
Synthesis, Crystal Structure and DFT Studies of 1,3-Dimethyl-5-propionylpyrimidine-2,4,6(1H,3H,5H)-trione

 ,
,  , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
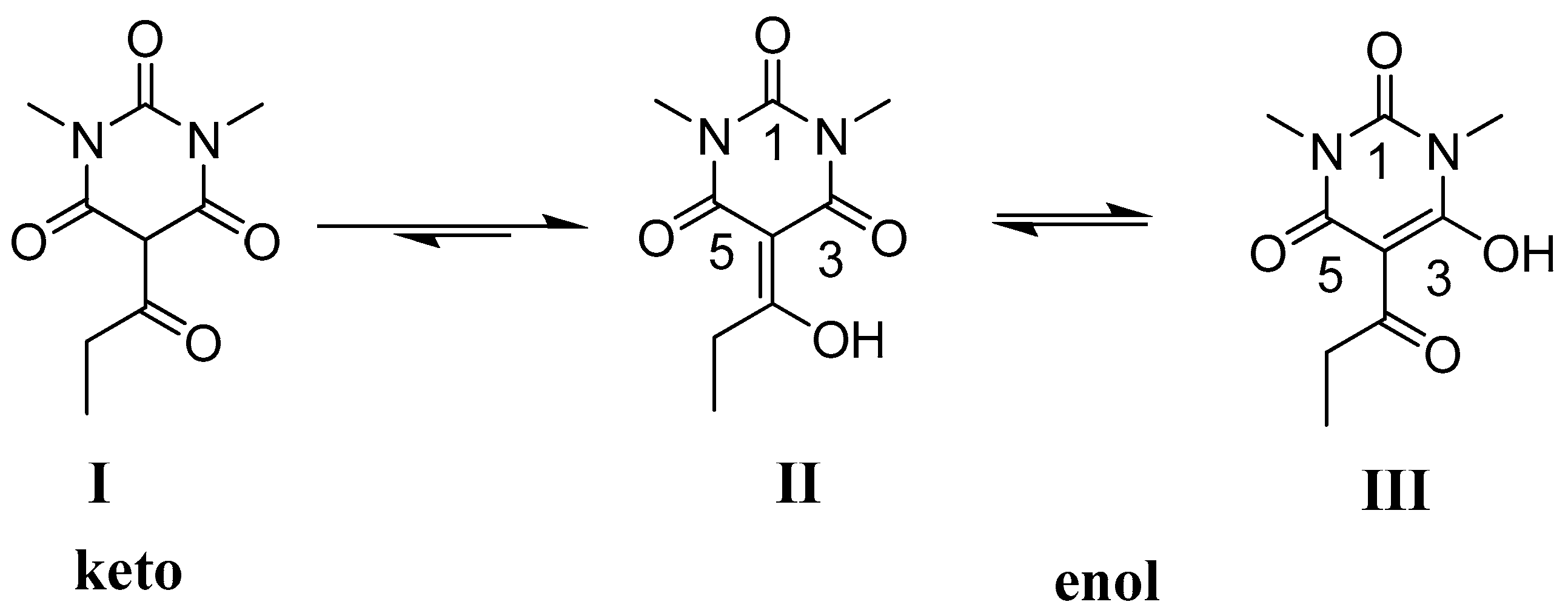
2.1. NMR Studies
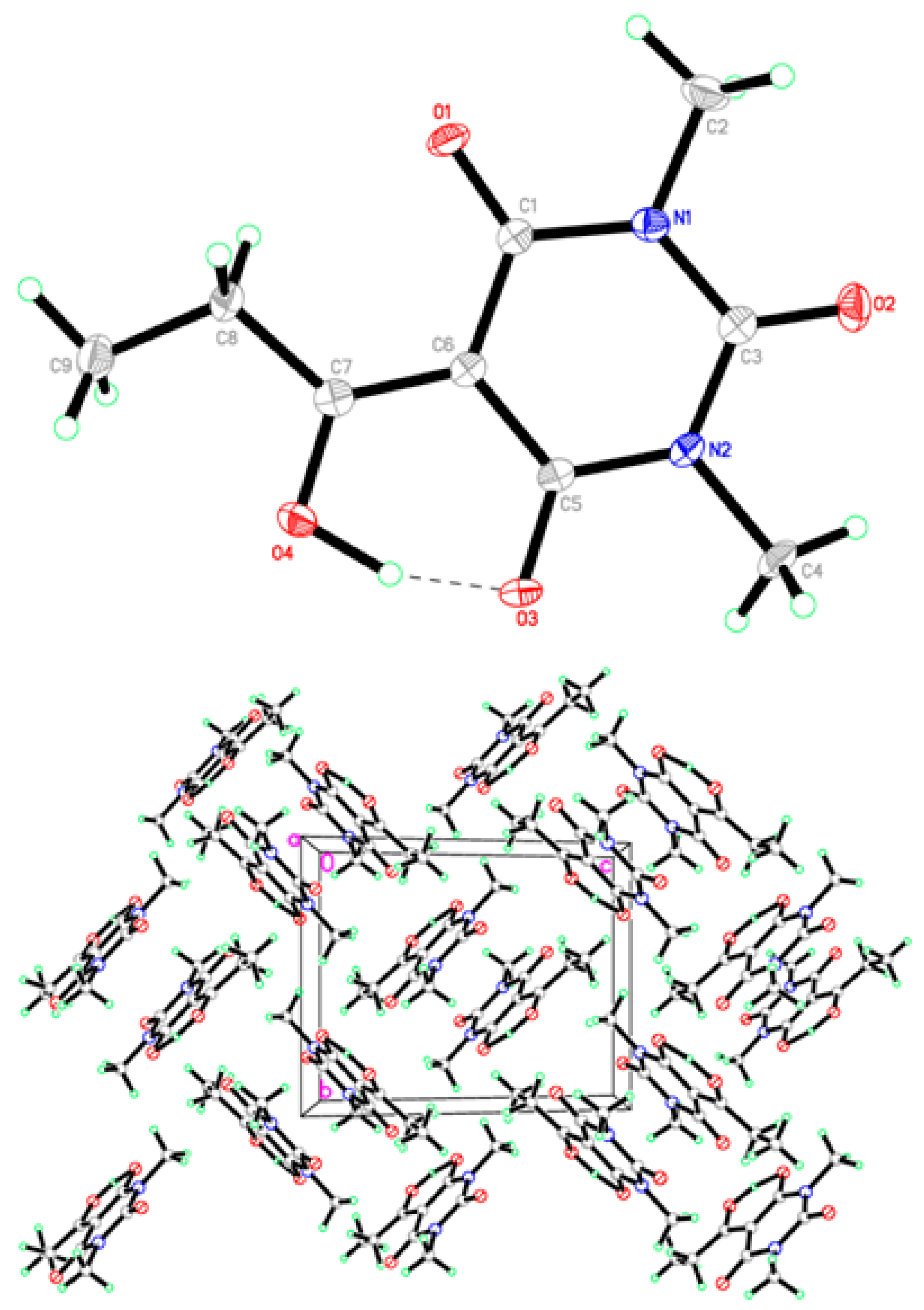
2.2. X-ray Structural Study and Theoretical Calculations
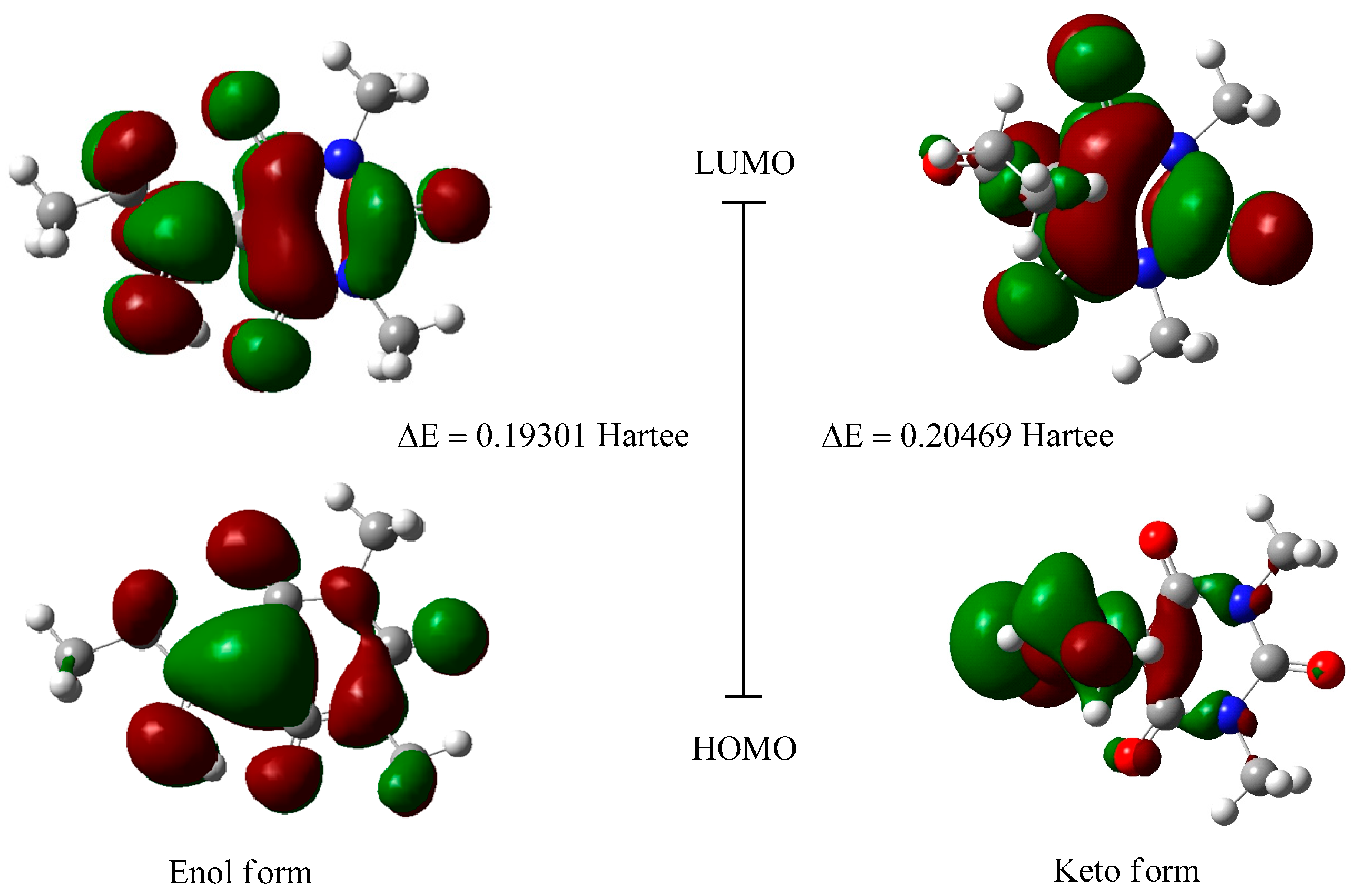
2.3. Frontier Orbital Energy Analysis and Molecular Total Energies
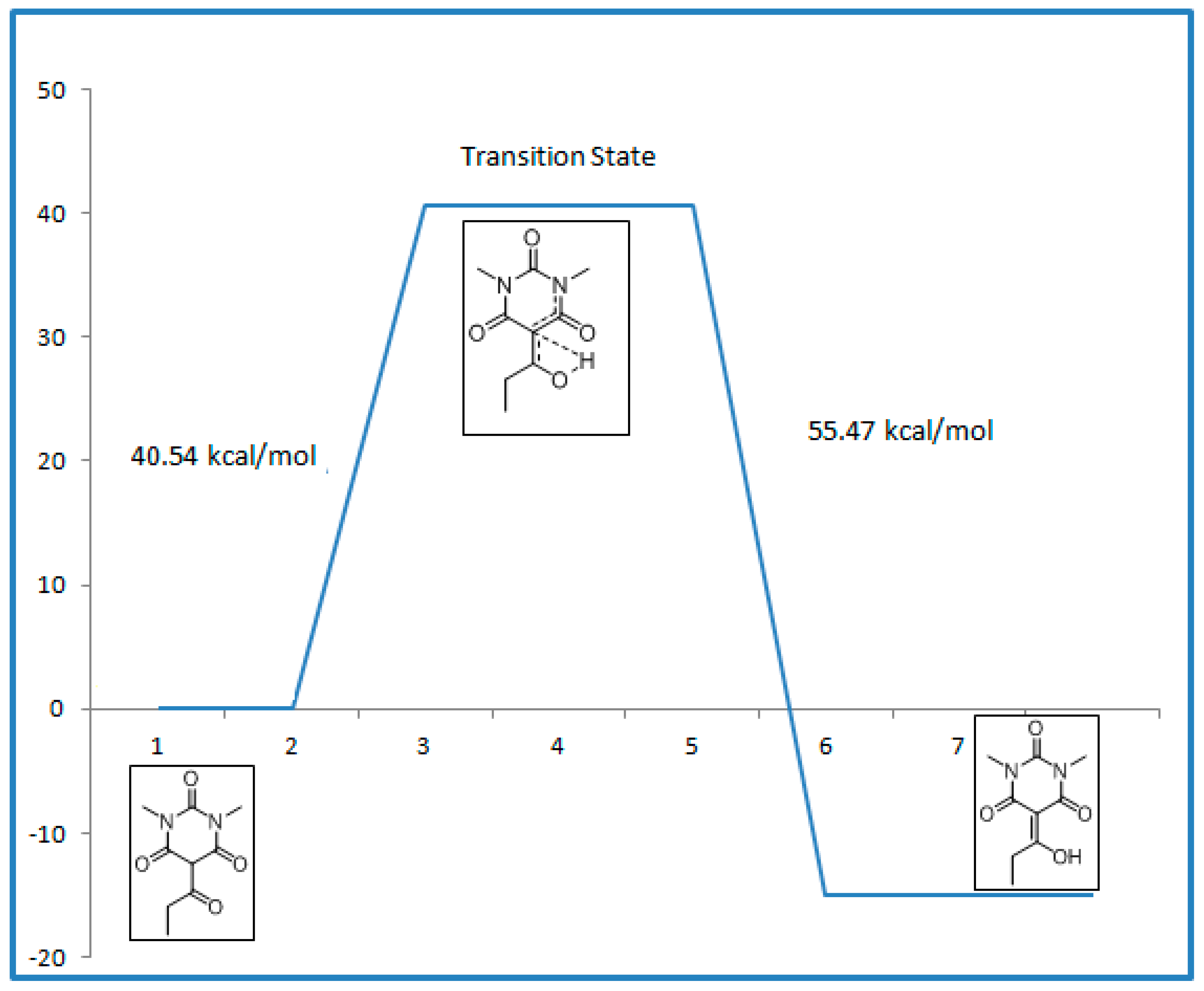
2.4. Transition State Calculations
3. Materials and Methods
3.1. General
3.2. General Procedure
3.3. Structure Determination
3.4. Theoretical Calculations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goodman, L.; Gilman, A. The Pharmacological Basis of Therapeutics; Mcgraw-Hill: New York, NY, USA, 1996; pp. 959–975. [Google Scholar]
- Olin, B. Central Nervous System Drugs, Sedatives and Hypnotics, Barbiturates, in Fact and Comparisons Drug Information; Facts and Comparisons: St. Louis, MO, USA, 1993; pp. 1398–1413. [Google Scholar]
- Brunton, L.; Lazo, J.; Parker, K.G. Gilman’s the Pharmacological Basis of Therapeutics, 11th ed.; Mc Graw-Hill: New York, NY, USA, 2005. [Google Scholar]
- Johns, M. Sleep and hypnotic drugs. Drugs 1975, 9, 448–478. [Google Scholar] [CrossRef] [PubMed]
- Whittle, S.R.; Turner, A.J. Differential effects of sedative and anticonvulsant barbiturates on specific [3 h] gaba binding to membrane preparations from rat brain cortex. Biochem. Pharmacol. 1982, 31, 2891–2895. [Google Scholar] [CrossRef]
- Brunner, H.; Ittner, K.P.; Lunz, D.; Schmatloch, S.; Schmidt, T.; Zabel, M. Highly enriched mixtures of methohexital stereoisomers by palladium-catalyzed allylation and their anaesthetic activity. Eur. J. Org. Chem. 2003, 2003, 855–862. [Google Scholar] [CrossRef]
- Lyons, K.E.; Pahwa, R. Pharmacotherapy of essential tremor. CNS Drugs 2008, 22, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Padmaja, A.; Reddy, G.S.; Venkata Nagendra Mohan, A.; Padmavathi, V. Michael adducts-source for biologically potent heterocycles. Chem. Pharm. Bull. 2008, 56, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Padmavathi, V.; Reddy, G.D.; Venkatesh, B.C.; Padmaja, A. One-pot synthesis of 5-{[bis (azolylmethylthio)] methylene} pyrimidine-2,4,6-(1H,3H,5H)-triones. Arch. Pharm. 2011, 344, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, C.; Fröscher, W. Low risk of development of substance dependence for barbiturates and clobazam prescribed as antiepileptic drugs: Results from a questionnaire study. CNS Neurosci. Ther. 2009, 15, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Maquoi, E.; Sounni, N.E.; Devy, L.; Olivier, F.; Frankenne, F.; Krell, H.-W.; Grams, F.; Foidart, J.-M.; Noël, A. Anti-invasive, antitumoral, and antiangiogenic efficacy of a pyrimidine-2,4,6-trione derivative, an orally active and selective matrix metalloproteinases inhibitor. Clin. Cancer Res. 2004, 10, 4038–4047. [Google Scholar] [CrossRef] [PubMed]
- Breyholz, H.J.; Wagner, S.; Faust, A.; Riemann, B.; Höltke, C.; Hermann, S.; Schober, O.; Schäfers, M.; Kopka, K. Radiofluorinated pyrimidine-2,4,6-triones as molecular probes for noninvasive mmp-targeted imaging. ChemMedChem 2010, 5, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, P.; Cataldo, D.; Endele, R.; Evrard, B.; Foidart, J.M.; Krell, H.W.; Zimmermann, G. Cyclodextrin Inclusions Complexes of Pyrimidine-2,4,6-Triones. Google Patents WO2005097058 A3, 17 August 2006. [Google Scholar]
- Richter, J.A.; Holtman, J.R. Barbiturates: Their in vivo effects and potential biochemical mechanisms. Prog. Neurobiol. 1982, 18, 275–319. [Google Scholar] [CrossRef]
- Neumann, D.M.; Cammarata, A.; Backes, G.; Palmer, G.E.; Jursic, B.S. Synthesis and antifungal activity of substituted 2,4,6-pyrimidinetrione carbaldehyde hydrazones. Biorg. Med. Chem. 2014, 22, 813–826. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Rees, C.W. Comprehensive Heterocyclic Chemistry; Pergamon Press: Oxford, UK, 1984. [Google Scholar]
- Perspicace, E.; Jouan-Hureaux, V.; Ragno, R.; Ballante, F.; Sartini, S.; La Motta, C.; Da Settimo, F.; Chen, B.; Kirsch, G.; Schneider, S. Design, synthesis and biological evaluation of new classes of thieno [3,2-d] pyrimidinone and thieno [1,2,3] triazine as inhibitor of vascular endothelial growth factor receptor-2 (vegfr-2). Eur. J. Med. Chem. 2013, 63, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Perretta, C.; Erickson, P.; Margosiak, S.; Almassy, R.; Lu, J.; Averill, A.; Yager, K.M.; Chu, S. Structure-based design, synthesis, and study of pyrazolo [1,5-a][1,3,5] triazine derivatives as potent inhibitors of protein kinase ck2. Bioorg. Med. Chem. Lett. 2007, 17, 4191–4195. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, E.T.; Lima, E.L. Reaction of 1,3-dimethyl-5-acetyl-barbituric acid (dab) with primary amines. Access to intermediates for selectively protected spermidines. Tetrahedron Lett. 2003, 44, 3621–3624. [Google Scholar] [CrossRef]
- Giziroglu, E.; Aygün, M.; Sarikurkcu, C.; Kazar, D.; Orhan, N.; Firinci, E.; Soyleyici, H.C.; Gokcen, C. Synthesis, characterization and antioxidant activity of new dibasic tridentate ligands: X-ray crystal structures of dmso adducts of 1,3-dimethyl-5-acetyl-barbituric acid o-hydroxybenzoyl hydrazone copper (ii) complex. Inorg. Chem. Commun. 2013, 36, 199–205. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 1987, 2, S1–S19. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Honarparvar, B.; Govender, T.; Maguire, G.E.; Soliman, M.E.; Kruger, H.G. Integrated approach to structure-based enzymatic drug design: Molecular modeling, spectroscopy, and experimental bioactivity. Chem. Rev. 2013, 114, 493–537. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.; Millam, J. Gaussview, version 5; Semichem Inc.: Shawnee Missions, KS, USA, 2009. [Google Scholar]
- Contreras, R.; Domingo, L.R.; Andrés, J.; Pérez, P.; Tapia, O. Nonlocal (pair site) reactivity from second-order static density response function: Gas-and solution-phase reactivity of the acetaldehyde enolate as a test case. J. Phys. Chem. A 1999, 103, 1367–1375. [Google Scholar] [CrossRef]
- Clare, B.W. Frontier orbital energies in quantitative structure-activity relationships: A comparison of quantum chemical methods. Theor. Chim. Acta 1994, 87, 415–430. [Google Scholar] [CrossRef]
- Clare, B.W. The relationship of charge transfer complexes to frontier orbital energies in qsar. J. Mol. Struct. Theochem. 1995, 331, 63–78. [Google Scholar] [CrossRef]
- Clare, B.W. Charge transfer complexes and frontier orbital energies in qsar: A congeneric series of electron acceptors. J. Mol. Struct. Theochem. 1995, 337, 139–150. [Google Scholar] [CrossRef]
- Heaton, C.; Miller, A.; Powell, R. Predicting the reactivity of fluorinated compounds with copper using semi-empirical calculations. J. Fluor. Chem. 2001, 107, 1–3. [Google Scholar] [CrossRef]
- Zhang, G.; Musgrave, C.B. Comparison of DFT methods for molecular orbital eigenvalue calculations. J. Phys. Chem. A 2007, 111, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Lewars, E. The concept of the potential energy surface. In Computational Chemistry: Chapter 2—Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Springer: Boston, MA, USA, 2003; pp. 9–41. [Google Scholar]
- Hendrickson, J.; Cram, D. Gs Hammond Organic Chemistry; McGraw-Hill: New York, NY, USA, 1970. [Google Scholar]
- Kruger, H.G. Ab initio mechanistic study of the protection of alcohols and amines with anhydrides. J. Mol. Struct. Theochem. 2002, 577, 281–285. [Google Scholar] [CrossRef]
- Singh, T.; Kruger, H.G.; Bisetty, K.; Power, T.D. Theoretical study on the formation of a pentacyclo-undecane cage lactam. Comput. Theor. Chem. 2012, 986, 63–70. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of shelx. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXTL-PC (Version 5.1); Siemens Analytical Instruments, Inc.: Madison, WI, USA, 1997. [Google Scholar]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Lengths | X-ray Crystal | DFT a | Bond Angles | X-ray Crystal | DFT a |
|---|---|---|---|---|---|
| O1-C1 | 1.218 (2) | 1.2215 | C2-N1-C3 | 116.2 (2) | 115.6509 |
| O2-C3 | 1.213 (3) | 1.2142 | C1-N1-C2 | 118.14 (17) | 118.7543 |
| O3-C5 | 1.264 (3) | 1.2489 | C1-N1-C3 | 125.7 (2) | 125.5948 |
| O4-C7 | 1.310 (3) | 1.3082 | C3-N2-C4 | 117.76 (19) | 118.4499 |
| N1-C1 | 1.404 (3) | 1.4113 | C3-N2-C5 | 123.31 (17) | 123.7613 |
| N1-C2 | 1.476 (3) | 1.4706 | C4-N2-C5 | 118.90 (19) | 117.7888 |
| N1-C3 | 1.383 (3) | 1.3878 | O1-C1-N1 | 119.39 (18) | 119.8531 |
| N2-C3 | 1.391 (3) | 1.4038 | N1-C1-C6 | 115.03 (17) | 115.4104 |
| N2-C5 | 1.372 (3) | 1.3781 | O1-C1-C6 | 125.58 (19) | 124.7364 |
| N2-C4 | 1.472 (3) | 1.4708 | O2-C3-N2 | 121.34 (18) | 121.7422 |
| N1-C3-N2 | 116.6 (2) | 116.5854 | |||
| O2-C3-N1 | 122.0 (2) | 121.6725 | |||
| N2-C5-C6 | 119.07 (18) | 118.5093 | |||
| O3-C5-N2 | 118.04 (18) | 118.5799 | |||
| O3-C5-C6 | 122.88 (19) | 122.9107 | |||
| O4-C7-C6 | 119.18 (18) | 120.1114 | |||
| O4-C7-C8 | 115.04 (17) | 114.6609 |
| -- | DFT (Enol Form) | DFT (Keto Form) |
|---|---|---|
| Etotal a | −760.87486944 | −760.84960093 |
| EHOMO | −0.27058 | −0.27366 |
| ELUMO | −0.07757 | −0.06897 |
| ΔE b | 0.19301 | 0.20469 |
| -- | Relative Energies (kcal/mol) |
|---|---|
| Keto form | 0 |
| Transition State | 40.54 |
| Enol form | −14.93 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, A.; Jad, Y.E.; Ghabbour, H.A.; De la Torre, B.G.; Kruger, H.G.; Albericio, F.; El-Faham, A. Synthesis, Crystal Structure and DFT Studies of 1,3-Dimethyl-5-propionylpyrimidine-2,4,6(1H,3H,5H)-trione. Crystals 2017, 7, 31. https://doi.org/10.3390/cryst7010031
Sharma A, Jad YE, Ghabbour HA, De la Torre BG, Kruger HG, Albericio F, El-Faham A. Synthesis, Crystal Structure and DFT Studies of 1,3-Dimethyl-5-propionylpyrimidine-2,4,6(1H,3H,5H)-trione. Crystals. 2017; 7(1):31. https://doi.org/10.3390/cryst7010031
Chicago/Turabian StyleSharma, Anamika, Yahya E. Jad, Hazem A. Ghabbour, Beatriz G. De la Torre, Hendrik G. Kruger, Fernando Albericio, and Ayman El-Faham. 2017. "Synthesis, Crystal Structure and DFT Studies of 1,3-Dimethyl-5-propionylpyrimidine-2,4,6(1H,3H,5H)-trione" Crystals 7, no. 1: 31. https://doi.org/10.3390/cryst7010031