NMR Crystallography at Fast Magic-Angle Spinning Frequencies: Application of Novel Recoupling Methods


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials Preparation and Structures
2.2. Fourier Transform Infrared and X-ray Crystallographic Studies
2.3. Solid-State Nuclear Magnetic Resonance
2.4. ssNMR Simulations
3. Results and Discussion
3.1. Characterisation
3.2. Chemical-Shift Assignment and Molecular Structure
3.3. H -H Distance Measurements
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ssNMR | Solid-state Nuclear Magnetic Resonance |
| ABZ | Albendazole |
| SERP | Selective Recoupling of Protons |
| HETCOR | Heteronuclear Correlation spectroscopy |
| MAS | Magic-Angle-Spinning |
| CPMAS | Cross-Polarisation under Magic-Angle-Spinning |
| CP | Cross-Polarisation |
References
- Holzer, W.; Claramunt, R.M.; López, C.; Alkorta, I.; Elguero, J. A study in desmotropy. Solid State Nucl. Magn. Reson. 2008, 34, 68–149. [Google Scholar] [CrossRef]
- Elguero, J. Polymorphism and desmotropy in heterocyclic crystal structures. Cryst. Growth Des. 2011, 11, 4731–4738. [Google Scholar] [CrossRef]
- Elder, D.P.; Patterson, J.E.; Holm, R. The solid-state continuum: A perspective on the interrelationships between different solid-state forms in drug substance and drug product: The solid-state continuum. J. Pharm. Pharmacol. 2014, 67, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Finta, Z.; Hermecz, I.; Heja, G.; Horvath, A.; Kiss, G.; Morvai, M.; Podanyi, B.; Sipos, J.; Szabo, A.; Vasvari, A.; et al. Sanofi-Aventis, Assignee. U.S. Patent 0249669, 25 October 2007. [Google Scholar]
- Bauer, M.; Harris, R.K.; Rao, R.C.; Apperley, D.C.; Rodger, C.A. NMR study of desmotropy in Irbesartan, a tetrazole-containing pharmaceutical compound. J. Chem. Soc. Perkin Trans. 1998, 2, 475–481. [Google Scholar] [CrossRef]
- Chattah, A.K.; Zhang, R.; Mroue, K.H.; Pfund, L.Y.; Longhi, M.R.; Ramamoorthy, A.; Garnero, C. Investigating Albendazole Desmotropes by Solid-State NMR Spectroscopy. Mol. Pharm. 2015, 12, 731–741. [Google Scholar] [CrossRef]
- Apperley, D.C.; Forster, A.H.; Fournier, R.; Harris, R.K.; Hodgkinson, P.; Lancaster, R.W.; Rades, T. Characterisation of indomethacin and nifedipine using variable-temperature solid-state NMR. Magn. Reson. Chem. 2005, 43, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Othman, A.; Evans, J.S.O.; Evans, I.R.; Harris, R.K.; Hodgkinson, P. Structural study of polymorphs and solvates of finasteride. J. Pharm. Sci. 2007, 960, 1380–1397. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Hodgkinson, P.; Zorin, V.; Dumez, J.N.; Elena-Herrmann, B.; Emsley, L.; Salager, E.; Stein, R.S. Computation and NMR crystallography of terbutaline sulfate. Magn. Reson. Chem. 2010, 48, S103–S112. [Google Scholar] [CrossRef] [PubMed]
- Mafra, L.; Santos, S.M.; Siegel, R.; Alves, I.; Paz, F.A.A.; Dudenko, D.; Spiess, H.W. Packing interactions in Hydrated and anhydrous forms of the antibiotic ciprofloxacin: A solid-state NMR, X-ray diffraction, and computer simulation study. J. Am. Chem. Soc. 2012, 134, 71–74. [Google Scholar] [CrossRef]
- Dudenko, D.V.; Yates, J.R.; Harris, K.D.M.; Brown, S.P. An NMR crystallography DFT-D approach to analyse the role of intermolecular hyderogen bonding and π-π interactions in driving cocrystallisation of indomethacin and nicotinamide. CrystEngComm 2013, 15, 8797–8807. [Google Scholar] [CrossRef]
- Santos, S.M.; Rocha, J.; Mafra, L. NMR Crystallography: Toward chemical shift-driven crystal structure determination of the β-lactum antibiotic amoxicillin. Crys. Growth Des. 2013, 13, 2390–2395. [Google Scholar] [CrossRef]
- Baias, M.; Dumez, J.N.; Svensson, P.H.; Schantz, S.; Day, G.M.; Emsley, L. De Novo determination of the crystal structure of a large drug molecule by crystal structure prediction-based powder NMR crystallography. J. Am. Chem. Soc. 2013, 135, 17501–17507. [Google Scholar] [CrossRef] [PubMed]
- Kalakewich, K.; Luliucci, R.; Harper, J.K. Establishing accurate high-resolution crystal structures in the absence of diffraction data and single crystals—An NMR approach. Cryst. Growth Des. 2013, 13, 5391–5396. [Google Scholar] [CrossRef]
- Rossini, A.J.; Widdlfield, C.M.; Zagdoun, A.; Lelli, M.; Schwarzwalder, M.; Coperet, C.; Lesage, A.; Emsley, L. Dynamics nuclear polarisation enhanced NMR spectroscopy for pharmaceutical formulations. J. Am. Chem. Soc. 2014, 136, 2324–2334. [Google Scholar] [CrossRef] [PubMed]
- Reddy, G.N.M.; Cook, D.S.; Luga, D.; Walton, R.I.; Marsh, A.; Brown, S.P. An NMR crystallography study of the hemihydrate of 2’,3’-O-isopropylidineguanosine. Solid State Nucl. Magn. Reson. 2015, 65, 41–48. [Google Scholar] [CrossRef]
- Mollica, G.; Dekhil, M.; Ziarelli, T.P.; Viel, S. Quantitative structural constraints for organic powders at natural isotopic abundance using dynamic nuclear polarisation solid-state NMR spectroscopy. Angew. Chem. Int. Ed. 2015, 54, 6028–6031. [Google Scholar] [CrossRef]
- Kalakewich, K.; Luliucci, R.; Mueller, K.T.; Eloranta, H.; Harper, J.K. Monitoring the refinement of crystal structures with 15N solid-state NMR shift tensor data. J. Chem. Phys. 2015, 143, 194702. [Google Scholar] [CrossRef]
- Lill Sten, O.N.; Widdifield, C.M.; Pettersen, A.; Ankarberg, A.S.; Lindkvist, M.; Aldred, P.; Gracin, S.; Shankland, N.; Shankland, K.; Schantz, S.; et al. Elucidating an amorphous form stabilization mechanism for Tenapanor Hydrochloride: Crystal structure analysis using X-ray diffraction, NMR crystallography and molecular modeling. Mol. Pharm. 2018, 15, 1476–1487. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.G.; Lalli, D.; Stanek, J.; Gowda, C.; Prakash, S.; Schwarzer, T.S.; Schubeis, T.; Castigilione, K.; Andreas, L.B.; Madhu, P.K.; et al. Selective. 1H -1H distance restraints in fully protonated proteins by fast magic-angle spinning solid-state NMR. J. Phys. Chem. Lett. 2017, 8, 2399–2405. [Google Scholar] [CrossRef]
- Duong, N.T.; Raran-Kurussi, S.; Nishiyama, Y.; Agarwal, V. Quantitative 1H -1H distances in protonated solids by frequency-selective recoupling at fast magic angle spinning NMR. J. Phys. Chem. Lett. 2018, 9, 5948–5954. [Google Scholar] [CrossRef] [PubMed]
- Malon, M.; Pandey, M.K.; Nishiyama, Y. Revealing the Local Proton Network through Three-Dimensional 13C/1H Double-Quantum/1H Single-Quantum and 1H Double-Quantum/13C/1H Single-Quantum Correlation Fast Magic-Angle Spinning Solid-State NMR Spectroscopy at Natural Abundance. J. Phys. Chem. B 2017, 121, 8123–8131. [Google Scholar] [CrossRef]
- Pranzo, M.B.; Cruickshank, D.; Corruzzi, M.; Caira, M.R.; Bettini, R. Enantiotropically related albendazole polymorphs. J. Pharm. Sci. 2010, 99, 3731–3742. [Google Scholar] [CrossRef]
- Alhalaweh, A.; Lou, B.; Bostrom, D.; Velaga, S.P. CSD Communication. Available online: https://www.ccdc.cam.ac.uk/Community/csd-communications/ (accessed on 15 December 2018).
- Laage, S.; Marchetti, A.; Sein, J.; Pierattelli, R.; Saas, H.J.; Grzesiek, S.; Lesage, A.; Pintacuda, G.; Emsley, L. Band-selective 1H -13C cross-polarisation in fast magic angle spinning solid-state NMR spectroscopy. J. Am. Chem. Soc. 2008, 130, 17216–17217. [Google Scholar] [CrossRef]
- Equbal, A.; Madhu, P.K.; Meier, B.H.; Nielsen, N.C.; Ernst, M.; Agarwal, V. Parameter independent low-power heteronuclear decoupling for fast magic-angle spinning solid-state NMR. J. Chem. Phys. 2017, 146, 084202. [Google Scholar] [CrossRef] [Green Version]
- Emsley, L.; Bodenhausen, G. Optimization of shaped selective pulses for NMR using a quaternion description of their overall propagators. J. Magn. Reson. 1992, 97, 135–148. [Google Scholar] [CrossRef]
- Bak, M.; Rasmussen, J.T.; Nielsen, N.C. SIMPSON: A general simulation program for solid-state NMR spectroscopy. J. Magn. Reson. 2000, 147, 296–330. [Google Scholar] [CrossRef] [PubMed]
- Tosner, Z.; Andersen, R.; Stevensson, B.; Eden, M.; Nielsen, N.C.; Vosegaard, T. Computer-intensive simulation of solid-state NMR experiments using SIMPSON. J. Magn. Reson. 2014, 246, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Bak, M.; Nielsen, N.C. REPULSION, A novel approach to efficient powder averaging in solid-state NMR. J. Magn. Reson. 1997, 125, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.H.; Reinstra, C.M. High-performance solvent suppression for proton-detected solid-state NMR. J. Magn. Reson. 2008, 192, 167–172. [Google Scholar] [CrossRef] [PubMed]








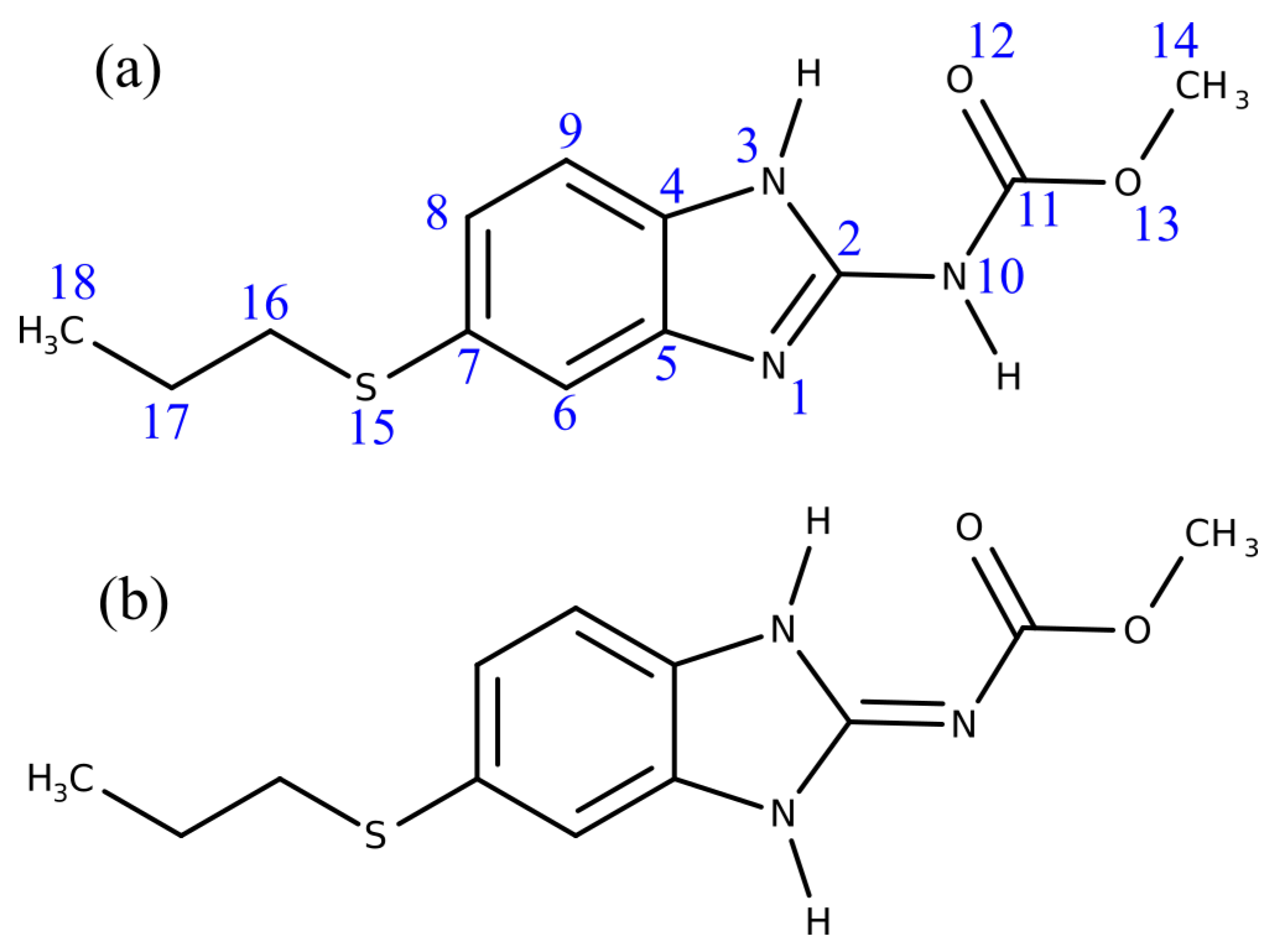{kind=link}
{kind=link}
{kind=link}
{kind=link}
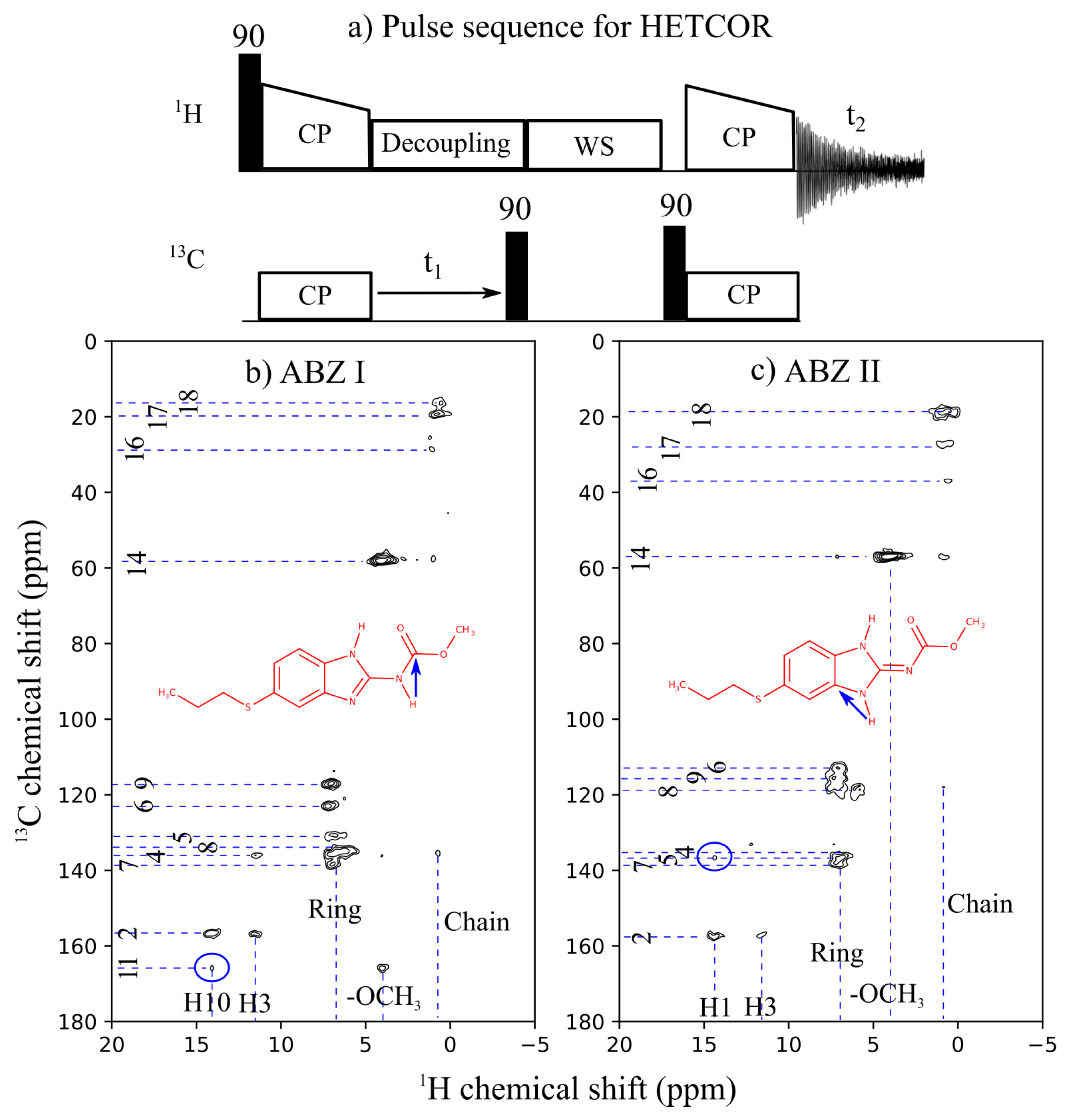{kind=link}
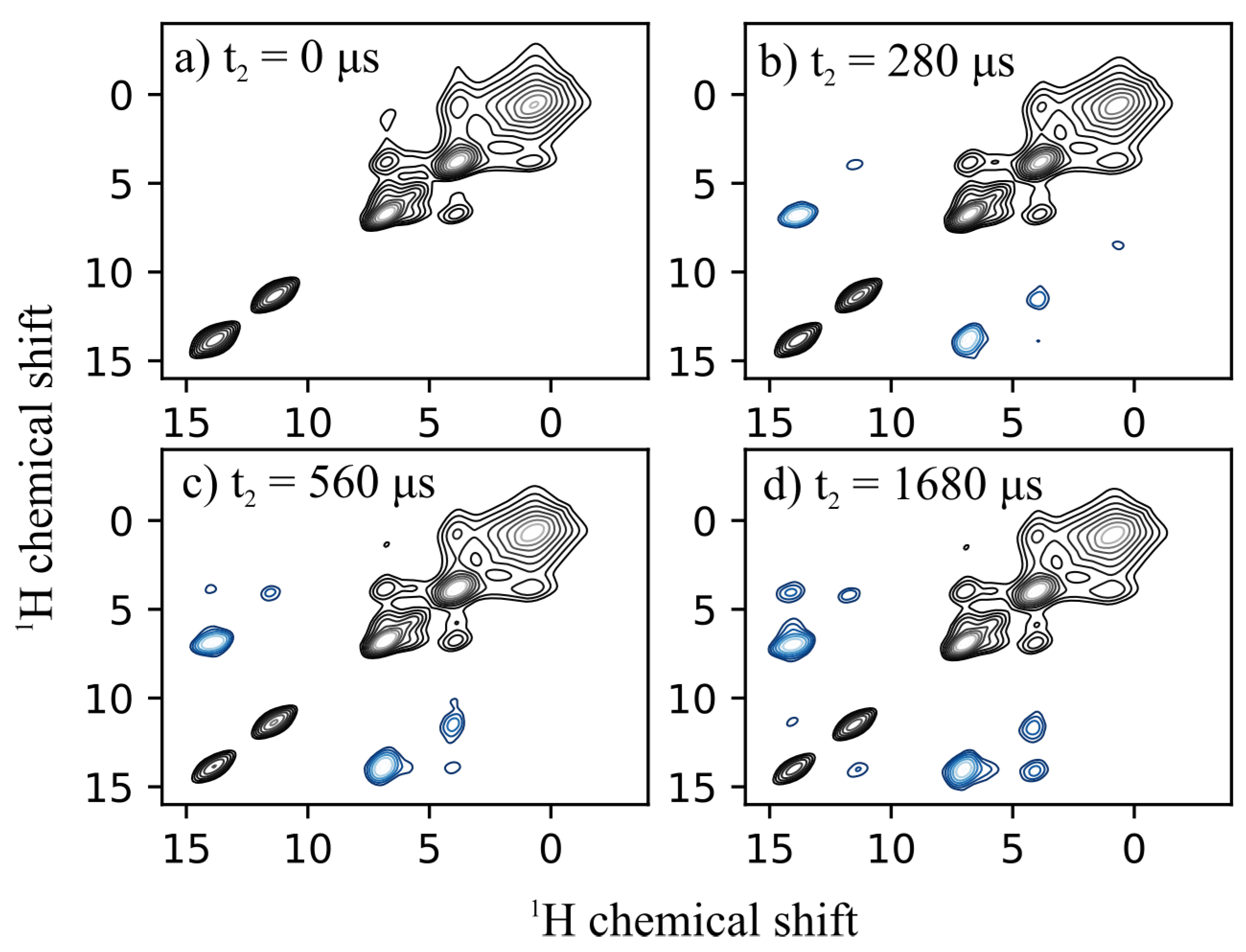{kind=link}
{kind=link}
| Protons | Intramolecular Distance (Å) | Intermolecular Distance (Å) |
|---|---|---|
| Albendazole Form I | ||
| H3–H10 | 3.6 | 6.63 |
| H3–H14 | 4.75, 4.82, 5.76 | 2.59, 2.85, 3.97 |
| H10–H14 | 4.12, 4.13, 4.21 | 7.16, 7.3, 8.57 |
| H10–H6 | 5.19 | 3.26 |
| Albendazole Form II | ||
| H3–H1 | 3.83 | 6.8 |
| H3–H14 | 4.85, 4.86, 5.84 | 2.55, 2.73, 3.88 |
| H1–H14 | 6.34, 6.36, 6.61 | 5.91, 5.97, 7.06 |
| H1–H6 | 2.81 | 7.23 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, M.G.; Mote, K.R.; Madhu, P.K. NMR Crystallography at Fast Magic-Angle Spinning Frequencies: Application of Novel Recoupling Methods. Crystals 2019, 9, 231. https://doi.org/10.3390/cryst9050231
Jain MG, Mote KR, Madhu PK. NMR Crystallography at Fast Magic-Angle Spinning Frequencies: Application of Novel Recoupling Methods. Crystals. 2019; 9(5):231. https://doi.org/10.3390/cryst9050231
Chicago/Turabian StyleJain, Mukul G., Kaustubh R. Mote, and Perunthiruthy K. Madhu. 2019. "NMR Crystallography at Fast Magic-Angle Spinning Frequencies: Application of Novel Recoupling Methods" Crystals 9, no. 5: 231. https://doi.org/10.3390/cryst9050231