
From Rate Measurements to Mechanistic Data for Condensed Matter Reactions: A Case Study Using the Crystallization of [Zn(OH2)6][ZnCl4]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
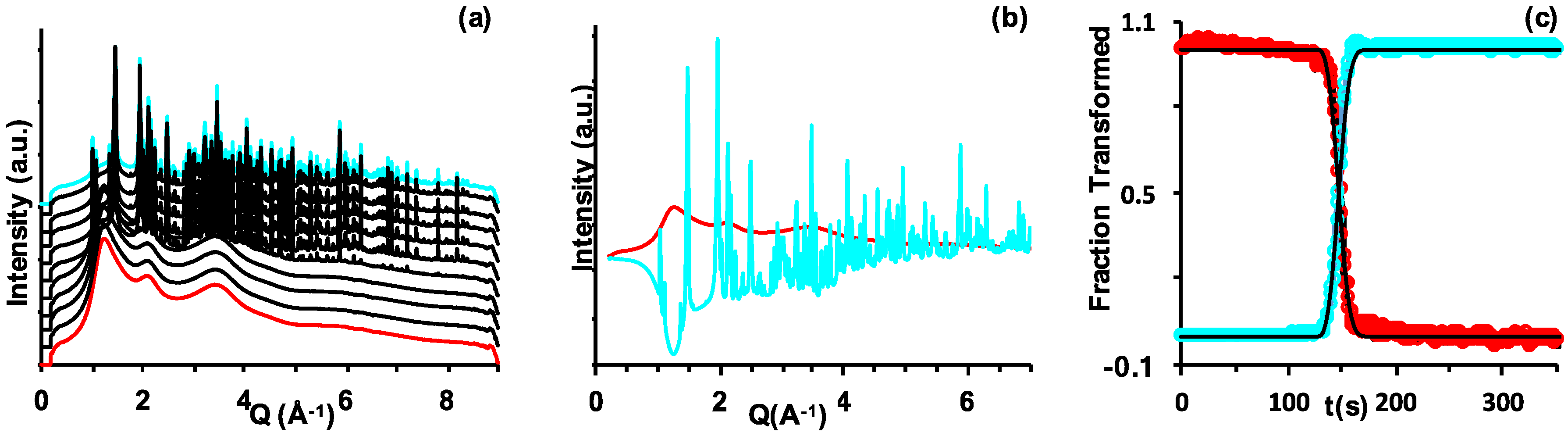
2.1. Temperature- and Time-Resolved X-ray Diffraction (TtXRD)
2.1.1. Bulk Sample Analysis
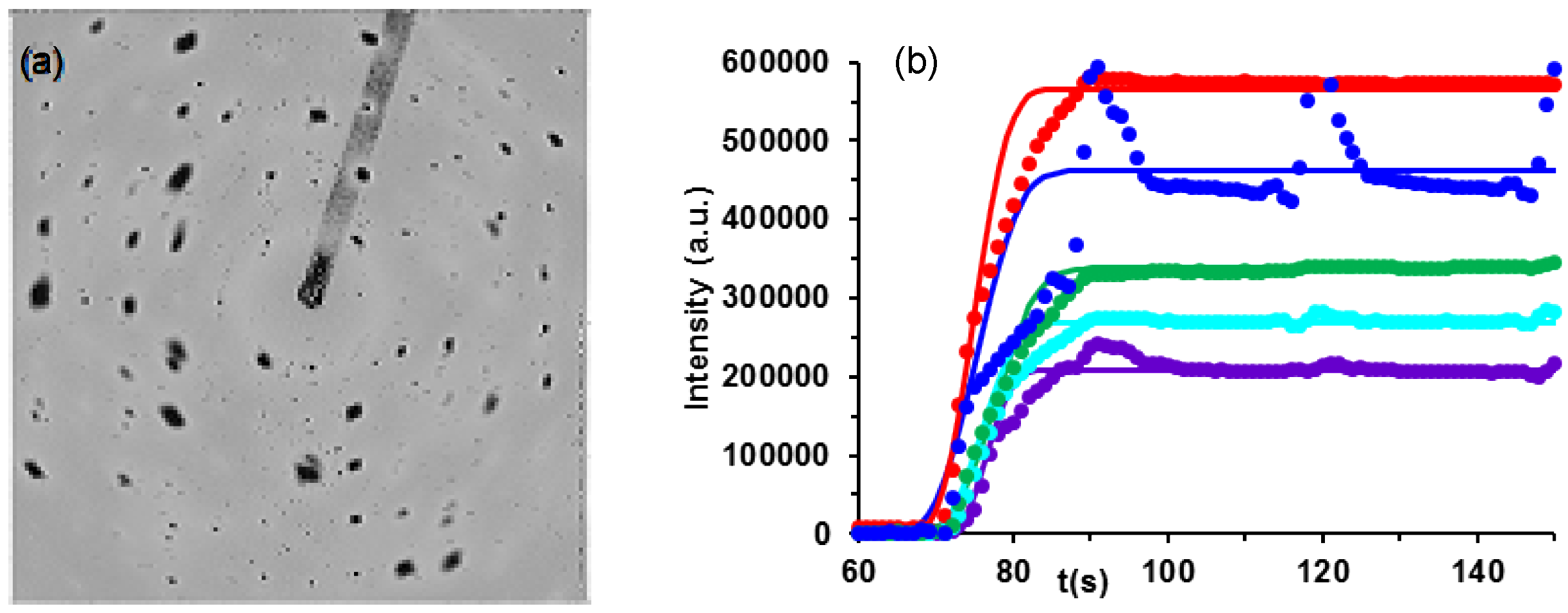
2.1.2. Individual Crystallite Analysis
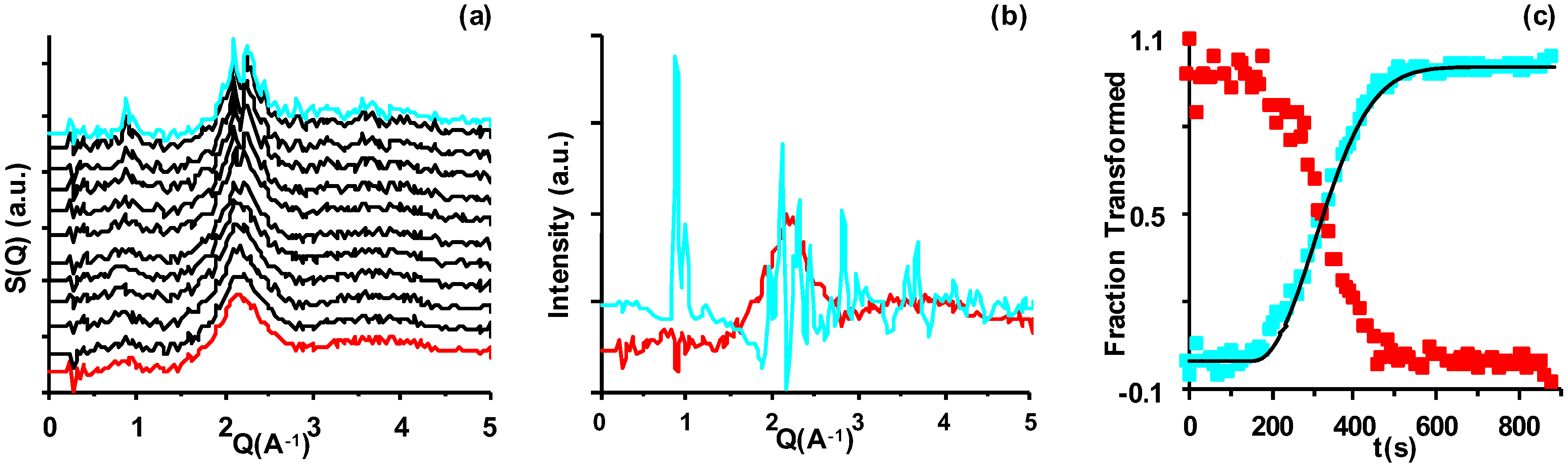
2.2. Temperature- and Time-Resolved Neutron Diffraction (TtND)
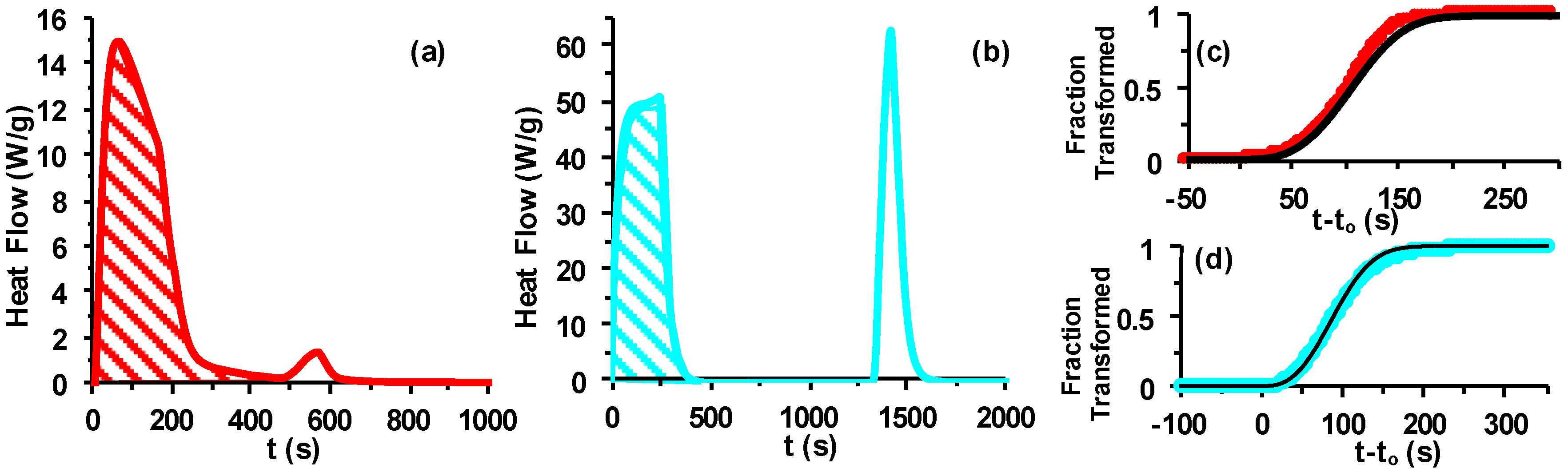
2.3. Differential Scanning Calorimetry (DSC)
2.4. Crystallization Video
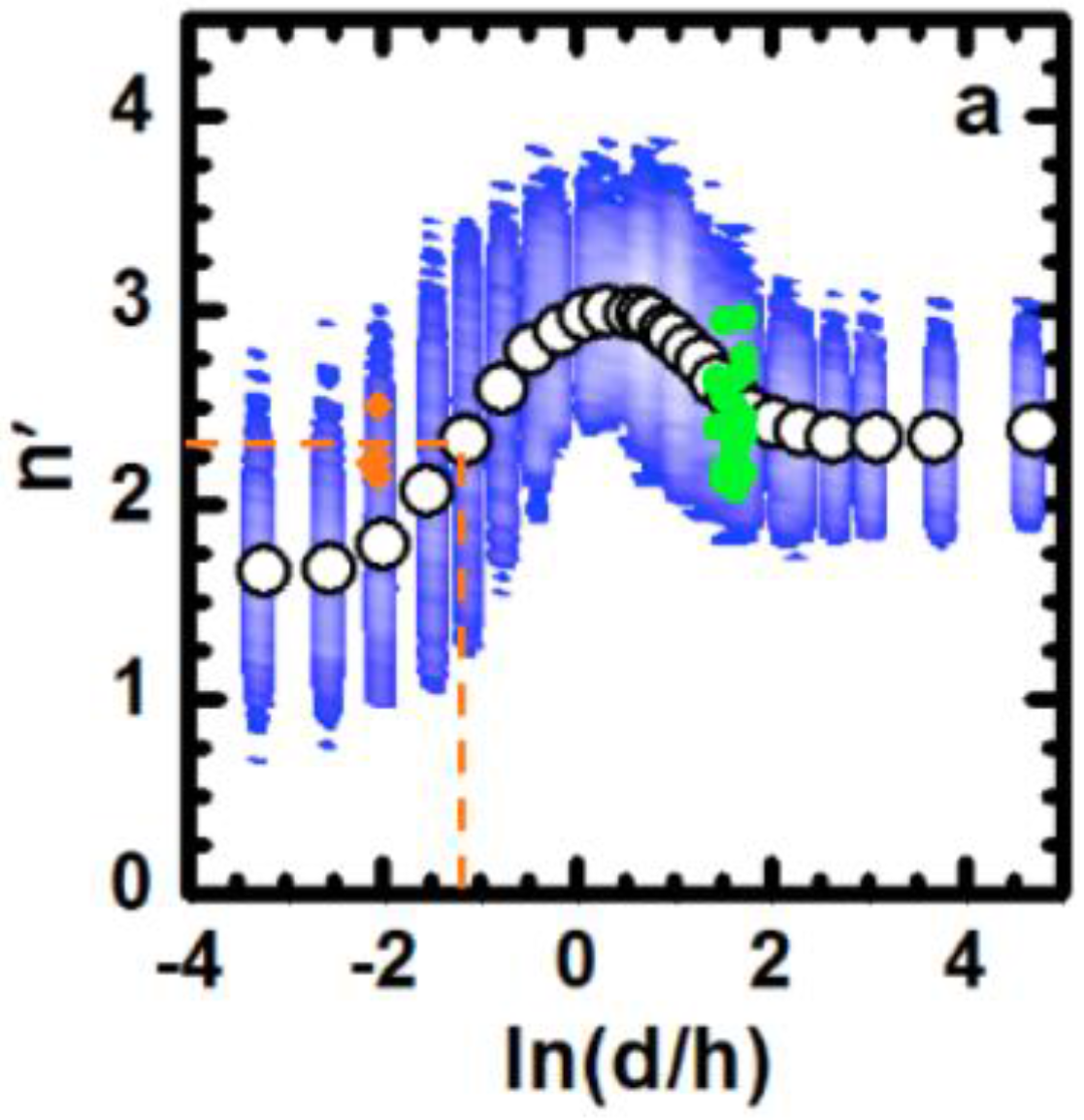
3. Discussion
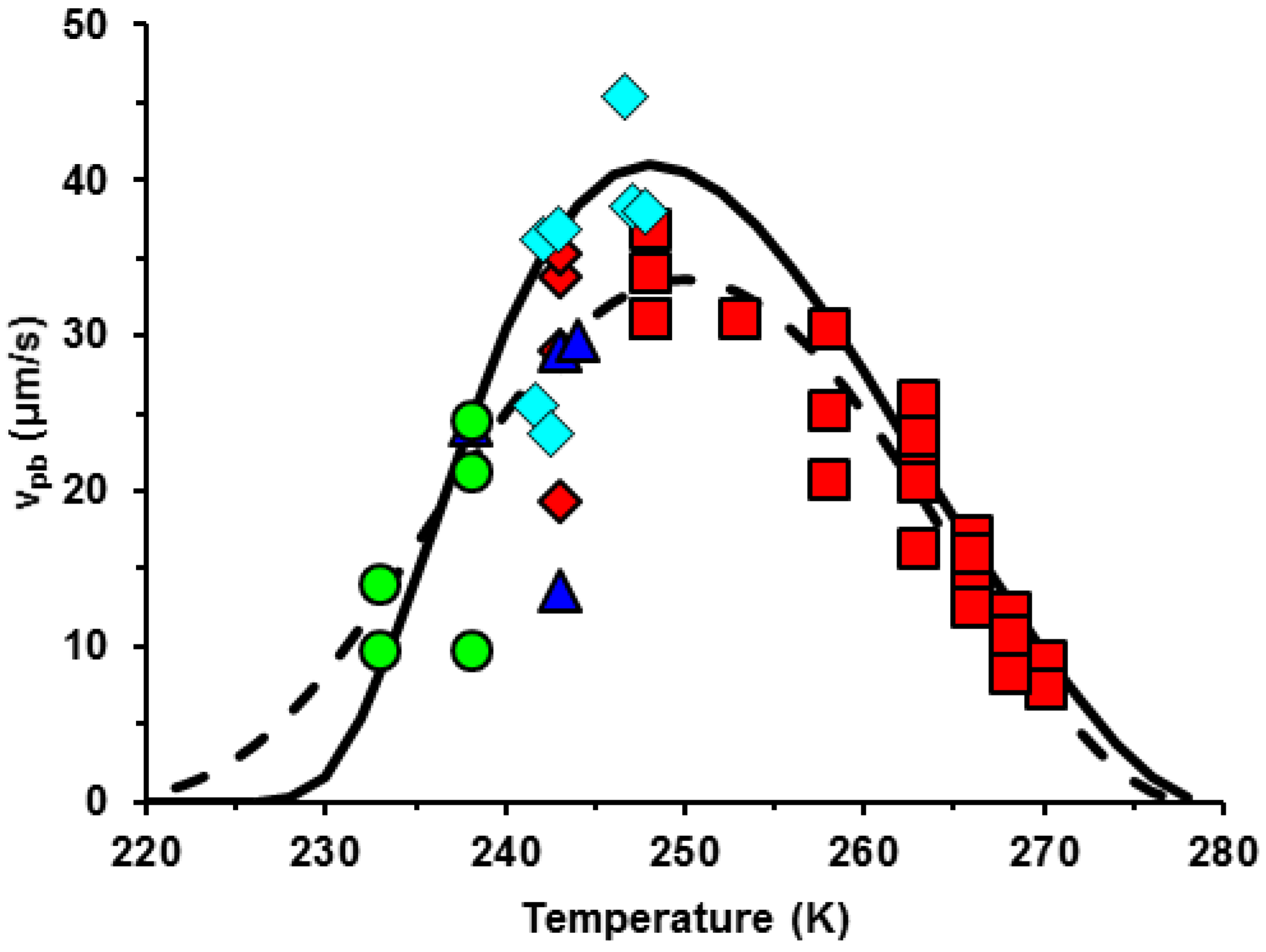
3.1. Velocity of the Phase Boundary
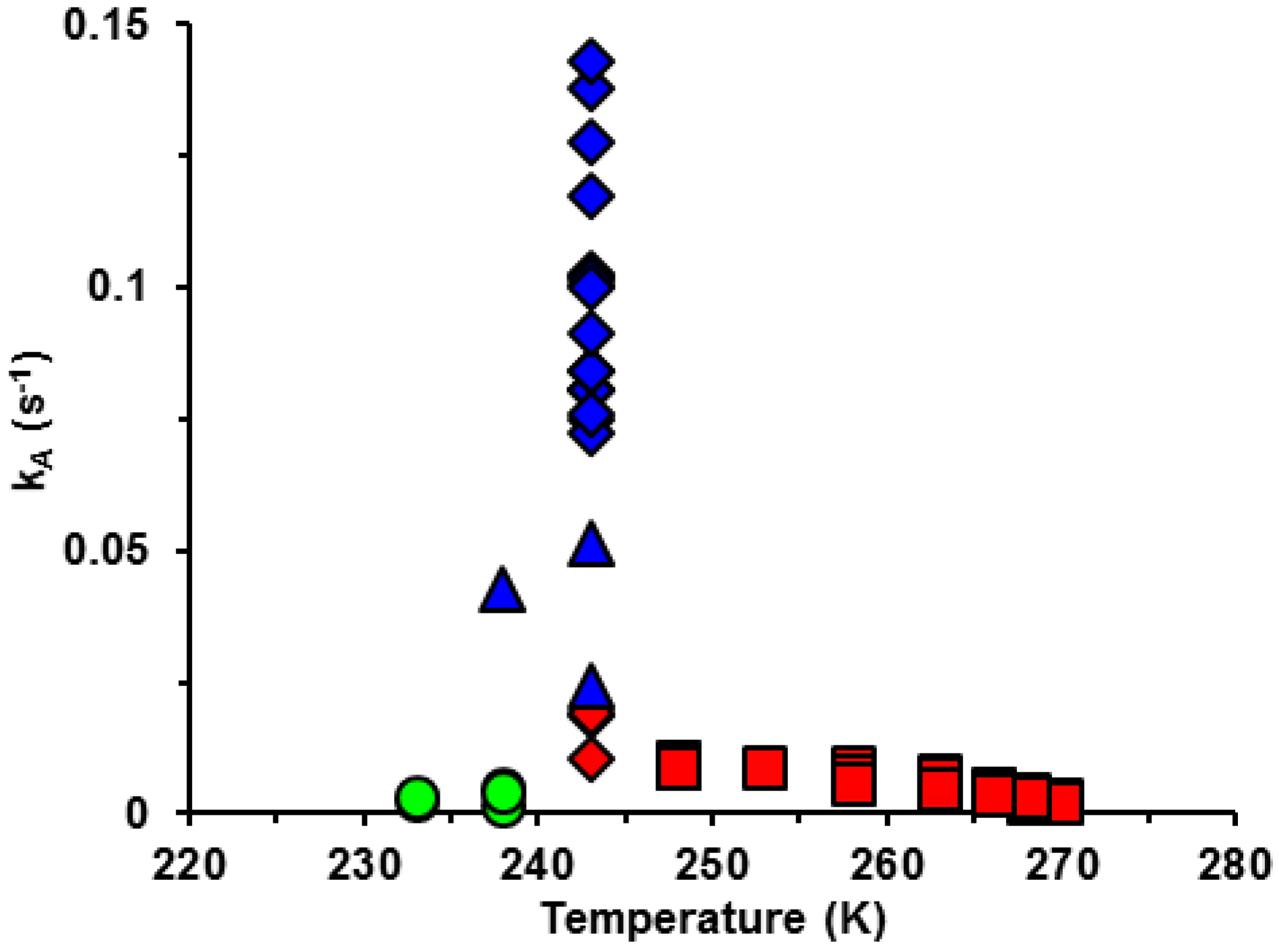
3.2. Activation Energy of Crystallization
4. Materials and Methods
4.1. Materials
4.2. Time-Temperature Resolved Synchrotron X-ray Diffraction (TtXRD)
4.3. Individual Crystallite Image Analysis
4.4. Time-Temperature Resolved Neutron Diffraction (TtND)
4.5. Isothermal Crystallization by DSC
4.6. Video Imaging of Crystallization
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maruyama, K.; Kagi, H.; Inoue, T.; Ohfuji, H.; Yoshino, T. In Situ Observation of Pressure-induced Crystallization from Amorphous Calcium Carbonate by Time-resolved X-ray Diffraction. Chem. Lett. 2015, 44, 434–436. [Google Scholar] [CrossRef]
- Dill, E.D.; Josey, A.A.; Folmer, J.C.W.; Hou, F.; Martin, J.D. Experimental Determination of the Crystallization Phase-Boundary Velocity in the Halozeotype CZX-1. Chem. Mater. 2013, 25, 3932–3940. [Google Scholar] [CrossRef]
- Kolmogorov, A.N. Statistical theory of crystallization of metals. Acad. Sci. Urss. Sci. 1937, 3, 355–359. [Google Scholar]
- Johnson, W.A.; Mehl, R.F. Reaction kinetics in processes of nucleation and growth. Trans. Am. Inst. Min. Metall. Eng. 1939, 135, 416–442. [Google Scholar]
- Avrami, M. Kinetics of Phase Change. I General Theory . J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of Phase Change. II: Transformation-Time Relations for Random Distribution of Nuclei. J. Chem. Phys. 1940, 8, 212–224. [Google Scholar] [CrossRef]
- Avrami, M. Granulation, Phase Change, and Microscructure Kinetics of Phase Change III. J. Chem. Phys. 1941, 9, 177–184. [Google Scholar] [CrossRef]
- Wilcox, R.J.; Losey, B.P.; Folmer, J.C.W.; Martin, J.D.; Zeller, M.; Sommer, R. Crystalline and Liquid Structure of Zinc Chloride Tri-hydrate: A unique ionic liquid. Inorg. Chem. 2015, 54, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Dill, E.D.; Folmer, J.C.W.; Martin, J.D. Simulating crystallization to elucidate the parameter space in the KJMA condensed phase transformation model. Chem. Mater. 2013, 25, 3941–3951. [Google Scholar] [CrossRef]
- Hou, F.; Martin, J.D.; Dill, E.D.; Folmer, J.C.; Josey, A.A. Transition Zone Theory of Crystal Growth and Viscosity. Chem. Mater. 2015, 27, 3526–3532. [Google Scholar] [CrossRef]
- Wilson, H.A. On the Velocity of Solidification and Viscosity of Super-cooled Liquids. Philos. Mag. 1900, 50, 238–250. [Google Scholar] [CrossRef]
- Henry, E.R.; Hofrichter, J. Singular Value Decomposition: Application to Analysis of Experimental Data. In Methods in Enzymology; Ludwig Brand, M.L.J., Ed.; Academic Press: San Diego, CA, USA, 1992; Volume 210, pp. 129–192. [Google Scholar]
- Dill, E.D.; Folmer, J.C.W.; Martin, J.D. RamDog: A Graphical User Interface Program to Interactively Analyze 2D Diffraction Images. ZENDO, 2014. Available online: https://zenodo.org/record/12268#.WGcPf3u3ySI (accessed on 30 December 2016). [CrossRef]
- Neuefeind, J.; Feygenson, M.; Carruth, J.; Hoffmann, R.; Chipley, K. The Nanoscale Ordered Materials Diffractometer NOMAD at the Spallation Neutron Source SNS. Nucl. Instrum. Methods Phys. Res. Sect. B 2012, 287, 68–78. [Google Scholar] [CrossRef]
- Lan, S.; Wei, X.; Zhou, J.; Lu, Z.P.; Wu, X.; Feygenson, M.; Neuefeind, J.; Wang, X.L. In-situ study of crystallization kinetics in ternary bulk metallic glass alloys with different glass forming abilities. Appl. Phys. Lett. 2014, 105, 201906. [Google Scholar] [CrossRef]
- Adam, G.; Gibbs, J.H. On Temperature Dependence of Cooperative Relaxation Properties in Glass-Forming Liquids. J. Chem. Phys. 1965, 43, 139–146. [Google Scholar] [CrossRef]
- Kauzmann, W. The Nature of the Glassy State and the Behavior of Liquids at Low Temperatures. Chem. Rev. 1948, 43, 219–256. [Google Scholar] [CrossRef]
- Chupas, P.J.; Chapman, K.W.; Lee, P.L. Applications of an amorphous silicon-based area detector for high-resolution, high-sensitivity and fast time-resolved pair distribution function measurements. J. Appl. Crystallogr. 2007, 40, 463–470. [Google Scholar] [CrossRef]
- Hammersley, A.P.; Svensson, S.O.; Hanfland, M.; Fitch, A.N.; Hausermann, D. Two-Dimensional Detector Software: From Real Detector to Idealized Image or Two-Theta Scan. High Press. Res. 1996, 14, 235–248. [Google Scholar] [CrossRef]
- Van der Walt, S.; Schönberger, J.L.; Nunez-Iglesias, J.; Boulogne, F.; Warner, J.D.; Yager, N.; Gouillart, E.; Yu, T. The Scikit-Image Contributors. Scikit-Image: Image processing in Python. PeerJ 2014, 2, e453. [Google Scholar] [CrossRef] [PubMed]
- Van der Walt, A.; Colbert, S.C.; Varoquaux, G. The NumPy Array: A Structure for Efficient Numerical Computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef]
- Jones, E.; Oliphant, E.; Peterson, P. SciPy: Open Source Scientific Tools for Python. 2001. Available online: http://www.scipy.org/ (accessed on 29 August 2016).
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]









© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hillis, B.G.; Losey, B.P.; Weng, J.; Ghaleb, N.; Hou, F.; Martin, J.D. From Rate Measurements to Mechanistic Data for Condensed Matter Reactions: A Case Study Using the Crystallization of [Zn(OH2)6][ZnCl4]. Crystals 2017, 7, 11. https://doi.org/10.3390/cryst7010011
Hillis BG, Losey BP, Weng J, Ghaleb N, Hou F, Martin JD. From Rate Measurements to Mechanistic Data for Condensed Matter Reactions: A Case Study Using the Crystallization of [Zn(OH2)6][ZnCl4]. Crystals. 2017; 7(1):11. https://doi.org/10.3390/cryst7010011
Chicago/Turabian StyleHillis, Berkley G., Bradley P. Losey, James Weng, Nezar Ghaleb, Feier Hou, and James D. Martin. 2017. "From Rate Measurements to Mechanistic Data for Condensed Matter Reactions: A Case Study Using the Crystallization of [Zn(OH2)6][ZnCl4]" Crystals 7, no. 1: 11. https://doi.org/10.3390/cryst7010011