
Sodium Intercalation in Nitrogen-Doped Graphene-Based Anode: A First-Principles Study

Abstract
:1. Introduction
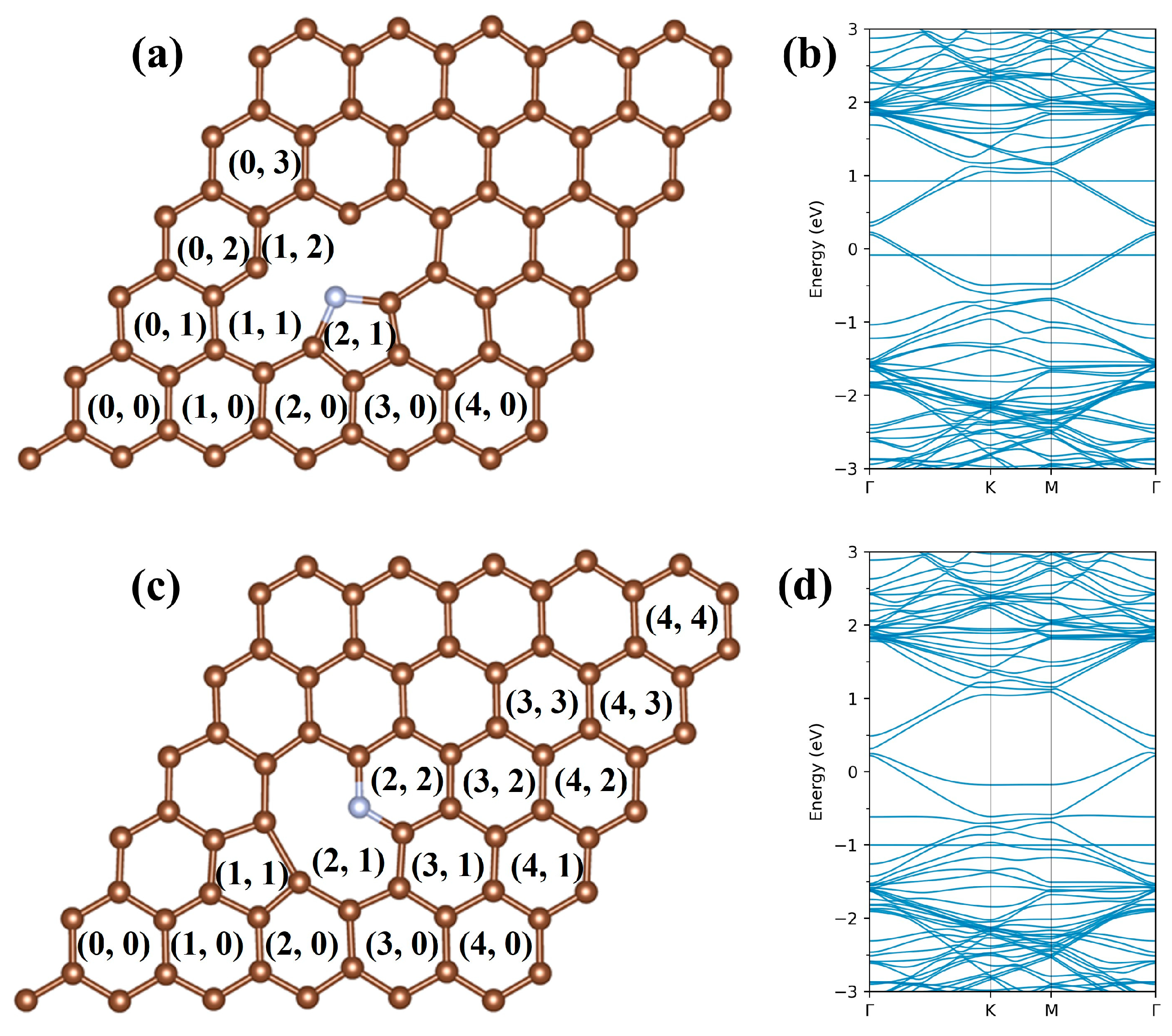
2. Computational Methods and Models
3. Results and Discussion
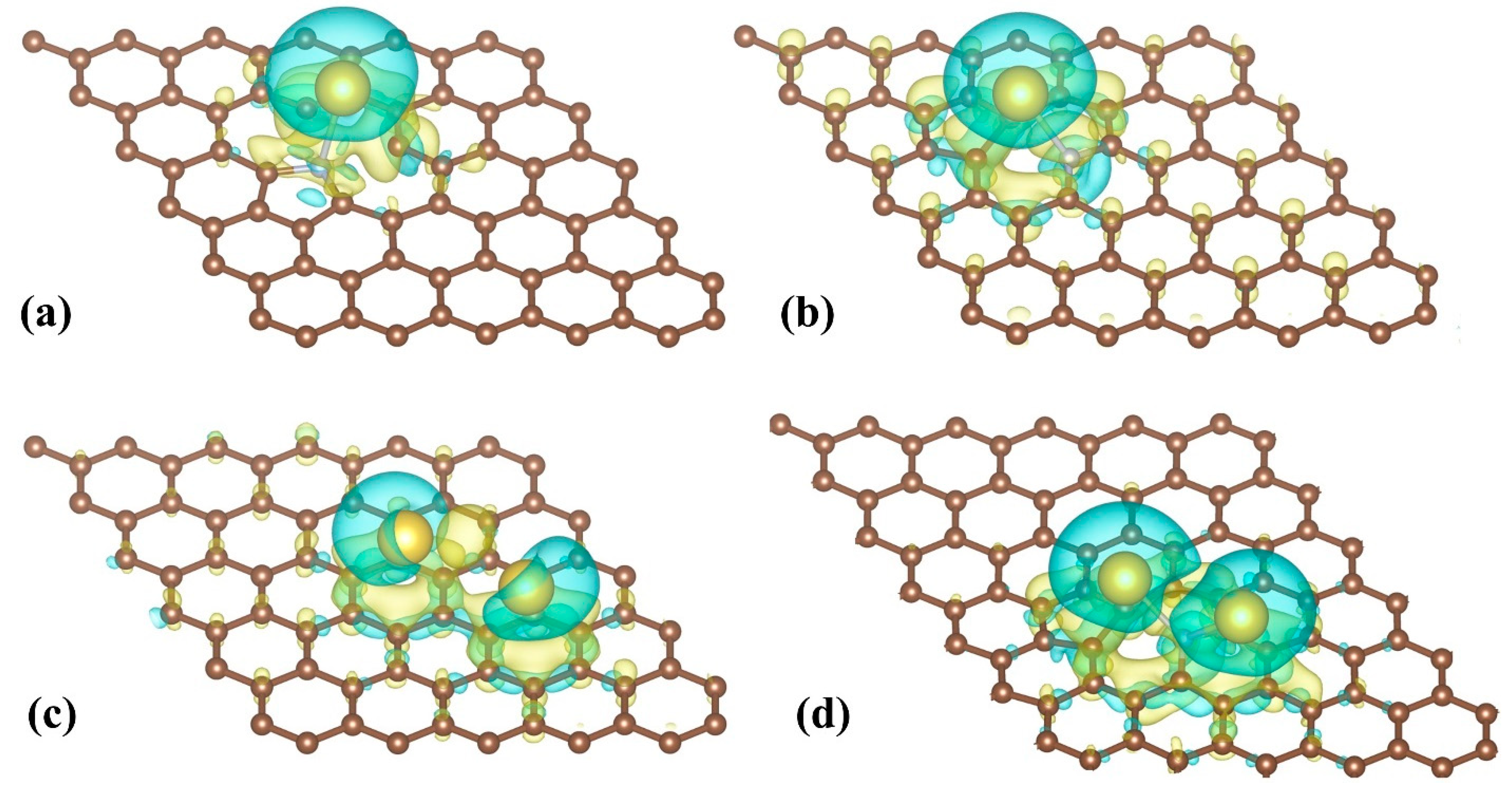
3.1. Single Na Adsorption
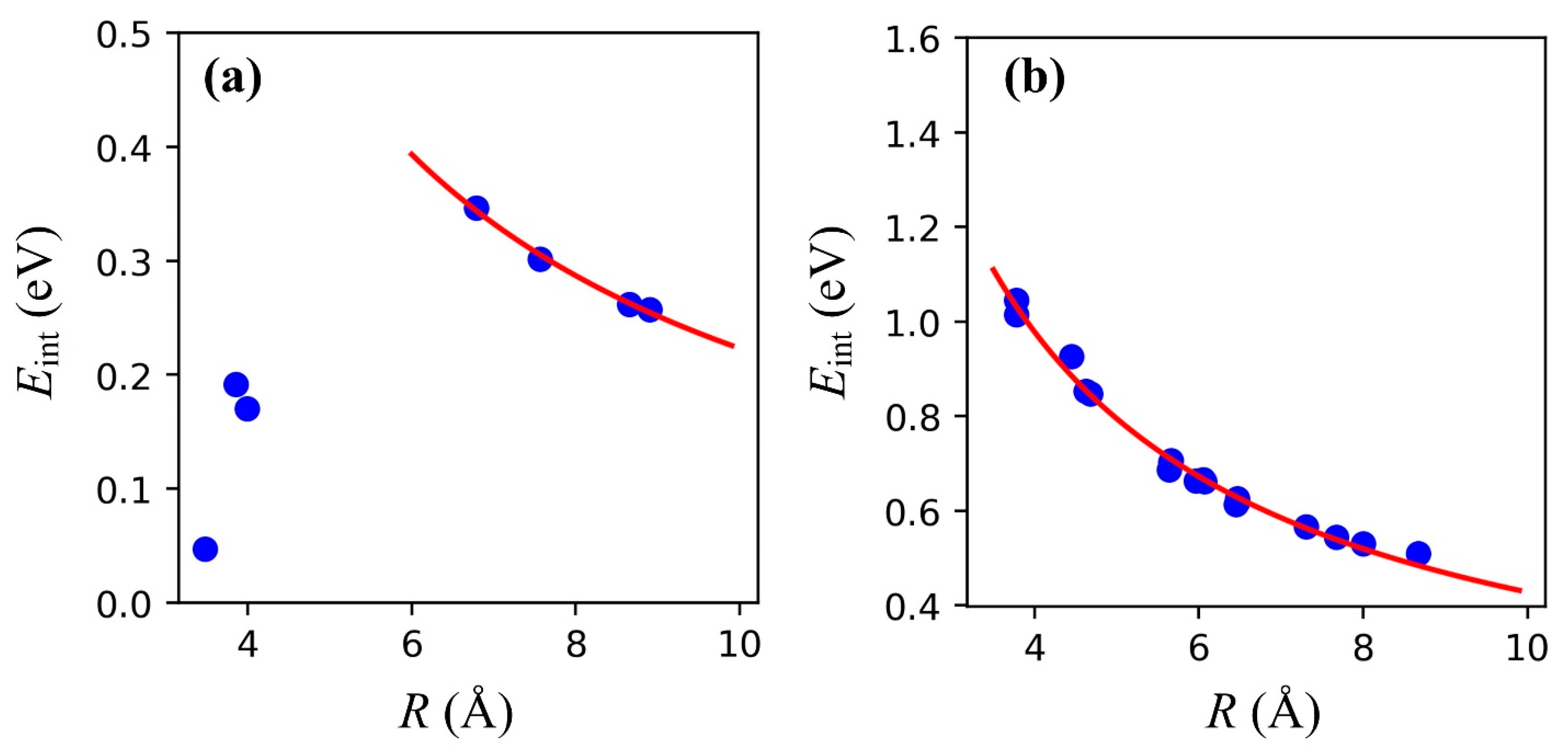
3.2. Double Na Adsorption and Na-Na Interactions
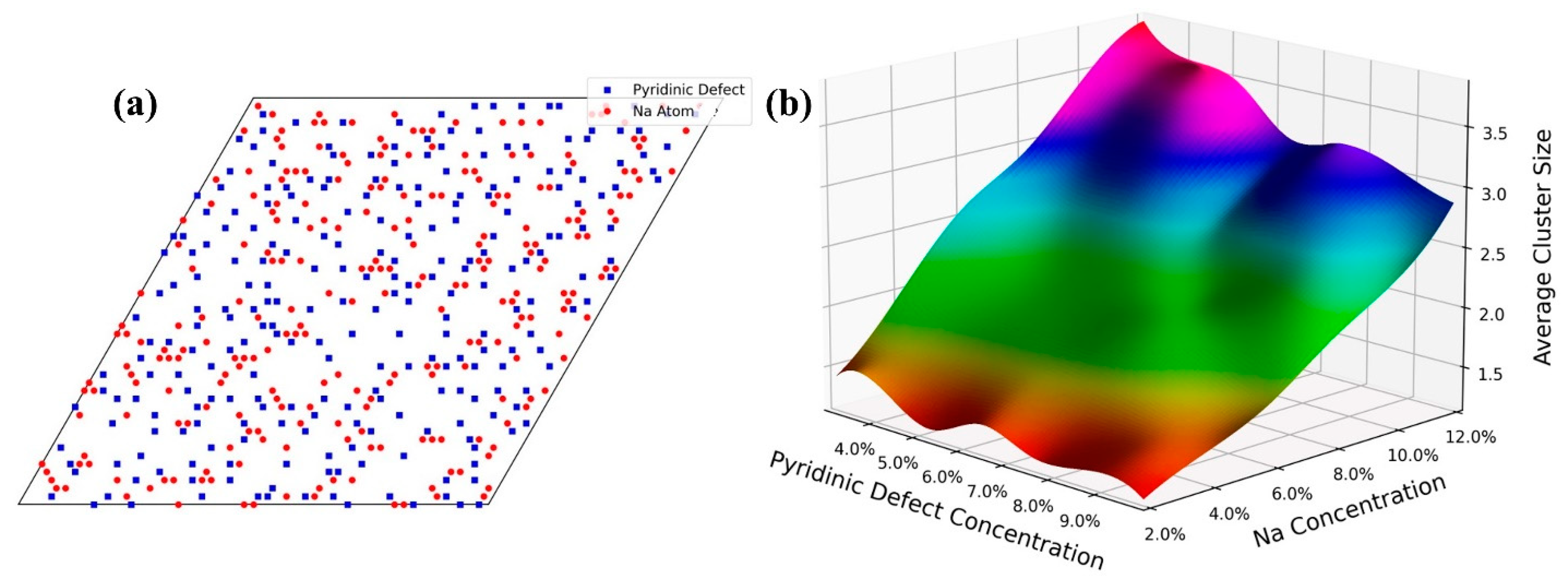
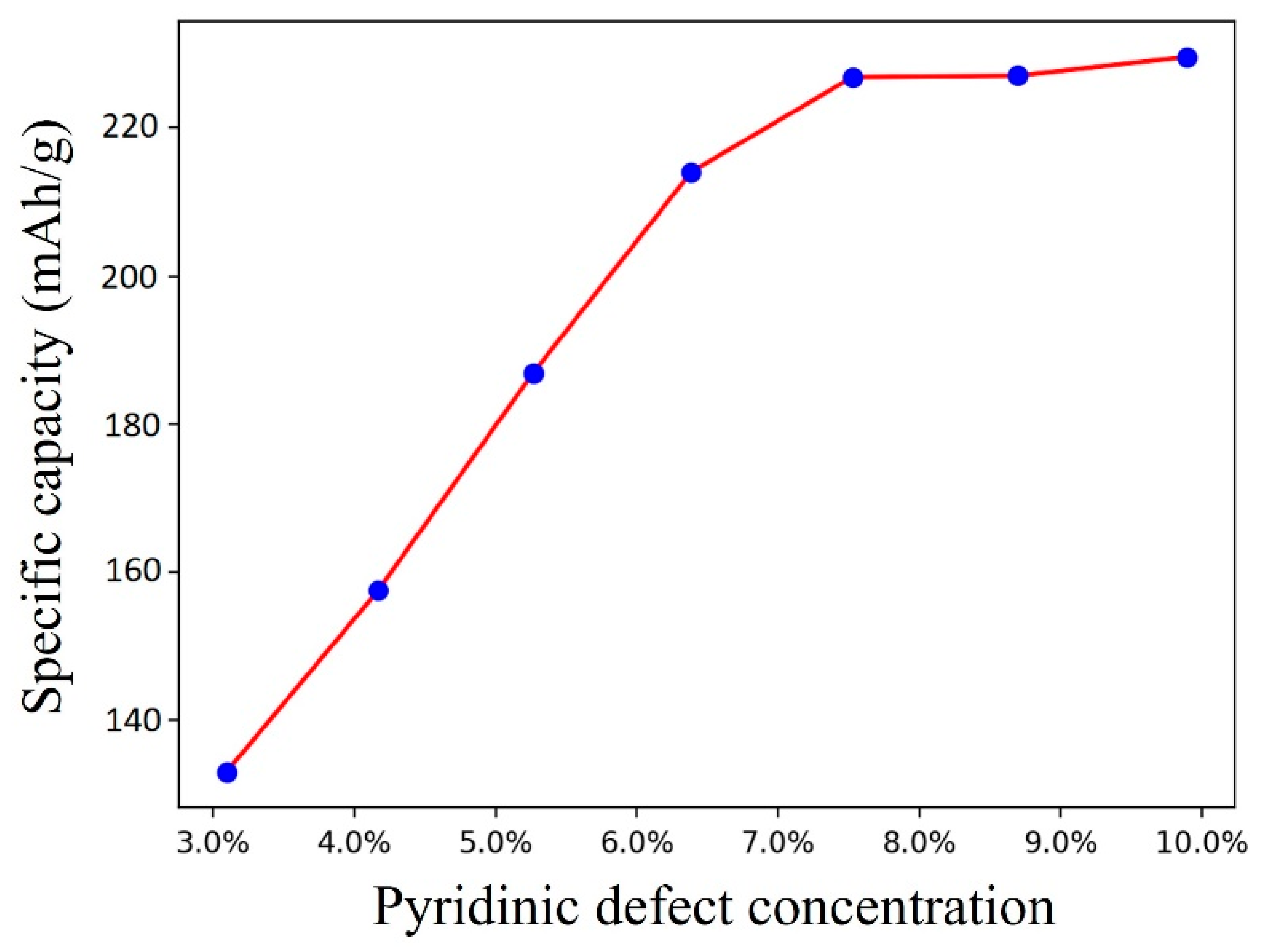
3.3. Na Distributions and Specific Capacity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tarascon, J.M.; Armand, M. Issues and Challenges Facing Rechargeable Lithium Batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef]
- Etacheri, V.; Marom, R.; Elazari, R.; Salitra, G.; Aurbach, D. Challenges in the Development of Advanced Li-Ion Batteries: A Review. Energy Environ. Sci. 2011, 4, 3243–3262. [Google Scholar] [CrossRef]
- Choi, J.W.; Aurbach, D. Promise and reality of post-lithium-ion batteries with high energy densities. Nat. Rev. Mater. 2016, 1, 16013. [Google Scholar] [CrossRef] [Green Version]
- Dunn, B.; Kamath, H.; Tarascon, J.M. Electrical energy storage for the grid: A battery of choices. Science 2011, 334, 928–935. [Google Scholar] [CrossRef] [Green Version]
- Vaalma, C.; Buchholz, D.; Weil, M.; Passerini, S. A cost and resource analysis of sodium-ion batteries. Nat. Rev. Mater. 2018, 3, 18013. [Google Scholar] [CrossRef]
- Kim, S.W.; Seo, D.H.; Ma, X.; Ceder, G.; Kang, K. Electrode Materials for Rechargeable Sodium-Ion Batteries: Potential Alternatives to Current Lithium-Ion Batteries. Adv. Energy Mater. 2012, 2, 710. [Google Scholar] [CrossRef]
- Slater, M.D.; Kim, D.; Lee, E.; Johnson, C.S. Sodium-Ion Batteries. Adv. Funct. Mater. 2013, 23, 947. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Research Development on Sodium-Ion Batteries. Chem. Rev. 2014, 114, 11636. [Google Scholar] [CrossRef]
- Kundu, D.; Talaie, E.; Duffort, V.; Nazar, L.F. The Emerging Chemistry of Sodium Ion Batteries for Electrochemical Energy Storage. Angew. Chem. Int. Ed. 2015, 54, 3431. [Google Scholar] [CrossRef] [PubMed]
- Abraham, K.M. How Comparable Are Sodium-Ion Batteries to Lithium-Ion Counterparts? ACS Energy Lett. 2020, 5, 3544–3547. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, W.; Lu, S.; Xiang, Y. Carbon Anode Materials: A Detailed Comparison between Na-ion and K-ion Batteries. Adv. Energy Mater. 2021, 11, 2003640. [Google Scholar] [CrossRef]
- Palomares, V.; Serras, P.; Villaluenga, I.; Hueso, K.B.; Carretero-Gonzalez, J.; Rojo, T. Na-ion batteries, recent advances and present challenges to become low cost energy storage systems. Energy Environ. Sci. 2012, 5, 5884–5901. [Google Scholar] [CrossRef]
- Hong, S.Y.; Kim, Y.; Park, Y.; Choi, A.; Choi, N.S.; Lee, K.T. Charge carriers in rechargeable batteries: Na ions vs. Li ions. Energy Environ. Sci. 2013, 6, 2067–2081. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Luo, W.; Shen, F.; Bommier, C.; Zhu, H.; Ji, X.; Hu, L. Na-Ion Battery Anodes: Materials and Electrochemistry. Acc. Chem. Res. 2016, 49, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Malyi, O.I.; Kostiantyn, S.; Kulish, V.V.; Tan, T.L.; Manzhos, S.; Persson, C. A computational study of Na behavior on graphene. Appl. Surf. Sci. 2015, 333, 235–243. [Google Scholar] [CrossRef]
- Wei, D.; Liu, Y.; Wang, Y.; Zhang, H.; Huang, L.; Yu, G. Synthesis of N-Doped Graphene by Chemical Vapor Deposition and Its Electrical Properties. Nano Lett. 2009, 9, 1752–1758. [Google Scholar] [CrossRef]
- Fan, M.; Feng, Z.Q.; Zhu, C.; Chen, X.; Chen, C.; Yang, J.; Sun, D. Recent progress in 2D or 3D N-doped graphene synthesis and the characterizations, properties, and modulations of N species. J. Mater. Sci. 2016, 51, 10323–10349. [Google Scholar] [CrossRef]
- Xu, H.; Ma, L. Nitrogen-doped graphene: Synthesis, characterizations and energy applications. J. Energy Chem. 2018, 27, 146–160. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Yang, W.; Ma, G.; Che, S.; Li, Y.; Jia, Y.; Chen, N.; Huang, G.; Li, Y. Edge-Nitrogen Enriched Porous Carbon Nanosheets Anodes with Enlarged Interlayer Distance for Fast Charging Sodium-Ion Batteries. Small 2022, 18, 2204375. [Google Scholar] [CrossRef]
- Ma, G.; Huang, K.; Zhuang, Q.; Ju, Z. Superior cycle stability of nitrogen-doped graphene nanosheets for Na-ion batteries. Mater. Lett. 2016, 174, 221–225. [Google Scholar] [CrossRef]
- Liu, H.; Jia, M.; Cao, B.; Chen, R.; Lv, X.; Tang, R.; Wu, F.; Xu, B. Nitrogen-doped carbon/graphene hybrid anode material for sodium-ion batteries with excellent rate capability. J. Power Sources 2016, 319, 195–201. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Zhang, L.; Xie, F.; Vasileff, A.; Qiao, S.Z. Graphitic Carbon Nitride (g-C3N4)-Derived N-Rich Graphene with Tuneable Interlayer Distance as a High-Rate Anode for Sodium-Ion Batteries. Adv. Mater. 2019, 31, 1901261. [Google Scholar] [CrossRef] [PubMed]
- Wasalathilake, K.C.; Ayoko, G.A.; Yan, C. Effects of heteroatom doping on the performance of graphene in sodium-ion batteries: A density functional theory investigation. Carbon 2018, 140, 276–285. [Google Scholar] [CrossRef]
- Lee, H.W.; Moon, H.S.; Hur, J.; Kim, I.T.; Park, M.S.; Yun, J.M.; Kim, K.H.; Lee, S.G. Mechanism of sodium adsorption on N-doped graphene nanoribbons for sodium ion battery applications: A density functional theory approach. Carbon 2017, 119, 492–501. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Huang, K. Statistic Mechanics, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1991. [Google Scholar]
- Mackay, D. Introduction to Monte Carlo Method. In Learning in Graphical Models; Jordan, M., Ed.; Kluwer Academic Press: Amsterdam, The Netherlands, 1998; pp. 175–204. [Google Scholar]
- Yu, M.; Trinkle, D.R. Accurate and efficient algorithm for bader charge integration. J. Chem. Phys. 2011, 134, 064111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reda, M.; Hansen, H.A.; Vegge, T. DFT study of stabilization effects on n-doped graphene for ORR catalysis. Catal. Today 2018, 312, 118–125. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pyrrolic site | (0, 0) | (0, 1) | (0, 2) | (0, 3) | (1, 0) | (1, 2) | (−1, 4) | (2, 0) | (2, 1) | (3, 0) |
| −1.91 | −1.99 | −2.12 | −2.03 | −1.99 | −2.70 | −1.89 | −2.05 | −2.71 | −1.97 | |
| Pyridinic site | (0, 0) | (1, 0) | (1, 1) | (2, 0) | (2, 1) | (2, 2) | (3, 0) | (3, 1) | (3, 2) | (3, 3) |
| −2.02 | −2.14 | −2.81 | −2.27 | −2.81 | −2.80 | −2.14 | −2.04 | −1.95 | −1.97 | |
| Pyridinic site | (4, 0) | (4, 1) | (4, 2) | (4, 3) | (4, 4) | |||||
| −2.03 | −2.00 | −2.02 | −2.04 | −2.04 |
| Site | (3, 3) | (4, 2) | (5, 2) | (5, 3) | (4, 4) | (3, 2) | (5, 1) | (6, 2) | (5, 4) | (2, 2) |
| −2.09 | −1.98 | −2.02 | −2.01 | −1.99 | −2.00 | −2.06 | −2.05 | −2.03 | −2.06 | |
| 0.66 | 1.04 | 0.85 | 0.85 | 1.02 | 0.93 | 0.70 | 0.66 | 0.69 | 0.67 | |
| Site | (7, 0) | (4, 1) | (6, 1) | (6, 3) | (3, 1) | (5, 0) | (6, 0) | (7, 1) | ||
| −2.08 | −2.06 | −2.08 | −2.06 | −2.07 | −2.08 | −2.09 | −2.09 | |||
| 0.51 | 0.71 | 0.62 | 0.61 | 0.63 | 0.57 | 0.54 | 0.53 |
| Neighbor Type | 1A | 2A | 2B | 3A | 3B | 4A | 4B |
|---|---|---|---|---|---|---|---|
| −1.14 | −1.12 | −1.10 | −1.07 | −1.12 | −1.16 | −1.17 | |
| 0.04 | 0.19 | 0.17 | 0.34 | 0.30 | 0.26 | 0.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, H.; Zhang, X. Sodium Intercalation in Nitrogen-Doped Graphene-Based Anode: A First-Principles Study. Crystals 2023, 13, 1011. https://doi.org/10.3390/cryst13071011
Ding H, Zhang X. Sodium Intercalation in Nitrogen-Doped Graphene-Based Anode: A First-Principles Study. Crystals. 2023; 13(7):1011. https://doi.org/10.3390/cryst13071011
Chicago/Turabian StyleDing, Henry, and Xu Zhang. 2023. "Sodium Intercalation in Nitrogen-Doped Graphene-Based Anode: A First-Principles Study" Crystals 13, no. 7: 1011. https://doi.org/10.3390/cryst13071011