
Colloidal Crystallization in 2D for Short-Ranged Attractions: A Descriptive Overview

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Simulations
3. Description of the Subprocesses
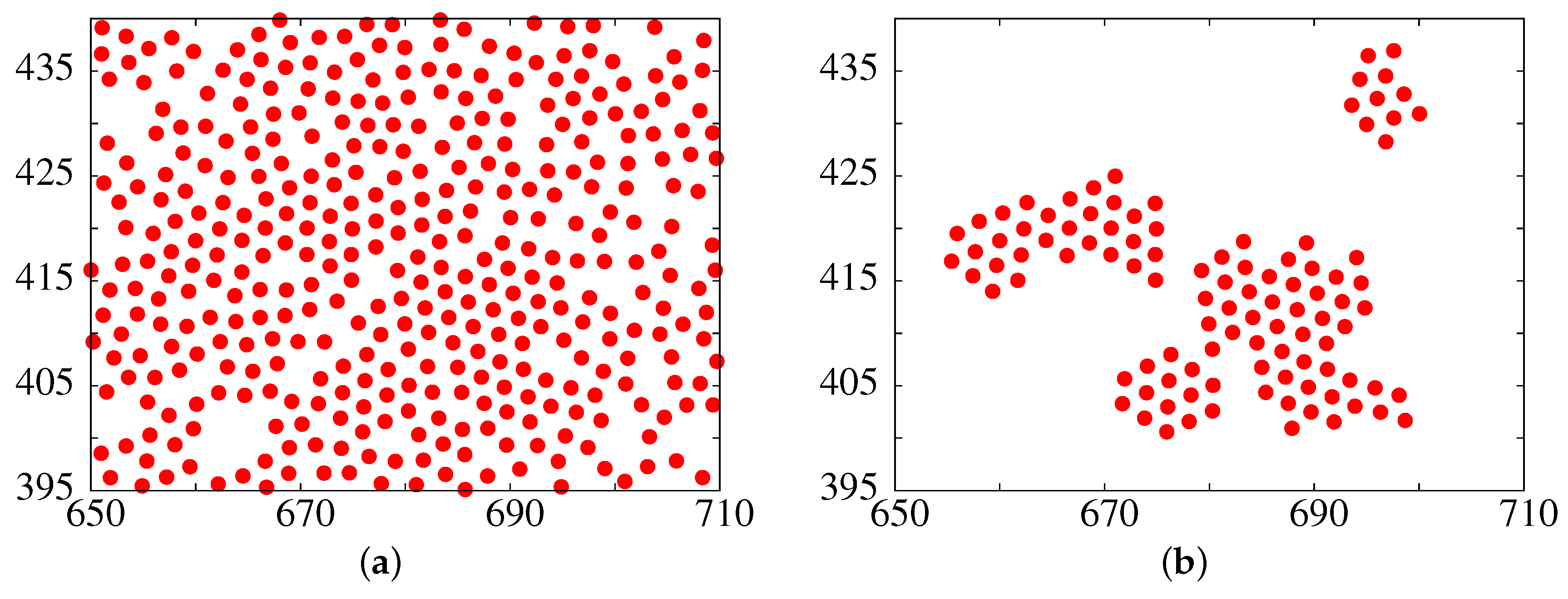
3.1. The Metastable Fluid-Fluid Phase Separation
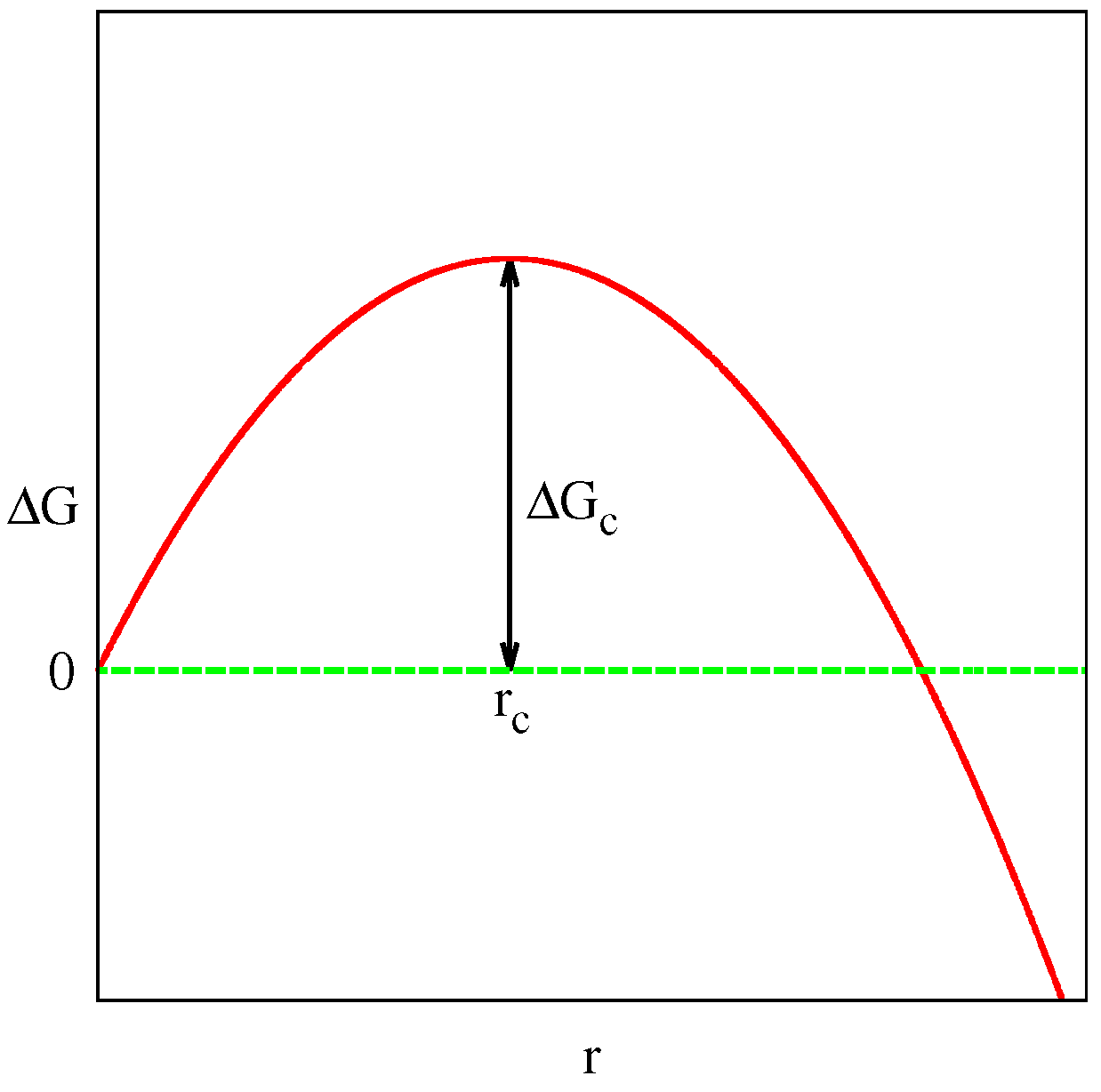
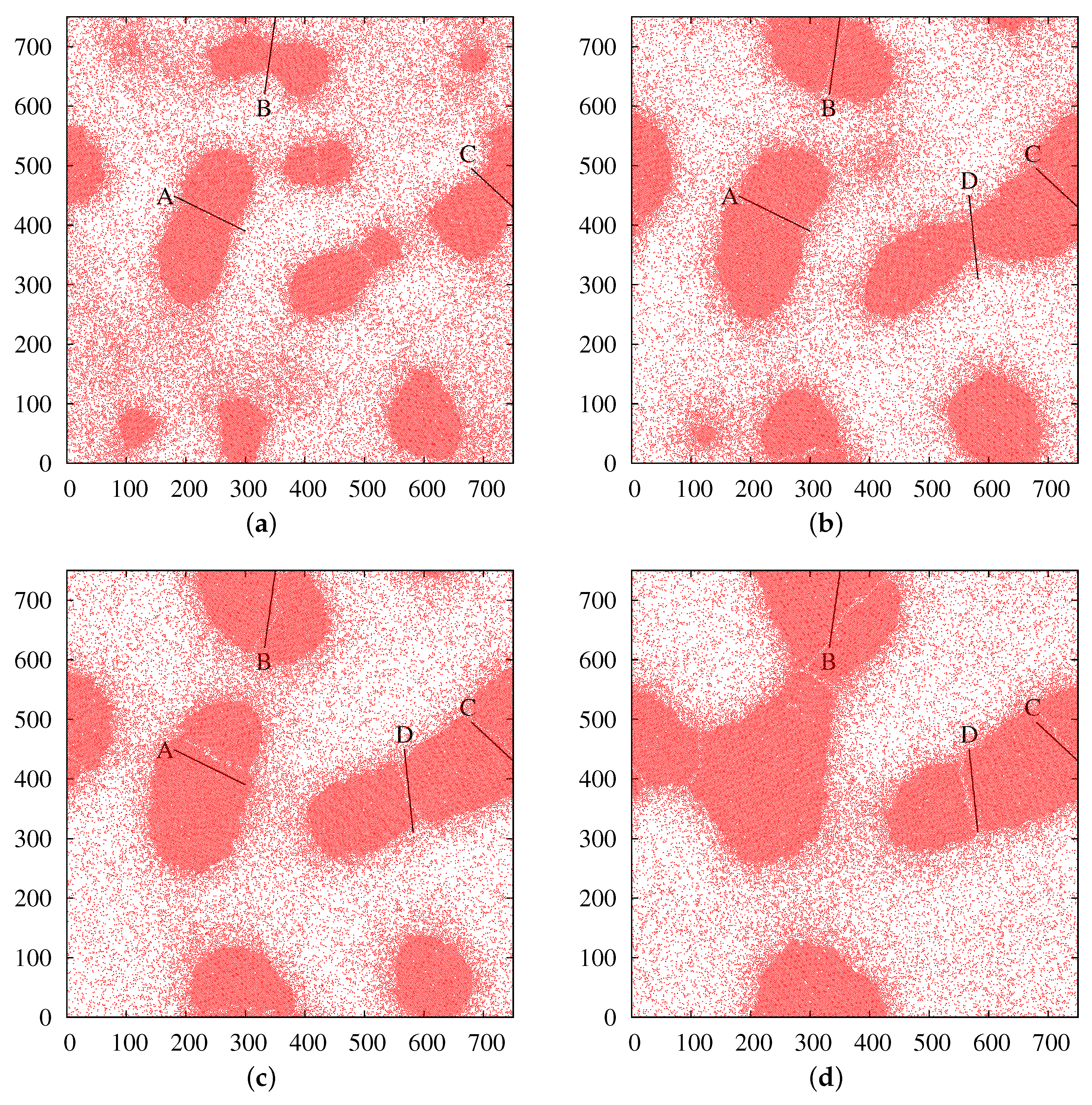
3.2. Crystalline Nucleation
3.3. Ostwald Ripening
- (i)
- At any instant of the ripening process, there exists a critical radius . Grains with a larger radius grow and smaller grains shrink. During the ripening process increases with time.
- (ii)
- In the long time limit, the grain size distribution becomes self-similar, when the sizes are scaled with .
- (iii)
- The critical radius is equal to the number-average of the limiting self-similar size distribution.
- (iv)
- The ripening rate, defined as , is constant and given by
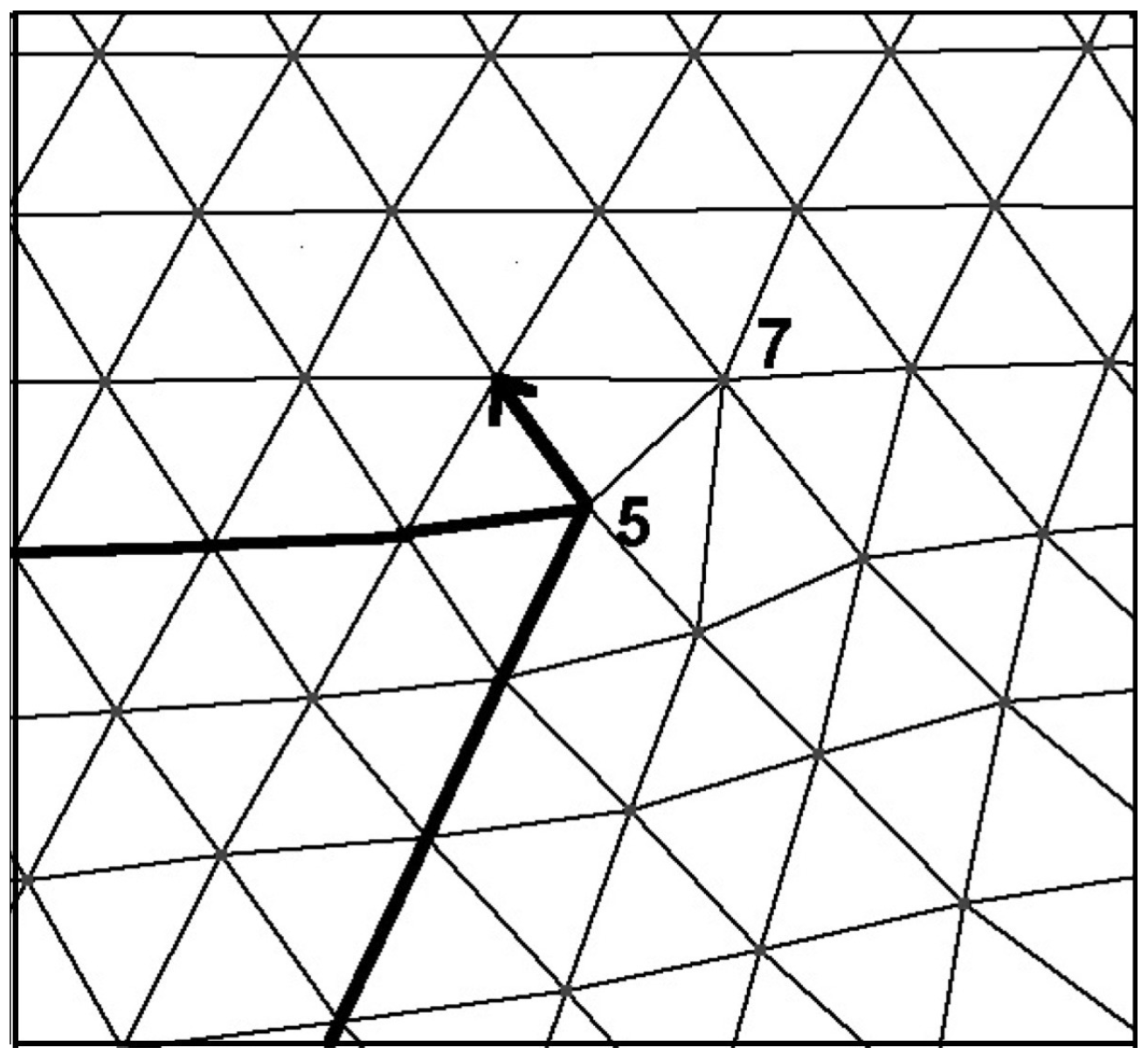
3.4. Grain Boundary Formation and Dynamics
3.5. Grain Coarsening by Dislocation Disappearance after Grain Boundary Healing
3.6. Some Theoretical Developments of Grain Coarsening
4. Discussion and Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Pieranski, P. Colloidal crystals. Contemp. Phys. 1983, 24, 25–73. [Google Scholar] [CrossRef]
- Gasser, U. Crystallization in three- and two-dimensional colloidal suspensions. J. Phys. Condens. Matter 2009, 21, 203101. [Google Scholar] [CrossRef] [PubMed]
- Schall, P.; Cohen, I.; Weitz, D.A.; Spaepen, F. Visualization of dislocation dynamics in colloidal crystals. Science 2004, 305, 1944–1948. [Google Scholar] [CrossRef] [PubMed]
- Schall, P.; Cohen, I.; Weitz, D.A.; Spaepen, F. Visualizing dislocation nucleation by indenting colloidal crystals. Nature 2006, 440, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S. Colloid model for atoms. Nat. Mater. 2006, 5, 253–254. [Google Scholar] [CrossRef] [PubMed]
- Schall, P. Laser difraction microscopy. Rep. Prog. Phys. 2009, 72, 076601. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, F.; Peng, Y.; Zheng, Z.; Han, Y. Imaging the homogeneous nucleation during the melting of superheated colloidal crystals. Science 2012, 338, 87–90. [Google Scholar] [CrossRef] [PubMed]
- See for example Crystallization of Nucleic Acids and Proteins: A Practical Approach, 2nd ed.; Ducruix, A.; Giegé, R. (Eds.) Oxford University Press: Oxford, UK, 2000.
- Pieranski, P. Two-dimensional interfacial colloidal crystals. Phys. Rev. Lett. 1980, 45, 569–572. [Google Scholar] [CrossRef]
- Grimes, C.C.; Adams, G. Evidence for a liquid-to-crystal phase transition in a classical, two-dimensional sheet of electrons. Phys. Rev. Lett. 1979, 42. [Google Scholar] [CrossRef]
- Onoda, G.Y. Direct observation of two-dimensional, dynamical clustering and ordering with colloids. Phys. Rev. Lett. 1985, 55, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y. Monte Carlo studies of two-dimensional melting: Dislocation vector systems. Phys. Rev. B 1982, 26, 6239–6253. [Google Scholar] [CrossRef]
- Chui, S.T. Grain-boundary theory of melting in two dimensions. Phys. Rev. Lett. 1982, 48, 933–935. [Google Scholar] [CrossRef]
- Tang, Y.; Armstrong, A.J.; Mockler, R.C.; O’Sullivan, W.J. Free-expansion melting of a colloidal monolayer. Phys. Rev. Lett. 1989, 62, 2401–2404. [Google Scholar] [CrossRef] [PubMed]
- Lansac, Y; Glaser, M.A.; Clark, N.A. Discrete elastic model for two-dimensional melting. Phys. Rev. E 2006, 73, 041501. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.A.; van Winkle, D.H. Experimental observation of two-stage melting in a classical two-dimensional screened coulomb system. Phys. Rev. Lett. 1987, 58, 1200–1203. [Google Scholar] [CrossRef] [PubMed]
- Kusner, R.E.; Mann, J.A.; Kerins, J.; Dahm, A.J. Two-stage melting of a two-dimensional colloidal lattice with dipole interactions. Phys. Rev. Lett. 1994, 73, 3113–3116. [Google Scholar] [CrossRef] [PubMed]
- Bladon, P.; Frenkel, D. Dislocation unbinding in dense two-dimensional crystals. Phys. Rev. Lett. 1995, 74, 2519–2522. [Google Scholar] [CrossRef] [PubMed]
- Zahn, K.; Lenke, R.; Maret, G. Two-stage melting of paramagnetic colloidal crystals in two dimensions. Phys. Rev. Lett. 1999, 82, 2721–2724. [Google Scholar] [CrossRef]
- Von Grünberg, H.H.; Keim, P.; Zahn, K.; Maret, G. Elastic behavior of a two-dimensional crystal near melting. Phys. Rev. Lett. 2004, 93, 255703. [Google Scholar] [CrossRef] [PubMed]
- Keim, P.; Maret, G.; von Grünberg, H.H. Frank’s constant in the hexatic phase. Phys. Rev. E 2007, 75, 031402. [Google Scholar] [CrossRef] [PubMed]
- Marcus, A.H.; Rice, S.A. Observations of first-order liquid-to-hexatic and hexatic-to-solid phase transitions in a confined colloidal suspension. Phys. Rev. Lett. 1996, 77, 2577–2580. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.J.; Chen, L.J. Phase transitions in two-dimensional colloidal particles at oil/water interfaces. J. Chem. Phys. 2007, 126, 034706. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.P.; Krauth, W. Two-step melting in two dimensions: First-order liquid-hexatic transition. Phys. Rev. Lett. 2011, 107, 155704. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.; Anderson, J.A.; Glotzer, S.C.; Isobe, M.; Bernard, E.P.; Krauth, W. Hard-disk equation of state: First-order liquid-hexatic transition in two dimensions with three simulation methods. Phys. Rev. E 2013, 87, 042134. [Google Scholar] [CrossRef] [PubMed]
- Kapfer, S.C.; Krauth, W. Two-dimensional melting: From liquid-hexatic coexistence to continuous transitions. Phys. Rev. Lett. 2015, 114, 035702. [Google Scholar] [CrossRef] [PubMed]
- Kosterlitz, J.M.; Thouless, D.J. Ordering, metastability and phase transitions in two-dimensional systems. J. Phys. C Solid State Phys. 1973, 6, 1181–1203. [Google Scholar] [CrossRef]
- Nelson, D.R.; Halperin, B.I. Dislocation-mediated melting in two dimensions. Phys. Rev. B 1979, 19, 2457–2484. [Google Scholar] [CrossRef]
- Young, A.P. Melting and the vector coulomb gas in two dimensions. Phys. Rev. B 1979, 19, 1855–1866. [Google Scholar] [CrossRef]
- Dillman, P.; Maret, G.; Keim, P. Polycrystalline solidification in a quenched 2D colloidal system. J. Phys. Condens. Matter 2008, 20, 404216. [Google Scholar] [CrossRef]
- Lekkerkerker, H.N.W.; Poon, W.C.K.; Pusey, P.N.; Stroobants, A.; Warren, P.B. Phase behaviour of colloid + polymer mixtures. Europhys. Lett. 1992, 20, 559–564. [Google Scholar] [CrossRef]
- Hagen, M.H.J.; Frenkel, D. Determination of phase diagrams for the hard-core attractive Yukawa system. J. Chem. Phys. 1994, 101, 4093–4097. [Google Scholar] [CrossRef]
- Ilett, S.M.; Orrock, A.; Poon, W.C.K.; Pusey, P.N. Phase behavior of a model colloid-polymer mixture. Phys. Rev. E 1995, 51, 1344–1352. [Google Scholar] [CrossRef]
- Asherie, N.; Lomakin, A.; Benedek, G.B. Phase diagram of colloidal solutions. Phys. Rev. Lett. 1996, 77, 4832–4835. [Google Scholar] [CrossRef] [PubMed]
- Hobbie, E.K. Metastability and depletion-driven aggregation. Phys. Rev. Lett. 1998, 81, 3996–3999. [Google Scholar] [CrossRef]
- Zhang, T.H.; Liu, X.Y. How does a transient amorphous precursor template crystallization. J. Am. Chem. Soc. 2007, 129, 13520–13526. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.R.; Dinsmore, A.D. Experimental evidence for two-step nucleation in colloidal crystallization. Phys. Rev. Lett. 2009, 102, 198302. [Google Scholar] [CrossRef] [PubMed]
- Berland, C.R.; Thurston, G.M.; Kondo, M.; Broide, M.L.; Pande, J.; Ogun, O.; Benedek, G.B. Solid-liquid phase boundaries of lens protein solutions. Proc. Natl. Acad. Sci. USA 1992, 89, 1214–1218. [Google Scholar] [CrossRef] [PubMed]
- Ten Wolde, P.R.; Frenkel, D. Enhancement of protein crystal nucleation by critical density fluctuations. Science 1997, 277, 1975–1978. [Google Scholar] [CrossRef] [PubMed]
- Talanquer, V.; Oxtoby, D.W. Crystal nucleation in the presence of a metastable critical point. J. Chem. Phys. 1998, 109, 223–227. [Google Scholar] [CrossRef]
- Galkin, O.; Vekilov, P.G. Control of protein crystal nucleation around the metastable liquid-liquid phase boundary. Proc. Natl. Acad. Sci. USA 2000, 97, 6277–6281. [Google Scholar] [CrossRef] [PubMed]
- Lomakin, A.; Asherie, N.; Benedek, G.B. Liquid-solid transition in nuclei of protein crystals. Proc. Natl. Acad. Sci. USA 2003, 100, 10254–10257. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Cates, M.E.; Lekkerkerker, H.N.W. Depletion force in colloidal systems. Phys. A 1995, 222, 10–24. [Google Scholar] [CrossRef]
- Rosenbaum, D.; Zamora, P.C.; Zukoski, C.F. Phase behavior of small attractive colloidal particles. Phys. Rev. Lett. 1996, 76, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Kelton, K.F. Crystal nucleation in liquids and glasses. In Solid State Physics; Ehrenreich, H., Turnbull, D., Eds.; Academic Press: San Diego, CA, USA, 1991; Volume 45, pp. 75–177. [Google Scholar]
- Gasser, U.; Weeks, E.R.; Schofield, A.; Pusey, P.N.; Weitz, D.A. Real-space imaging of nucleation and growth in colloidal crystallization. Science 2001, 292, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Rubin-Zuzic, M.; Morfill, G.E.; Ivlev, A.; Pompl, R.; Klumov, B.A.; Bunk, W.; Thomas, H.M.; Rothermel, H.; Havnes, O.; Fouquet, A. Kinetic Develpment of crystallization fronts in complex plasmas. Nat. Phys. 2006, 2, 181–185. [Google Scholar] [CrossRef]
- Yau, S.T.; Vekilov, P.G. Quasi-planar nucleus structure in apoferritin crystallization. Nature 2000, 406, 494–497. [Google Scholar] [PubMed]
- Pan, A.C.; Chandler, D. Dynamics of nucleation in the Ising model. J. Phys. Chem. B 2004, 108, 19681–19686. [Google Scholar] [CrossRef]
- Ostwald, W. Studien uber die Bildung und Umwandlung fester Korper. Z. Phys. Chem. 1897, 22, 289–330. [Google Scholar]
- Penn, R.L.; Banfield, J.F. Morphology development and crystal growth in nanocrystalline aggregates under hydrotermal conditions: Insights from titania. Geochim. Cosmochim. Acta 1999, 63, 1549–1557. [Google Scholar] [CrossRef]
- Madras, G.; McCoy, B.J. Growth and ripening kinetics of crystalline polymorphs. Cryst. Growth Des. 2003, 3, 981–990. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, H.; Banfield, J.F. Two-stage crystal-growth kinetics observed during hydrotermal coarsening of nanocrystalline ZnS. Nano Lett. 2003, 3, 373–378. [Google Scholar] [CrossRef]
- Streets, A.M.; Quake, S.R. Ostwald ripening of clusters during protein crystallization. Phys. Rev. Lett. 2010, 104, 178102. [Google Scholar] [CrossRef] [PubMed]
- Iacopini, S.; Palberg, T.; Schöpe, H.J. Ripening-dominated crystallization in polydisperse hard-sphere-like colloids. Phys. Rev. E 2009, 79, 010601. [Google Scholar] [CrossRef] [PubMed]
- Stavans, J. The evolution of cellular structures. Rep. Prog. Phys. 1993, 56, 733–789. [Google Scholar] [CrossRef]
- Gokhale, S.; Nagamanasa, K.H.; Ganapathy, R.; Sood, A.K. Grain growth and grain boundary dynamics in colloidal polycrystals. Soft Matter 2013, 9, 6634–6644. [Google Scholar] [CrossRef]
- Edwards, T.D.; Yang, Y.; Beltran-Villegas, D.J.; Bevan, M.A. Colloidal crystal grain boundary formation and motion. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Nagamanasa, K.H.; Gokhale, S.; Ganapathy, R.; Sood, A.K. Confined glassy dynamics at grain boundaries in colloidal crystals. Proc. Natl. Acad. Sci. USA 2011, 108, 11323–11326. [Google Scholar] [CrossRef] [PubMed]
- Skinner, T.O.E.; Aarts, D.G.A.L.; Dullens, R.P.A. Supercooled dynamics of grain boundary particles in two-dimensional colloidal crystals. J. Chem. Phys. 2011, 135, 124711. [Google Scholar] [CrossRef] [PubMed]
- Trautt, Z.T.; Upmanyu, M.; Karma, A. Interface mobility from interface random walk. Science 2006, 314, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Skinner, T.O.E.; Aarts, D.G.A.L.; Dullens, R.P.A. Grain-boundary fluctuations in two-dimensional colloidal crystals. Phys. Rev. Lett. 2010, 105, 168301. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.A.; Easterling, K.E. Phase Transformations in Metals and Alloys, 2nd ed.; CRC Press: Boca Raton, FL, USA, 1992. [Google Scholar]
- Kikuchi, K.; Yoshida, M.; Maekawa, T.; Watanabe, H. Metropolis Monte Carlo method as a numerical technique to solve the Fokker-Planck equation. Chem. Phys. Lett. 1991, 185, 335–338. [Google Scholar] [CrossRef]
- Kikuchi, K.; Yoshida, M.; Maekawa, T.; Watanabe, H. Metropolis Monte Carlo method for Brownian dynamics simulation generalized to include hydrodynamics interactions. Chem. Phys. Lett. 1992, 196, 57–61. [Google Scholar] [CrossRef]
- Yoshida, M.; Kikuchi, K. Metropolis Monte Carlo Brownian dynamics simulation of the ion atmosphere polarization around a rodlike polyion. J. Phys. Chem. 1994, 98, 10303–10306. [Google Scholar] [CrossRef]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar]
- Honeycutt, J.D.; Andersen, H.C. The effect of periodic boundary conditions on homogeneous nucleation observed in computer simulations. Chem. Phys. Lett. 1984, 108, 535–538. [Google Scholar] [CrossRef]
- Honeycutt, J.D.; Andersen, H.C. Small system size artifacts in the molecular dynamics simulation of homogeneous crystal nucleation in supercooled atomic liquids. J. Phys. Chem. 1986, 90, 1585–1589. [Google Scholar] [CrossRef]
- González, A.E.; Ixtlilco-Cortés, L. Fractal structure of the crystalline-nuclei boundaries in 2D colloidal crystallization: Computer simulations. Phys. Lett. A 2012, 376, 1375–1379. [Google Scholar] [CrossRef]
- Halperin, B.I.; Nelson, D.R. Theory of two-dimensional melting. Phys. Rev. Lett. 1978, 41, 121–124. [Google Scholar] [CrossRef]
- Fraser, D.P.; Zuckermann, M.J.; Mouritsen, O.G. Simulation technique for hard-disk models in two dimensions. Phys. Rev. A 1990, 42, 3186–3195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaster, A. Computer simulation of the two-dimensional melting transition using hard disks. Phys. Rev. E 1999, 59, 2594–2602. [Google Scholar] [CrossRef]
- Terao, T.; Nakayama, T. Crystallization in a quasi-two-dimensional colloidal system at an air-water interface. Phys. Rev. E 1999, 60, 7157–7162. [Google Scholar] [CrossRef]
- Huerta, A.; Naumis, G.G.; Wasan, D.T.; Henderson, D.; Trokhymchuk, A. Attraction driven disorder in a hard-core colloidal monolayer. J. Chem. Phys. 2004, 120, 1506–1510. [Google Scholar] [CrossRef] [PubMed]
- Dillman, P.; Maret, G.; Keim, P. Two-dimensional colloidal systems in time-dependent magnetic fields. Eur. Phys. J. Spec. Top. 2013, 222, 2941–2959. [Google Scholar] [CrossRef]
- Lutsko, J.F.; Nicolis, G. Theoretical evidence for a dense fluid precursor to crystallization. Phys. Rev. Lett. 2006, 96, 046102. [Google Scholar] [CrossRef] [PubMed]
- Mandelbrot, B.B. The Fractal Geometry of Nature; W. H. Freeman & Co.: San Francisco, FL, USA, 1988. [Google Scholar]
- González, A.E.; Martínez-López, F.; Moncho-Jordá, A.; Hidalgo-Álvarez, R. Two-dimensional colloidal aggregation: Concentration effects. J. Colloid Interface Sci. 2002, 246, 227–234. [Google Scholar] [CrossRef] [PubMed]
- González, A.E.; Martínez-López, F.; Moncho-Jordá, A.; Hidalgo-Álvarez, R. Concentration effects on two- and three-dimensional colloidal aggregation. Phys. A 2002, 314, 235–245. [Google Scholar] [CrossRef]
- Lifshitz, I.M.; Slyozov, V.V. The kinetics of precipitation from supersaturated solid solutions. J. Phys. Chem. Solids 1961, 19, 35–50. [Google Scholar] [CrossRef]
- Wagner, C. Theorie de alterung von niederschlägen durch umlösen. Ber. Bunsen-Ges. Phys. Chem. 1961, 65, 581–591. [Google Scholar]
- Söhnel, O.; Garside, J. Precipitation; Butterworth-Heinemann: Oxford, UK, 1992. [Google Scholar]
- Ng, J.D.; Lorber, B.; Witz, J.; Theóbald-Dietrich, A.; Kern, D.; Giegé, R. The crystallization of biological macromolecules from precipitates: Evidence for Ostwald ripening. J. Cryst. Growth 1996, 168, 50–62. [Google Scholar] [CrossRef]
- Ståhl, M.; Åslund, B.; Rasmuson, Å.C. Aging of reaction-crystallized benzoic acid. Ind. Eng. Chem. Res. 2004, 43, 6694–6702. [Google Scholar] [CrossRef]
- Finsy, R. On the critical radius in Ostwald ripening. Langmuir 2004, 20, 2975–2976. [Google Scholar] [CrossRef] [PubMed]
- Qing-bo, W.; Finsy, R.; Hai-bo, X.; Xi, L. On the critical radius in generalized Ostwald ripening. J. Zhejiang Univ. Sci. B 2005, 6, 705–707. [Google Scholar]
- Job, G.; Herrmann, F. Chemical potential—A quantity in search of recognition. Eur. J. Phys. 2006, 27, 353–371. [Google Scholar] [CrossRef]
- Brailsford, A.D.; Wynblatt, P. The dependence of Ostwald ripening kinetics on particle volume fraction. Acta Metall. 1979, 27, 489–497. [Google Scholar] [CrossRef]
- Voorhees, P.W.; Glicksman, M.E. Solution to the multi-particle diffusion problem with applications to Ostwald ripening—I. Theory. Acta Metall. 1984, 32, 2001–2011. [Google Scholar] [CrossRef]
- Marqusee, J.A.; Ross, J. Theory of Ostwald ripening: Competitive growth and its dependence on volume fraction. J. Chem. Phys. 1984, 80, 536–543. [Google Scholar] [CrossRef]
- Tokuyama, M.; Kawasaki, K. Statistical-mechanical theory of coarsening of spherical droplets. Phys. A 1984, 123, 386–411. [Google Scholar] [CrossRef]
- Enomoto, Y.; Tokuyama, M.; Kawasaki, K. Finite volume fraction effects on Ostwald ripening. Acta Metall. 1986, 34, 2119–2128. [Google Scholar] [CrossRef]
- Yao, J.H.; Elder, K.R.; Guo, H.; Grant, M. Theory and simulation of Ostwald ripening. Phys. Rev. B 1993, 47. [Google Scholar] [CrossRef]
- Baldan, A. Progress in Ostwald ripening theories and their applications to nickel-based superalloys. J. Mater. Sci. 2002, 37, 2171–2202. [Google Scholar] [CrossRef]
- Rosehain, W.; Ewen, D. The intercrystalline cohesion of metals. J. Inst. Met. 1913, 10, 119–149. [Google Scholar]
- Zhang, H.; Srolovitz, D.J.; Douglas, J.F.; Warren, J.A. Grain boundaries exhibit the dynamics of glass-forming liquids. Proc. Natl. Acad. Sci. USA 2009, 106, 7735–7740. [Google Scholar] [CrossRef] [PubMed]
- Janssens, K.G.F.; Olmsted, D.; Holm, E.A.; Foiles, S.M.; Plimpton, S.J.; Derlet, P.M. Computing the mobility of grain boundaries. Nat. Mater. 2006, 5, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, J.J.; Asta, M.; Karma, A. Atomistic and continuum modeling of dendritic solidification. Mater. Sci. Eng. R 2003, 41, 121–163. [Google Scholar] [CrossRef]
- Hernández-Guzmán, J.H.; Weeks, E.R. The equilibrium intrinsic crystal-liquid interface of colloids. Proc. Natl. Acad. Sci. USA 2009, 106, 15198–15202. [Google Scholar] [CrossRef] [PubMed]
- Sides, S.W.; Grest, G.S.; Lacasse, M.D. Capillary waves at liquid-vapor interfaces: A molecular dynamics simulation. Phys. Rev. E 1999, 60, 6708–6713. [Google Scholar] [CrossRef]
- Aarts, D.G.A.; Schmidt, M.; Lekkerkerker, H.N.W. Direct visual observation of thermal capillary waves. Science 2004, 304, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.P.A.; Fisher, D.S.; Weeks, J.D. Agreement of capillary-wave theory with exact results for the interface profile of the two-dimensional Ising model. Phys. Rev. Lett. 1982, 48. [Google Scholar] [CrossRef]
- Hapke, T.; Patzold, G.; Heermann, D.W. Surface tension of amorphous polymer films. J. Chem. Phys. 1998, 109, 10075–10081. [Google Scholar] [CrossRef]
- Li, J.C.M. Possibility of subgrain rotation during recrystallization. J. Appl. Phys. 1962, 33, 2958–2965. [Google Scholar] [CrossRef]
- Harris, K.E.; Singh, V.V.; King, A.H. Grain rotation in thin films of gold. Acta Mater. 1998, 46, 2623–2633. [Google Scholar] [CrossRef]
- Nabarro, F.R.N. Theory of Crystal Dislocations; Oxford Univ. Press: London, UK, 1967. [Google Scholar]
- Weertman, J.; Weertman, J.R. Elementary Dislocation Theory; Oxford Univ. Press: Oxford, UK, 1992. [Google Scholar]
- Hirth, J.P.; Lothe, J. Theory of Dislocations, 2nd ed.; Krieger Publishing Co.: Malabar, FL, USA, 1992. [Google Scholar]
- Hull, D.; Bacon, D.J. Introduction to Dislocations, 5th ed.; Butterworth-Heinemann: Oxford, UK, 2011. [Google Scholar]
- Anderson, M.P.; Srolovitz, D.J.; Grest, G.S.; Sahni, P.S. Computer simulation of grain growth—I. Kinetics. Acta Metall. 1984, 32, 783–791. [Google Scholar] [CrossRef]
- Srolovitz, D.J.; Anderson, M.P.; Sahni, P.S.; Grest, G.S. Computer simulation of grain growth—II. Grain size distribution, topology, and local dynamics. Acta Metall. 1984, 32, 793–802. [Google Scholar] [CrossRef]
- Rollet, A.D.; Srolovitz, D.J.; Anderson, M.P. Simulation and theory of abnormal grain growth—Anisotropic grain boundary energies and mobilities. Acta Metall. 1989, 37, 1227–1240. [Google Scholar] [CrossRef]
- Landau, D.P. Monte Carlo studies of critical and multicritical phenomena. In Applications of the Monte Carlo Method in Statistical Physics; Binder, K., Ed.; Springer: Berlin, Germany, 1984; pp. 93–124. [Google Scholar]
- Kawasaki, K.; Nagai, T.; Nakashima, K. Vertex models for two-dimensional grain growth. Philos. Mag. B 1989, 60, 399–421. [Google Scholar] [CrossRef]
- Weygand, D.; Bréchet, Y.; Lépinoux, J. A vertex dynamics simulation of grain growth in two dimensions. Philos. Mag. B 1998, 78, 329–352. [Google Scholar] [CrossRef]
- Raabe, D. Cellular automata in materials science with particular reference to recrystallization simulation. Annu. Rev. Mater. Res. 2002, 32, 53–76. [Google Scholar] [CrossRef]
- Doherty, R.D.; Hughes, D.A.; Humphreys, F.J.; Jonas, J.J.; Juul Jensen, D.; Kassner, M.E.; King, W.E.; McNelley, T.R.; McQueen, H.J.; Rollet, A.D. Current issues in recrystallization: A review. Mater. Sci. Eng. A 1997, 238, 219–274. [Google Scholar] [CrossRef]
- Raabe, D.; Becker, R.C. Coupling of a crystal plasticity finite-element model with a probabilistic cellular automaton for simulating primary static recrystallization in aluminum. Model.Simul. Mater. Sci. Eng. 2000, 8, 445–462. [Google Scholar] [CrossRef]
- Raabe, D. Yield surface simulation for partially recrystallized aluminum polycrystals on the basis of spatially discrete data. Comp. Mater. Sci. 2000, 19, 13–26. [Google Scholar] [CrossRef]
- Chen, L.-Q. Phase-field models for microstructure evolution. Annu. Rev. Mater. Res. 2002, 32, 113–140. [Google Scholar] [CrossRef]
- Kobayashi, R.; Warren, J.A.; Carter, W.C. Vector-valued phase field model for crystallization and grain boundary formation. Phys. D 1998, 119, 415–423. [Google Scholar] [CrossRef]
- Kobayashi, R.; Warren, J.A.; Carter, W.C. A continuum model of grain boundaries. Phys. D 2000, 140, 141–150. [Google Scholar] [CrossRef]
- Warren, J.A.; Kobayashi, R.; Lobkovsky, A.E.; Carter, W.C. Extending phase field models of solidification to polycrystalline materials. Acta Mater. 2003, 51, 6035–6058. [Google Scholar] [CrossRef]
- Krill, C.E., III; Chen, L.-Q. Computer simulation of 3-D grain growth using a phase field model. Acta Mater. 2002, 50, 3057–3073. [Google Scholar] [CrossRef]
- Kobayashi, R.; Warren, J.A. Modeling the formation and dynamics of polycrystals in 3D. Phys. A 2005, 356, 127–132. [Google Scholar] [CrossRef]
- Kim, S.G.; Kim, D.I.; Kim, W.T.; Park, Y.B. Computer simulations of two-dimensional and three-dimensional ideal grain growth. Phys. Rev. E 2006, 74, 061605. [Google Scholar] [CrossRef] [PubMed]
- Bjerre, M.; Tarp, J.M.; Angheluta, L.; Mathiesen, J. Rotation-induced grain growth and stagnation in phase-field crystal models. Phys. Rev. E 2013, 88, 020401. [Google Scholar] [CrossRef] [PubMed]
- Gránásy, L.; Pusztai, T.; Warren, J.A. Modelling polycrystalline solidification using phse field theory. J. Phys. Condens. Matter 2004, 16, R1205–R1235. [Google Scholar] [CrossRef]
- Singer-Loginova, I.; Singer, H.M. The phase field technique for modeling multiphase materials. Rep. Prog. Phys. 2008, 71, 106501. [Google Scholar] [CrossRef]
- Hansen, J.P.; McDonald, I.R. Theory of Simple Liquids; Academic Press: London, UK, 1986. [Google Scholar]
- Becker, R.; Döring, W. Kinetische behandlung der keimbildung in übersättigten Dämpfen. Ann. Phys. 1935, 24, 719–752. [Google Scholar] [CrossRef]
- Turnbull, D.; Fisher, J.C. Rate of nucleation in condensed systems. J. Chem. Phys. 1949, 17, 71–73. [Google Scholar] [CrossRef]
- Binder, K.; Stauffer, D. Statistical theory of nucleation, condensation and coagulation. Adv. Phys. 1976, 25, 343–396. [Google Scholar] [CrossRef]
- Pusey, P.N.; van Megen, W. Phase behaviour of concentrated suspensions of nearly hard colloidal spheres. Nature 1986, 320, 340–342. [Google Scholar] [CrossRef]
- Zhu, J.; Li, M.; Rogers, R.; Meyer, W.; Ottewill, R.H.; STS-73 Space Shuttle Crew; Russel, W.B.; Chaikin, P.M. Crystallization of hard-sphere colloids in microgravity. Nature 1987, 387, 883–885. [Google Scholar]
- Auer, S.; Frenkel, D. Prediction of absolute crystal-nucleation rate in hard sphere colloids. Nature 2001, 409, 1020–1023. [Google Scholar] [CrossRef] [PubMed]
- Anderson, V.J.; Lekkerkerker, H.N. Insights into phase transition kinetics from colloid science. Nature 2002, 416, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Cacciuto, A.; Auer, S.; Frenkel, D. Onset of heterogeneous crystal nucleation in colloidal suspensions. Nature 2004, 428, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Auer, S.; Frenkel, D. Numerical simulations of crystal nucleation in colloids. Adv. Polym. Sci. 2005, 173, 149–207. [Google Scholar]
- Schilling, T.; Schöpe, H.J.; Oettel, M.; Opletal, G.; Snook, I. Precursor-mediatedi crystallization process in suspensions of hard spheres. Phys. Rev. Lett. 2010, 105, 025701. [Google Scholar] [CrossRef] [PubMed]
- Deutschländer, S.; Dillmann, P.; Maret, G.; Keim, P. Kibble-Zurek mechanism in colloidal monolayers. Proc. Natl. Acad. Sci. USA 2015, 112, 6925–6930. [Google Scholar] [CrossRef] [PubMed]
- Swygenhoven, H.V. Grain boundaries and dislocations. Science 2002, 296, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Cherkaoui, M.; Capolungo, L. Atomistic and Continuun Modeling of Nanocrystalline Materials: Deformation Mechanisms and Scale Transition; Springer: Berlin, Germany, 2009. [Google Scholar]





















© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, A.E. Colloidal Crystallization in 2D for Short-Ranged Attractions: A Descriptive Overview. Crystals 2016, 6, 46. https://doi.org/10.3390/cryst6040046
González AE. Colloidal Crystallization in 2D for Short-Ranged Attractions: A Descriptive Overview. Crystals. 2016; 6(4):46. https://doi.org/10.3390/cryst6040046
Chicago/Turabian StyleGonzález, Agustín E. 2016. "Colloidal Crystallization in 2D for Short-Ranged Attractions: A Descriptive Overview" Crystals 6, no. 4: 46. https://doi.org/10.3390/cryst6040046