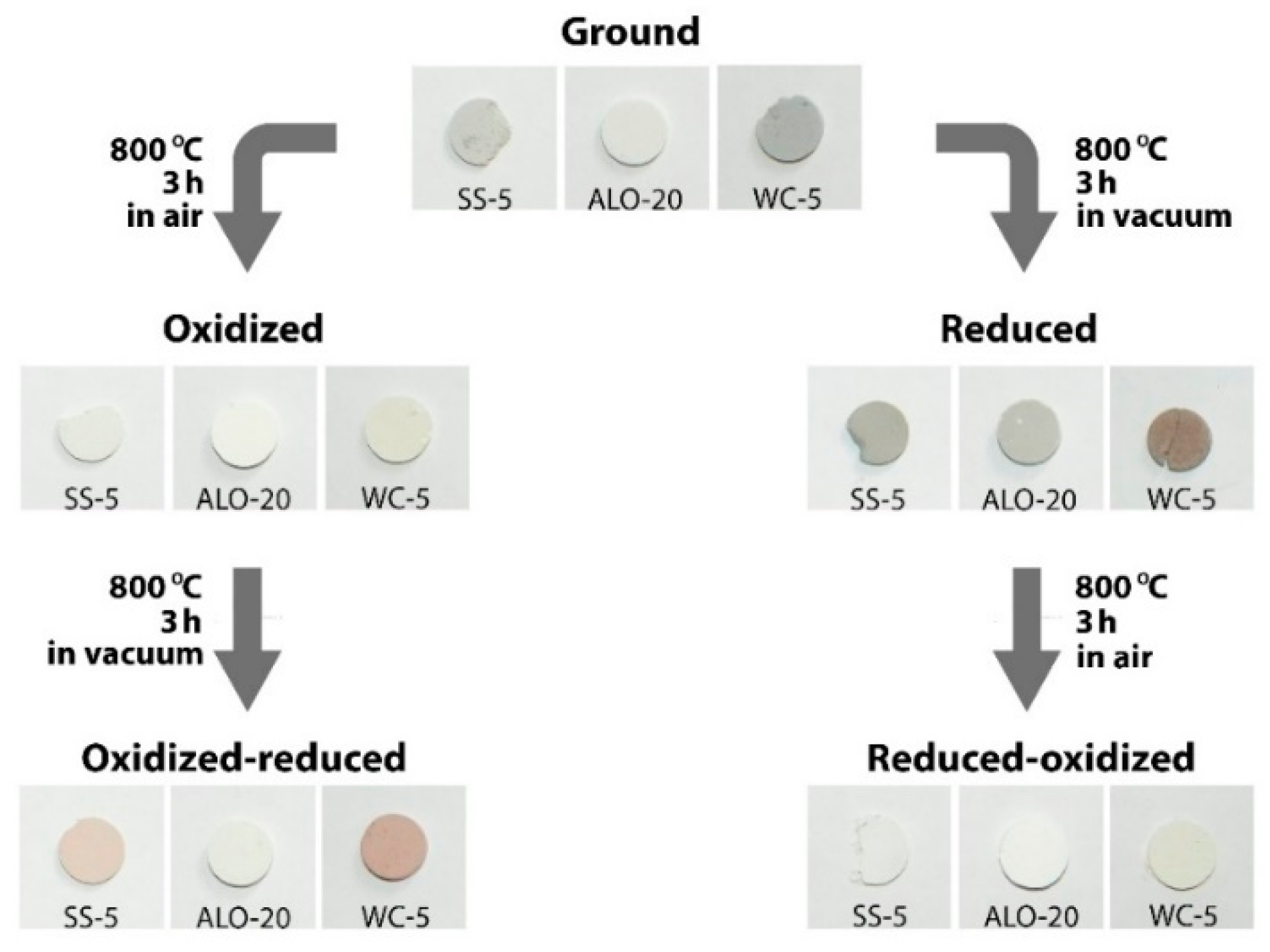
Equation (1) does not account for the redox processes indicated by color changes seen during ball-milling and annealing treatments. The colors of samples ground in alumina, stainless steel and tungsten carbide vials varied from light gray to dark gray (see
Scheme 1), evidenced by their optical reflection spectra (
Figure 4a)—the darker the sample, the lower its reflection in the whole spectral range. As mentioned above, the samples underwent a change concerning the oxidation state of niobium during the grinding process. This partial reduction could be compensated by oxidizing the sample applying a heat-treatment in air at 800 °C. The oxidative annealing resulted in white color for the powder ground in stainless steel vial (
Scheme 1). Upon subsequent reduction, the sample became brownish, while the pellet pressed from the as-ground powder became gray when reduced directly. The oxidation process resulted in a white color even in the case of the previously reduced sample. As an example, the optical reflection spectra of the as-ground and annealed samples ball-milled in stainless steel vial are shown in
Figure 4b. Similar effects were observed for powders ground in the other two vials: the change of color was less evidenced for alumina vial but was stronger for tungsten carbide vial.
In
Figure 4a no distinctive feature characteristic for Fe
2+ having an absorption band near 500 nm can be discerned for any of the as-ground samples. The comparison of reduced samples also shows very small differences in this respect. Instead, differences in amplitude of the whole spectrum dominate. Still some change of coloration induced by the redox treatments may be related to the iron contamination at least partly coming from the starting material. For the preparation of LiNbO
3 high- purity raw materials with less than 2 ppm, Fe was used. Since the effective distribution coefficient of Fe between the molten and solid lithium niobate of congruent composition is around 1, it is not expected to be enriched in the crucible residue during the growth process. On the other hand, even 10 ppm iron does not induce a sizable increase of the optical absorption. According to Phillips et al. [
32] the difference in the absorption coefficient at about 500 nm between oxidized and reduced LN containing about 0.5 mol% Fe is less than 4 cm
−1, i.e. less than 0.001 cm
−1/ppm Fe, which cannot cause dominant coloration changes in our case.
3.3. Discussion of the Mechanochemical Reaction Including Redox Processes
Congruent LiNbO
3 crystals are strongly Li deficient and can be described by the Li
1−5xNb
1+xO
3 formula, where x ≈ 0.01. The excess Nb ions occupy Li sites and are charge compensated by Li vacancies. The antisite Nb
Li ions may trap electrons, forming small polarons. Moreover, Nb
Li—Nb
Nb pairs, consisting of an antisite and its regular nearest-neighbor along the ferroelectric c axis, are capable to form stable bipolarons (for a review see [
33]). The strongly localized polaron/bipolaron models can evidently be applied for the redox processes in LN nanocrystals.
Already before the appearance of the LiNb
3O
8 phase the LN particles underwent partial reduction as a result of ball-milling. During reduction, oxygen gas and lithium oxide are formed, the latter leaving the sample only upon annealing treatments. This leads to the appearance of various polarons with elementary cell loss at the surface [
33,
34]
where the Nb
Nb4+ + Nb
Li4+ pair makes a bipolaron. The remaining electrons may also form either a further bipolaron or two Nb
Li4+ polarons on pre-existing Nb
Li antisite defects in the congruent bulk. Their broad absorption bands near 500 nm and 760 nm and the disordered structure result in uniform gray color as observed for the as-ground samples (see
Scheme 1 and reflection spectra in
Figure 4a). In this stage the surface is disordered and consists of strongly subcongruent lithium niobate, while Li
2O forms a different phase.
The structure of the particles consisting of a large number of grains can be understood to result from an interplay of disturbed ferroelectric surface fields. The grains may be assumed to be monodomain regions of LN pairwise attracted by the strong electric fields acting on most blank surfaces of this ferroelectric. Structural damage does not allow exact fitting of the attached surfaces, resulting also in crystallographic misorientation of the grains in touch. During ball milling, the particles constantly break up and recoalesce in different arrangements, while reduction may only proceed on surfaces where oxygen evaporation is possible for a sufficiently long period. Li2O segregation on such exposed grain surfaces may finally shield the electric fields and hamper further attachments with neighboring grains. During prolonged grinding, this may result in a structure where a large part of the Li2O phase is on the external surface of the particles.
Oxidation of the samples leads to the evaporation of the segregated Li
2O phase and in parallel the disappearance of all polarons. The latter recovers the white color by reverting the reduction described by Equation (2) and promotes the formation of the LiNb
3O
8 phase according to Equation (1) whereby a further LN formula unit is used up:
as observed by XRD, Raman and optical reflection measurements prominently for the oxidized samples. The LiNb
3O
8 phase may form an epitaxial layer on the LiNbO
3 surface as described by Semiletov et al. [
35].
In all other preparation stages various mixtures of bipolarons and polarons are present mainly absorbing in the blue-green and red range, respectively (
Figure 4b). While the as-ground state has a balanced mixture, its direct reduction leads to a larger bipolaron portion (less reflection in the blue-green region, see the curve with lowest reflection in
Figure 4b. The same reduction, if preceded by oxidation, reproduces only bipolarons but very few Nb
Li4+ polarons (high reflection only in the red region, see blue curve). Some additional structure observed near 350 nm and 670 nm in samples having an oxidizing step in their history might be attributed to absorption related to the LiNb
3O
8 phase. As shown by Sugak et al. [
36] the coloration is formed near the crystal surface and its distribution depends on annealing temperature. Annealing is assumed to attack the exposed surface of the particles without essentially changing their deeper structure. It should be noted that the large formation enthalpy of oxygen vacancies in LN compared to that of similar defects of the cation sublattice prevents the diffusion of oxygen within the bulk, while diffusing cations may easily occupy the empty Li sites amply available in congruent LN (see [
33] and references therein).
Reaction (2) is an equivalent version of Equation (2) in Sugak et al.’s paper, separately showing near-surface formation of polarons by Nb displacement to a Li site on the one hand, and electrons available for diffusion to more deeply situated antisites causing similar coloration on the other hand. This distinction, together with the overlooked fact that elementary cells are lost upon reduction, resolves the problems of Sugak et al. about unrealistic properties of coloration allegedly following from their Equation (2). In contrast to the opinion of Sugak et al., reduction-oxidation cycles are not completely reversible processes due to possible Li oxide loss especially at higher temperatures and in closely stoichiometric LiNbO
3, the latter being also much more resistant to reduction. Thermal reduction was shown to increase off-stoichiometry which, in turn, leads to larger density [
37,
38], quantitatively supporting Equation (2). All this gives full support to the cationic model of coloration excluding any diffusion of oxygen in the bulk. A further argument for reaction (2) specifically in our case is the expected higher density of Li-poor grain kernels, taken into account that they are produced by mechanical pressing exerted by the vials.
3.4. Quantitative Determination of the Degree of Decomposition During Ball-Milling
The quantity of lithium oxide segregated at the particle surfaces during ball-milling was determined by coulometric titration in the as-ground samples. No similar measurements were attempted for the annealed samples since the Li
2O has a fairly large volatility at the annealing temperature. All titration curves exhibited a single neutralization step as the acid was produced in-situ by the current passing through the cell. This indicates that the primary decomposition product was Li
2O and no significant amount of Li
2CO
3 was present, despite long storing times of several weeks in air elapsed after grinding prior to the titration procedure. The presence of carbonate should have led to a two-stage neutralization process, first leading to bicarbonate formation, but this was never observed. Although the decomposition product detected by the Raman measurement was lithium carbonate, this is no counterargument, as the Raman intensity of the various lithium-containing compounds may be very different and hence, the sensitivity of the Raman measurement may not be comparable for the various possible phases. The weight of dissolved Li
2O (
mOX) was calculated with the following equation:
where
M is the molar weight of Li
2O,
Q is the charge passed until the equivalence point,
F is the Faraday-constant (96485 C/mol), while the number in the denominator indicates that the hydrolysis of 1 mol of Li
2O results in 2 mols of hydroxide ions. Four measurements were performed for each batch ground in different vials. The measured Li
2O mass ratios
wox, expressed as weight percent of the as-ground powder are given in
Table 3. The uncertainty of the measurements is given as the standard deviation of the consecutive titration results.
The total surface of particles in the sample is proportional to 1/
R, where
R is the average particle radius as measured by DLS. The values of
wOX in
Table 3 indeed increase monotonously with 1/
R, though a fully quantitative trend cannot be established. No similar trend related to the inverse grain radius 1/
r can be seen in the given range of
r values obtained by XRD. These observations can be understood if segregation mainly occurs on the outer particle surfaces where both Li
2O and O
2 may freely leave, giving rise to a niobium-rich layer. However, it is also possible that part of the newly created surfaces, together with part of the Li
2O formed, gets buried during later stages of milling and cannot be dissolved for titration.
The quantitative determination of the lithium oxide loss enables us to estimate the thickness of the lithium triniobate layer in the oxidized samples. We use a simplified model of compact, uniform, spherical LN particles of unmodified composition covered by a uniformly thick LiNb3O8 phase. The shell thickness is calculated with the assumption that only the Li2O equivalent to this outer shell could be dissolved and analyzed by titration.
From the reaction indicated in Equation (1) it follows that
where
n stands for the molar quantity of the relevant materials, while the indices OX and LTN refer to the lithium oxide segregate and the lithium triniobate shell of the particles, respectively. For the weight of the particle shell we obtain
where
M is the molar weight. The weight ratio of the lithium oxide in the ground material,
wOX, is as follows:
The weight of each particle component can be expressed with the geometric parameter of the core-shell structure,
d being the shell thickness and
ρ the density:
The higher order terms with respect to
dLTN have been neglected since
dLTN ≪
R. The weight ratio of the lithium oxide is then
Again, the
dLTN ≪
R relation justifies the neglection of the second term in the denominator, leading to
By assuming that the Li
2O leaving the particle also forms a similar shell in the as-ground sample, for the thickness of this shell we calculate
where we take
2.01 g/cm
3,
= 4.65 g/cm
3 and
= 4.975 g/cm
3 [
39]. The values of
dLTN and
dOX are also included in
Table 3 and correspond to a layer thickness of at most a few unit cells.
The same amount of segregate (either Li2O or LiNb3O8), if spread evenly on all grain boundaries, would result in a much thinner layer. Neither the corresponding LiNb3O8 layer would be seen as XRD peaks nor would the equivalent amount of Li2O be readily soluble due to its hindered accessibility.
This finding gives further support to our previous assumption that the processes described by Equations (2) and (3) essentially occur on the outer surfaces. Accordingly, particle and grain size reduction proceeds as long as surfaces freshly broken up during ball milling have enough time to pile up a non-ferroelectric surface layer preventing them from stable recoalescing. Below a certain size limit, depending on the detailed properties of the milling system, this becomes impossible as recoalescence becomes too fast. The thickness of the outer segregate layer apparently has a narrow range defined by a similar requirement of sufficient atmospheric contact of the polar surface.
The proposed formation of the core-shell structure would require direct experimental evidence. However, the particle size achieved by the ball-milling process was too large for a direct transmission electron microscopic study of the particles.
Finally, we remark that the given description corresponds to the surface-screening mechanisms in ferroelectric thin films reviewed by Kalinin [
40]. In particular, very similar processes seem to occur in prospective lithium-ion batteries using LiNb
3O
8 as an anode material [
41].

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}