Mechanical Properties, Electronic Structures, and Debye Temperature of NixBy Compounds Obtained by the First Principles Calculations

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Methods
3. Results and Discussion
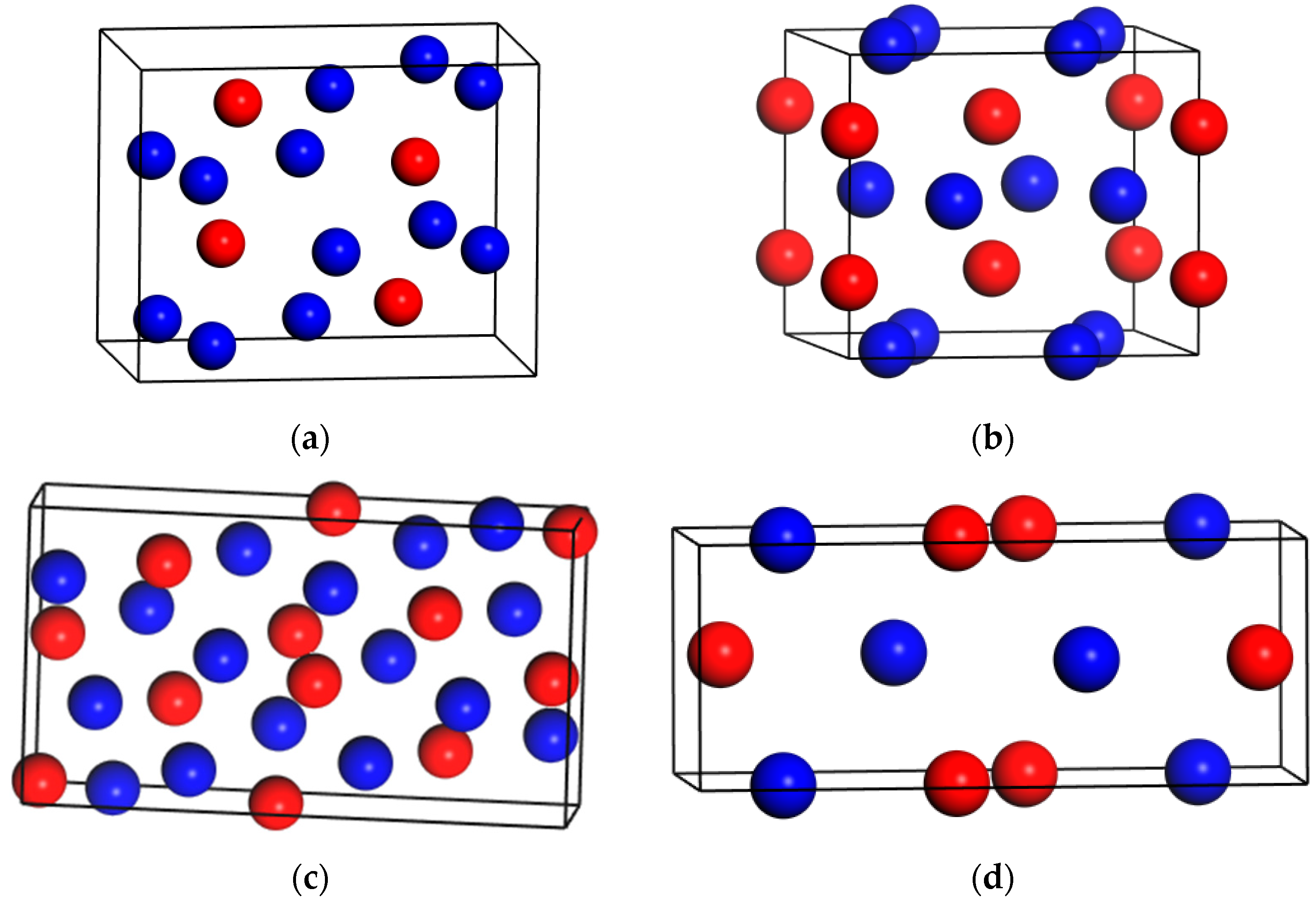
3.1. Stability
3.2. Mechanical Properties
3.2.1. Elastic Constant and Elastic Modulus
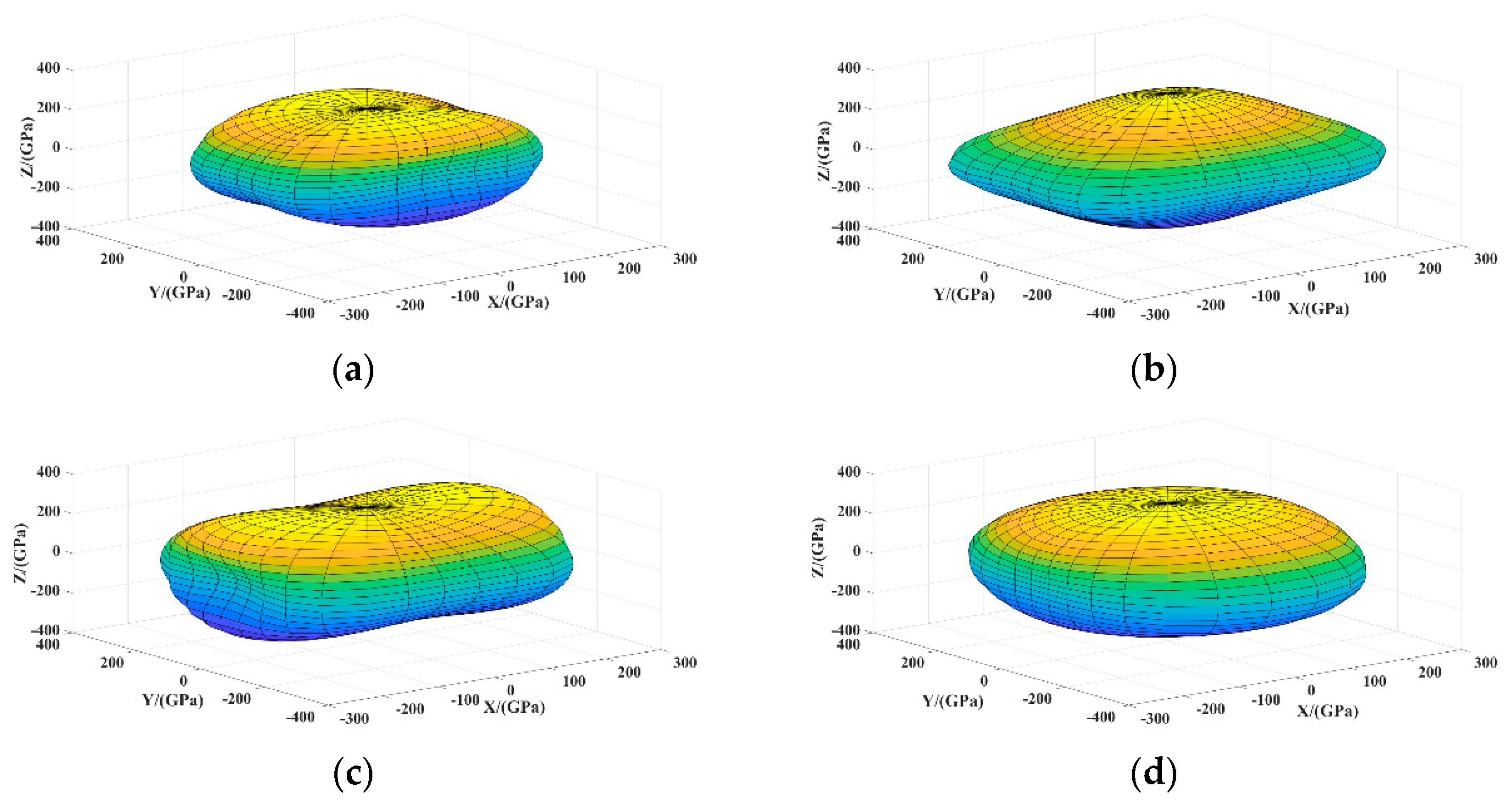
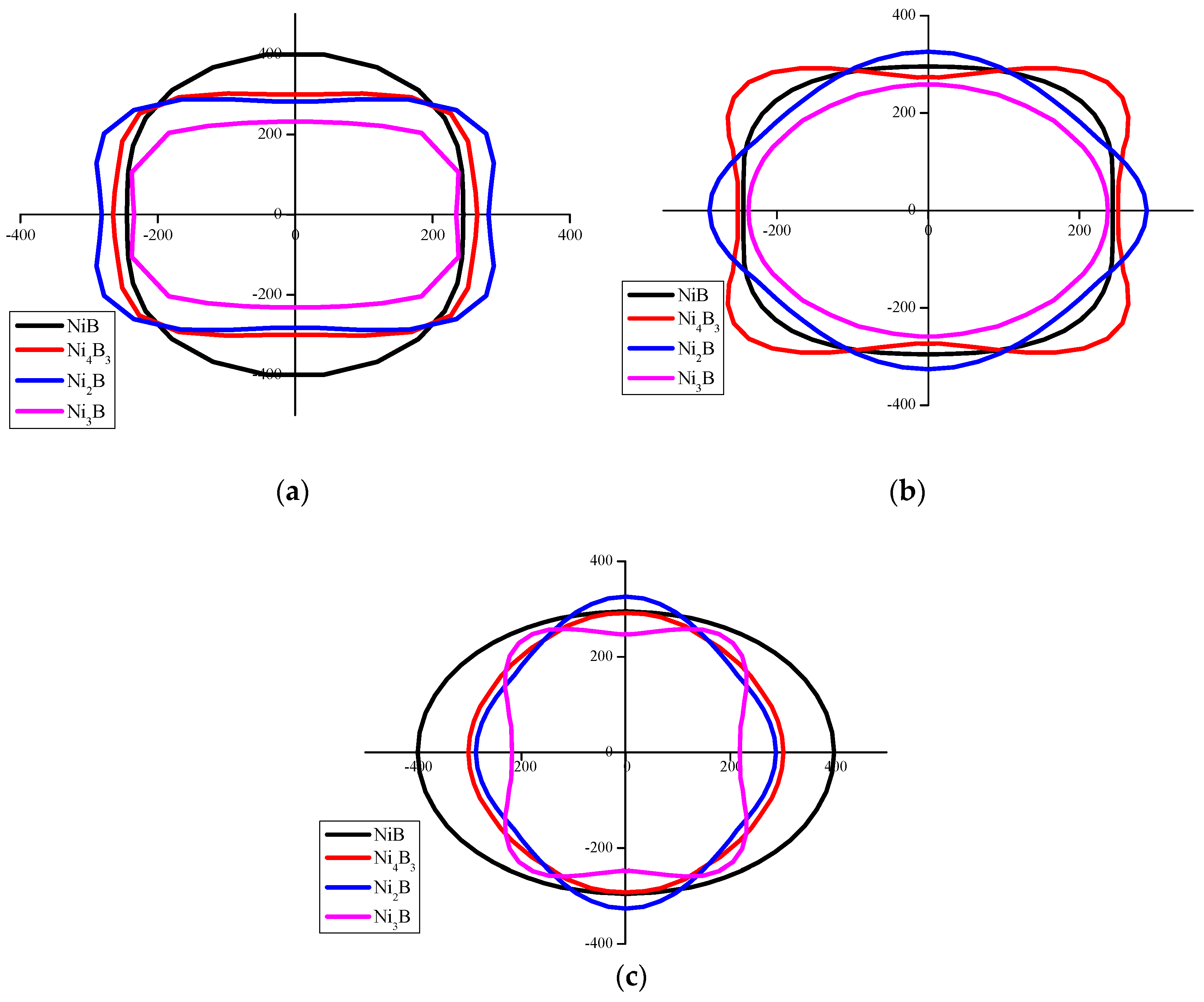
3.2.2. Anisotropy
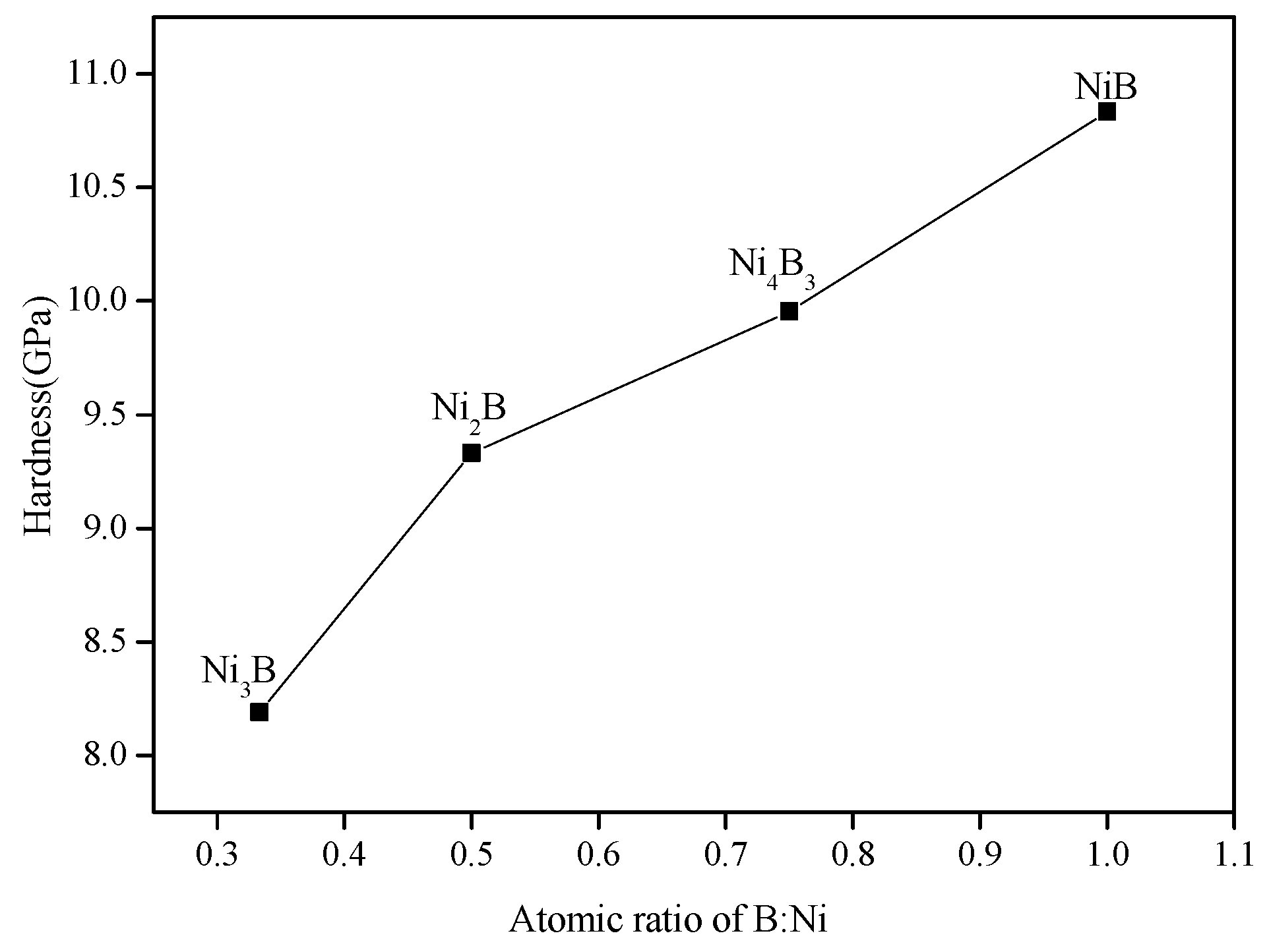
3.2.3. Hardness
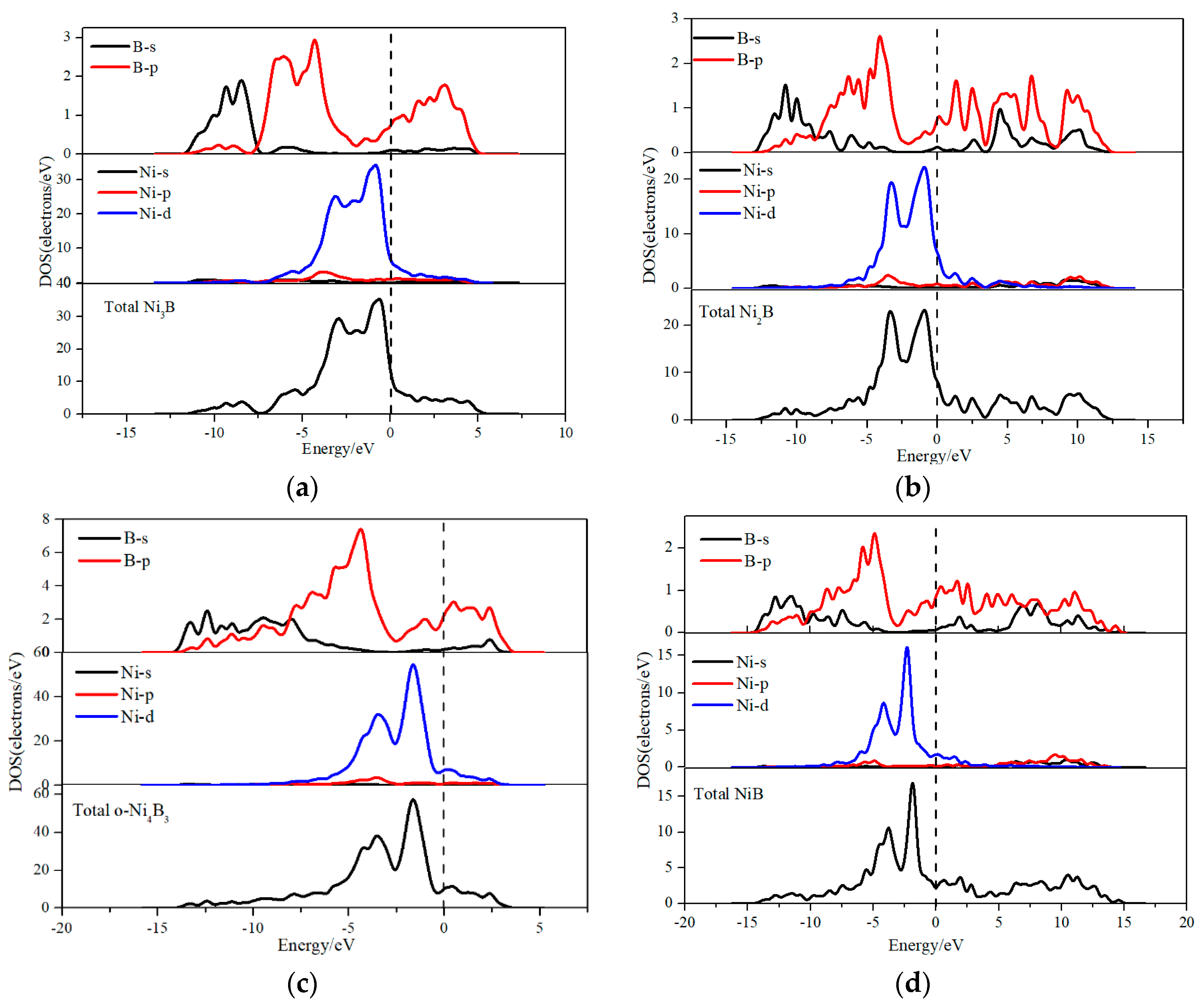
3.3. Electronic Structures
3.4. Debye Temperature
4. Conclusions
- (1)
- The calculated lattice parameters are consistent with the predecessor calculation data. The formation energy of all NixBy compounds is negative, which indicates that all NixBy compounds have stable structures.
- (2)
- NixBy compounds have mechanical stability. NiB has the largest bulk modulus, shear modulus and Young’s modulus and the smallest Poisson’s ratio, which imply that the hardness of NiB is higher than other NixBy compounds. NixBy compounds exhibit anisotropic characteristics, and Ni4B3 had the greatest anisotropy. The mechanical properties of the NixBy compounds increase with the increase of the B atomic ratio.
- (3)
- NixBy compounds exhibit p-d hybridization and they exhibit the metal bond and the covalent bond.
- (4)
- NiB has largest Debye temperature (7681.8 K), which indicates that NiB has the highest thermodynamic stability. Debye temperature of the NixBy compounds increase with the increase of the B atomic ratio.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Farahmand, P.; Liu, S.; Zhang, Z.; Kovacevic, R. Laser cladding assisted by induction heating of Ni–WC composite enhanced by nano-WC and La2O3. Ceram. Int. 2014, 40, 15421–15438. [Google Scholar] [CrossRef]
- Wang, K.; Fu, H.; Li, Y.; Lei, Y.; Wei, S.; Su, Z. Effect of power on microstructure and properties of laser cladding NiCrBSi composite coating. Trans. Inst. Met. Finish. 2017, 95, 328–336. [Google Scholar]
- Chen, C.H.; Bai, Y.; Chen, W.; Ye, X.C. Boron Influence on Structures and Properties in Nickel-Based Alloys. Appl. Mech. Mater. 2013, 395–396, 251–258. [Google Scholar] [CrossRef]
- González, R.; García, M.A.; Peñuelas, I.; Cadenas, M.; Fernández, M.D.R.; Battez, A.H.; Felgueroso, D. Microstructural study of NiCrBSi coatings obtained by different processes. Wear 2007, 263, 619–624. [Google Scholar] [CrossRef]
- Ma, Q.; Li, Y.; Wang, J.; Liu, K. Investigation on cored-eutectic structure in Ni60/WC composite coatings fabricated by wide-band laser cladding. J. Alloys Compd. 2015, 645, 151–157. [Google Scholar] [CrossRef]
- Wu, Z.J.; Ge, S.H.; Zhang, M.H.; Li, W.; Mu, S.C.; Tao, K.Y. Controlled Synthesis of Supported Nickel Boride Catalyst Using Electroless Plating. J. Phys. Chem. C 2007, 111, 8587–8593. [Google Scholar] [CrossRef]
- Choi, J.W.; Hwang, G.H.; Han, W.K.; Kang, S.G. Phase transformation of Ni-B, Ni-P diffusion barrier deposited electrolessly on Cu interconnect. Appl. Surf. Sci. 2006, 253, 2171–2178. [Google Scholar] [CrossRef]
- Eraslan, S.; Ürgen, M. Oxidation behavior of electroless Ni-P, Ni-B and Ni-W-B coatings deposited on steel substrates. Surf. Coat. Technol. 2015, 265, 46–52. [Google Scholar] [CrossRef]
- Anik, M.; Körpe, E.; Şen, E. Effect of coating bath composition on the properties of electroless nickel–boron films. Surf. Coat. Technol. 2008, 202, 1718–1727. [Google Scholar] [CrossRef]
- Wang, K.; Chang, B.; Chen, J.; Fu, H.; Lin, Y.; Lei, Y. Effect of Molybdenum on the Microstructures and Properties of Stainless Steel Coatings by Laser Cladding. Appl. Sci. 2017, 7, 1065. [Google Scholar] [CrossRef]
- Hou, X.; Du, D.; Wang, K.; Hong, Y.; Chang, B. Microstructure and Wear Resistance of Fe-Cr-Mo-Co-CB Amorphous Composite Coatings Synthesized by Laser Cladding. Metals 2018, 8, 622. [Google Scholar] [CrossRef]
- Li, Q.; Lei, Y.; Fu, H. Growth mechanism, distribution characteristics and reinforcing behavior of (Ti, Nb)C particle in laser cladded Fe-based composite coating. Appl. Surf. Sci. 2014, 316, 610–616. [Google Scholar] [CrossRef]
- Wang, K.; Chang, B.; Lei, Y.; Fu, H.; Lin, Y. Effect of Cobalt on Microstructure and Wear Resistance of Ni-Based Alloy Coating Fabricated by Laser Cladding. Metals 2017, 7, 551. [Google Scholar] [CrossRef]
- Liu, Y.; Xing, J.; Li, Y.; Sun, L.; Wang, Y. A first principles study of adhesion and electronic structure at Fe (110)/graphite (0001) interface. Appl. Surf. Sci. 2017, 405, 497–502. [Google Scholar] [CrossRef]
- Shein, I.R.; Medvedeva, N.I.; Ivanovskii, A.L. Electronic and structural properties of cementite-type M3X (M = Fe, Co, Ni; X = C or B) by first principles calculations. Physica B 2006, 371, 126–132. [Google Scholar] [CrossRef]
- Zhou, C.T.; Xing, J.D.; Xiao, B.; Feng, J.; Xie, X.J.; Chen, Y.H. First principles study on the structural properties and electronic structure of X2B (X = Cr, Mn, Fe, Co, Ni, Mo and W) compounds. Comp. Mater. Sci. 2009, 44, 1056–1064. [Google Scholar] [CrossRef]
- Zhou, Y.; Xiang, H.; Feng, Z.; Li, Z. Electronic Structure and Mechanical Properties of NiB: A Promising Interphase Material for Future UHTCf/UHTC Composites. J. Am. Ceram. Soc. 2016, 99, 2110–2119. [Google Scholar] [CrossRef]
- Wang, K.; Li, Y.; Fu, H.; Lei, Y.; Su, Z.; Ma, P. A study of laser cladding NiCrBSi/Mo composite coatings. Surf. Eng. 2018, 34, 267–275. [Google Scholar]
- Liu, K.; Fan, H.; Ren, P.; Yang, C. Structural, electronic and optical properties of BiFeO3 studied by first-principles. J. Alloys Compd. 2011, 509, 1901–1905. [Google Scholar] [CrossRef]
- Skylaris, C.K. A benchmark for materials simulation. Science 2016, 351, 1394–1395. [Google Scholar] [CrossRef] [PubMed]
- Ravi, C. First-principles study of crystal structure and stability of AlMgSi(Cu) precipitates. Acta Mater. 2004, 52, 4213–4227. [Google Scholar] [CrossRef]
- Connétable, D.; Thomas, O. First-principles study of the structural, electronic, vibrational, and elastic properties of orthorhombic NiSi. Phys. Rev. B 2009, 79, 094101. [Google Scholar] [CrossRef]
- Schaefer, Z.L.; Ke, X.; Schiffer, P.; Schaak, R.E. Direct Solution Synthesis, Reaction Pathway Studies, and Structural Characterization of Crystalline Ni3B Nanoparticles. J. Phys. Chem. C 2008, 112, 19846–19851. [Google Scholar] [CrossRef]
- Kong, Y.; Xiong, W.; Guo, H.; Sun, W.; Du, Y.; Zhou, Y. Elastic and thermodynamic properties of the Ni–B system studied by first-principles calculations and experimental measurements. Calphad 2010, 34, 245–251. [Google Scholar] [CrossRef]
- Fujimori, M.; Nakata, T.; Nakayama, T.; Nishibori, E.; Kimura, K.; Takata, M.; Sakata, M. Peculiar covalent bonds in alpha-rhombohedral boron. Phys. Rev. Lett. 1999, 82, 4452–4455. [Google Scholar] [CrossRef]
- Liu, Y.H.; Chong, X.Y.; Jiang, Y.H.; Zhou, R.; Feng, J. Mechanical properties and electronic structures of Fe-Al intermetallic. Physica B 2017, 506, 1–11. [Google Scholar] [CrossRef]
- Xiao, B.; Feng, J.; Zhou, C.T.; Jiang, Y.H.; Zhou, R. Mechanical properties and chemical bonding characteristics of Cr7C3 type multicomponent carbides. J. Appl. Phys. 2011, 109, 023507. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Y.; Xiao, B.; Min, T.; Fan, Z.; Ma, S.; Xu, L. Theoretical study on the stability, elasticity, hardness and electronic structures of W–C binary compounds. J. Alloys Compd. 2010, 502, 28–37. [Google Scholar] [CrossRef]
- Maibam, J.; Indrajit Sharma, B.; Bhattacharjee, R.; Thapa, R.K.; Brojen Singh, R.K. Electronic structure and elastic properties of scandium carbide and yttrium carbide: A first principles study. Physcia B 2011, 406, 4041–4045. [Google Scholar] [CrossRef]
- Feng, J.; Xiao, B.; Chen, J.; Du, Y.; Yu, J.; Zhou, R. Stability, thermal and mechanical properties of PtxAly compounds. Mater. Des. 2011, 32, 3231–3239. [Google Scholar] [CrossRef]
- Qi, C.J.; Jiang, Y.H.; Liu, Y.Z.; Zhou, R. Elastic and electronic properties of XB2 (X = V, Nb, Ta, Cr, Mo, and W) with AlB2 structure from first principles calculations. Ceram. Int. 2014, 40, 5843–5851. [Google Scholar] [CrossRef]
- Feng, J.; Xiao, B.; Zhou, R.; Pan, W.; Clarke, D.R. Anisotropic elastic and thermal properties of the double perovskite slab–rock salt layer Ln2SrAl2O7 (Ln = La, Nd, Sm, Eu, Gd or Dy) natural superlattice structure. Acta Mater. 2012, 60, 3380–3392. [Google Scholar] [CrossRef]
- Pathak, A.; Mehta, K.K.; Singh, A.K. A first principles calculation of Ni-16Cr and Ni-16Mo alloys. J. Appl. Res. Technol. 2017, 15, 78–82. [Google Scholar] [CrossRef]
- Sun, S.; Fu, H.; Lin, J.; Guo, G.; Lei, Y.; Wang, R. The stability, mechanical properties, electronic structures and thermodynamic properties of (Ti, Nb)C compounds by first-principles calculations. J. Mater. Res. 2018, 33, 495–506. [Google Scholar] [CrossRef]
- Ozisik, H.; Deligoz, E.; Colakoglu, K.; Surucu, G. Structural and mechanical stability of rare-earth diborides. Chin. Phys. B 2013, 22, 369–376. [Google Scholar] [CrossRef]
- Liu, Y.; Xing, J.; Fu, H.; Li, Y.; Sun, L.; Lv, Z. Structural stability, mechanical properties, electronic structures and thermal properties of XS (X = Ti, V, Cr, Mn, Fe, Co, Ni) binary compounds. Phys. Lett. A 2017, 381, 2648–2657. [Google Scholar] [CrossRef]
- Özışık, H.; Çiftci, Y.Ö.; Çolakoğlu, K.; Deligöz, E. The structural, elastic and vibrational properties of the DyX (X = P, As) compounds. Phys. Scr. 2011, 83, 035601. [Google Scholar] [CrossRef]
- Lee, E.; Lee, B.J. Modified embedded-atom method interatomic potential for the Fe-Al system. J. Phys. Condens. Matter. 2010, 22, 175702. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Chong, X.; Jiang, Y.; Feng, J. Effects of alloying elements such as Ti, Zr and Hf on the mechanical and thermodynamic properties of Pd-Base superalloy. J. Alloys Compd. 2017, 710, 589–599. [Google Scholar] [CrossRef]
- Feng, J.; Xiao, B.; Zhou, R.; Pan, W. Anisotropy in elasticity and thermal conductivity of monazite-type REPO4 (RE = La, Ce, Nd, Sm, Eu and Gd) from first-principles calculations. Acta Mater. 2013, 61, 7364–7383. [Google Scholar] [CrossRef]
- Nye, J.F. Physical Properties of Crystals: Their Representation by Tensors and Matrices; Oxford University Press: Oxford, UK, 1985. [Google Scholar]
- Liu, Y.; Jiang, Y.; Feng, J.; Zhou, R. Elasticity, electronic properties and hardness of MoC investigated by first principles calculations. Physcia B 2013, 419, 45–50. [Google Scholar] [CrossRef]
- Sun, S.; Liu, Y.; Fu, H.; Guo, X.; Ma, S.; Lin, J.; Guo, G.; Lei, Y.; Wang, R. First Principles Study of Mechanical Properties and Electronic Structures of Vanadium-Doped TiC and TiN. Adv. Eng. Mater. 2018, 20, 1800295. [Google Scholar] [CrossRef]
- Chen, X.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Deligoz, E.; Ciftci, Y.O.; Jochym, P.T.; Colakoglu, K. The first principles study on PtC compound. Mater. Chem. Phys. 2008, 111, 29–33. [Google Scholar] [CrossRef]
- Chen, C.L.; Lu, W.; He, L.L.; Ye, H.Q. First-principles study of deformation-induced phase transformations in Ti-Al intermetallics. J. Mater. Res. 2009, 24, 1662–1666. [Google Scholar] [CrossRef]
- Feng, J.; Xiao, B.; Wan, C.L.; Qu, Z.X.; Huang, Z.C.; Chen, J.C.; Zhou, R.; Pan, W. Electronic structure, mechanical properties and thermal conductivity of Ln2Zr2O7 (Ln = La, Pr, Nd, Sm, Eu and Gd) pyrochlore. Acta Mater. 2011, 59, 1742–1760. [Google Scholar] [CrossRef]
- Liu, L.; Xu, G.; Wang, A.; Wu, X.; Wang, R. First-principles investigations on structure stability, elastic properties, anisotropy and Debye temperature of tetragonal LiFeAs and NaFeAs under pressure. J. Phys. Chem. Solids 2017, 104, 243–251. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Space Group | Crystal Type | Lattice Constants (Å) | Cell Angles (°) | Formation | ||||
|---|---|---|---|---|---|---|---|---|---|
| a | b | c | α | β | γ | Energy (kJ/mol) | |||
| Ni | Fm-3m | cubic | 3.530 | 3.530 | 3.530 | 90.0 | 90.0 | 90.0 | |
| 3.520 a | 3.520 a | 3.520 a | |||||||
| Ni3B | Pnma | orthorhombic | 4.417 | 6.671 | 5.199 | 90.0 | 90.0 | 90.0 | −32.819 |
| 4.429 b | 6.659 b | 5.108 b | |||||||
| Ni2B | I4/mcm | tetragonal | 4.991 | 4.286 | 4.991 | 90.0 | 90.0 | 90.0 | −35.848 |
| 4.991 c | 4.284 c | 4.991 c | |||||||
| Ni4B3 | Pnma | orthorhombic | 3.005 | 6.603 | 12.011 | 90.0 | 90.0 | 90.0 | −36.026 |
| 2.981 d | 6.568 d | 11.954 d | |||||||
| NiB | Cmcm | orthorhombic | 2.991 | 2.982 | 7.338 | 90.0 | 90.0 | 90.0 | −32.547 |
| 2.927 e | 2.963 e | 7.394 e | |||||||
| B | R-3m | rhombohedral | 4.899 | 4.899 | 12.551 | 90.0 | 90.0 | 120.0 | |
| 4.924 f | 4.924 f | 12.609 f | |||||||
| Compounds | Elastic Constants | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 | |
| Ni3B | 348.6 | 184.1 | 161.7 | 340.8 | 184.4 | 363.4 | 132.6 | 96.7 | 112.4 |
| Ni2B | 405.7 | 207.6 | 165.5 | 408.6 | 167.1 | 416.0 | 102.9 | 102.8 | 138.9 |
| Ni4B3 | 379.3 | 201.1 | 210.2 | 417.4 | 169.2 | 401.5 | 110.8 | 147.8 | 140.5 |
| NiB | 352.3 | 170.0 | 194.7 | 508.5 | 178.1 | 419.9 | 137.2 | 111.2 | 129.4 |
| Compounds | BV | BR | B | GV | GR | G | E | υ | B/G |
|---|---|---|---|---|---|---|---|---|---|
| Ni3B | 234.8 | 234.7 | 234.8 | 103.2 | 99.8 | 101.5 | 266.2 | 0.311 | 2.313 |
| Ni2B | 256.7 | 256.4 | 256.6 | 114.9 | 112.7 | 113.8 | 297.5 | 0.307 | 2.254 |
| Ni4B3 | 262.1 | 262.1 | 262.1 | 121.0 | 116.5 | 118.8 | 309.2 | 0.308 | 2.276 |
| NiB | 262.9 | 258.4 | 260.7 | 124.7 | 120.9 | 122.8 | 318.5 | 0.296 | 2.122 |
| Compounds | AU | AB | AG | A1 | A2 | A3 |
|---|---|---|---|---|---|---|
| Ni3B | 0.167 | 0.000 | 0.016 | 1.365 | 1.153 | 1.400 |
| Ni2B | 0.099 | 0.001 | 0.010 | 0.838 | 0.839 | 1.392 |
| Ni4B3 | 0.196 | 0 | 0.019 | 1.230 | 1.231 | 1.425 |
| NiB | 0.174 | 0.009 | 0.015 | 1.433 | 0.777 | 0.994 |
| Species | Atoms | s | p | d | Total Electrons | Charge | |
| Ni3B | B | 1.12 | 2.46 | 3.58 | −0.58 | ||
| Ni | 0.35 | 0.73 | 8.72 | 9.81 | 0.19 | ||
| Ni2B | B | 1.07 | 2.45 | 3.52 | −0.52 | ||
| Ni | 0.30 | 0.72 | 8.72 | 9.74 | 0.26 | ||
| Ni4B3 | B | 1.00 | 2.50 | 3.53 | −0.53 | ||
| Ni | 0.24 | 0.63 | 8.73 | 9.60 | 0.40 | ||
| NiB | B | 0.99 | 2.55 | 3.54 | −0.54 | ||
| Ni | 0.18 | 0.55 | 8.73 | 9.46 | 0.54 | ||
| Species | Bond | (Å) | |||||
| Ni3B | B-Ni | 2.11 | 0.28 | ||||
| Ni-Ni | 2.56 | −0.01 | |||||
| Ni2B | B-B | 2.14 | 0.65 | ||||
| B-Ni | 2.14 | 0.15 | |||||
| Ni-Ni | 2.59 | −0.11 | |||||
| Ni4B3 | B-B | 2.33 | 0.76 | ||||
| B-Ni | 2.30 | 0.17 | |||||
| Ni-Ni | 2.93 | −0.13 | |||||
| NiB | B-B | 1.78 | 1.45 | ||||
| B-Ni | 2.15 | 0.28 | |||||
| Ni-Ni | 2.61 | −0.14 | |||||
| Compounds | vs/m∙s−1 | v1/m∙s−1 | vm/m∙s−1 | ΘD/K |
|---|---|---|---|---|
| Ni3B | 3505.6 | 6693.7 | 3921.2 | 553.4 |
| Ni2B | 3663.8 | 6939.4 | 4095.9 | 600.7 |
| Ni4B3 | 3958.1 | 7447.8 | 4422.9 | 645.1 |
| NiB | 4052.4 | 7532.9 | 4524.4 | 681.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.; Du, D.; Chang, B.; Hong, Y.; Ju, J.; Sun, S.; Fu, H. Mechanical Properties, Electronic Structures, and Debye Temperature of NixBy Compounds Obtained by the First Principles Calculations. Crystals 2018, 8, 451. https://doi.org/10.3390/cryst8120451
Wang K, Du D, Chang B, Hong Y, Ju J, Sun S, Fu H. Mechanical Properties, Electronic Structures, and Debye Temperature of NixBy Compounds Obtained by the First Principles Calculations. Crystals. 2018; 8(12):451. https://doi.org/10.3390/cryst8120451
Chicago/Turabian StyleWang, Kaiming, Dong Du, Baohua Chang, Yuxiang Hong, Jiang Ju, Shuting Sun, and Hanguang Fu. 2018. "Mechanical Properties, Electronic Structures, and Debye Temperature of NixBy Compounds Obtained by the First Principles Calculations" Crystals 8, no. 12: 451. https://doi.org/10.3390/cryst8120451