

Synthesis and Structure Characterization of Three Pharmaceutical Compounds Based on Tinidazole

Abstract
:1. Introduction
2. Materials and Methods
2.1. General Materials and Methods
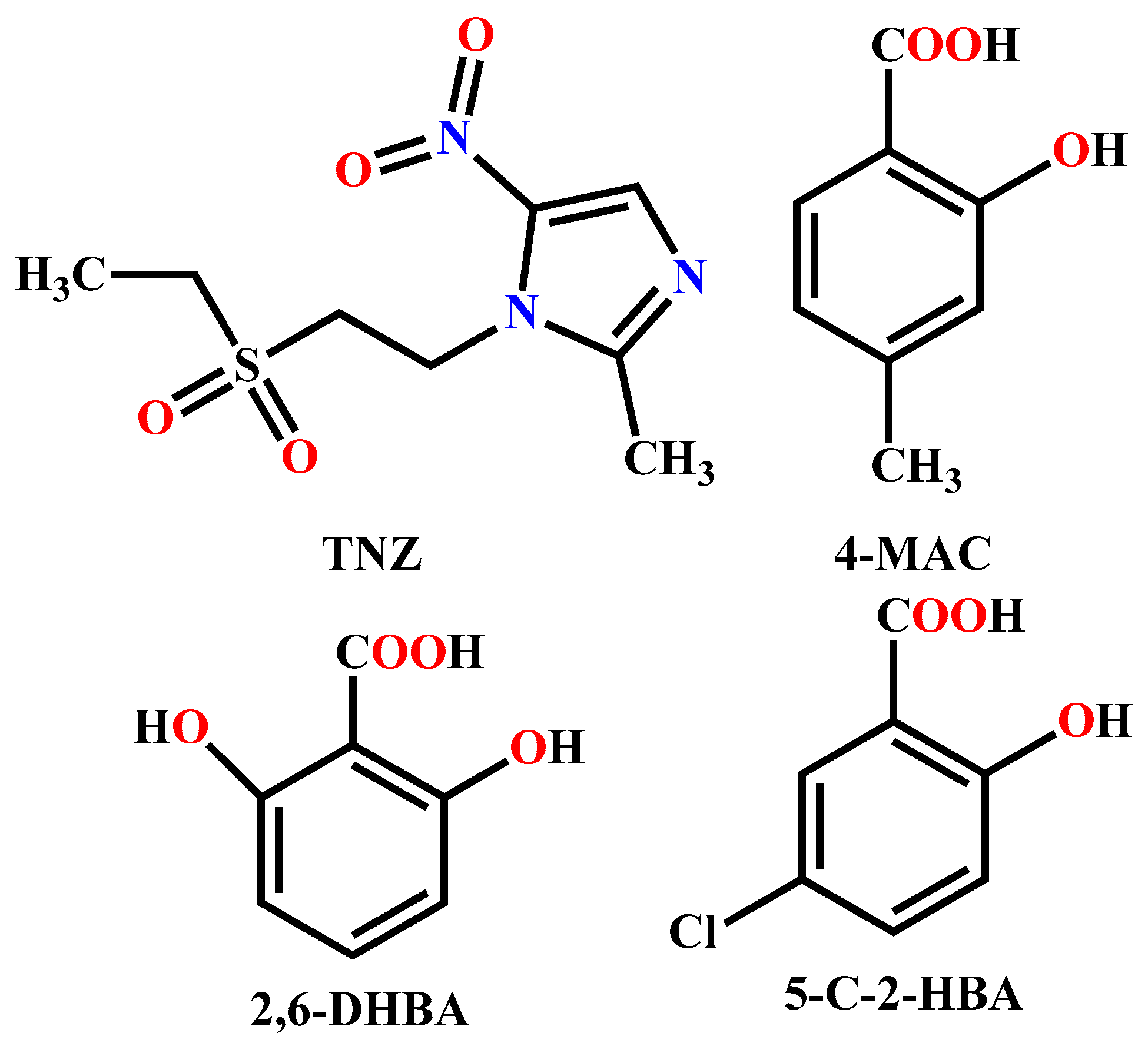
2.2. Syntheses of the Compounds 1–3
2.3. X-ray Crystallography
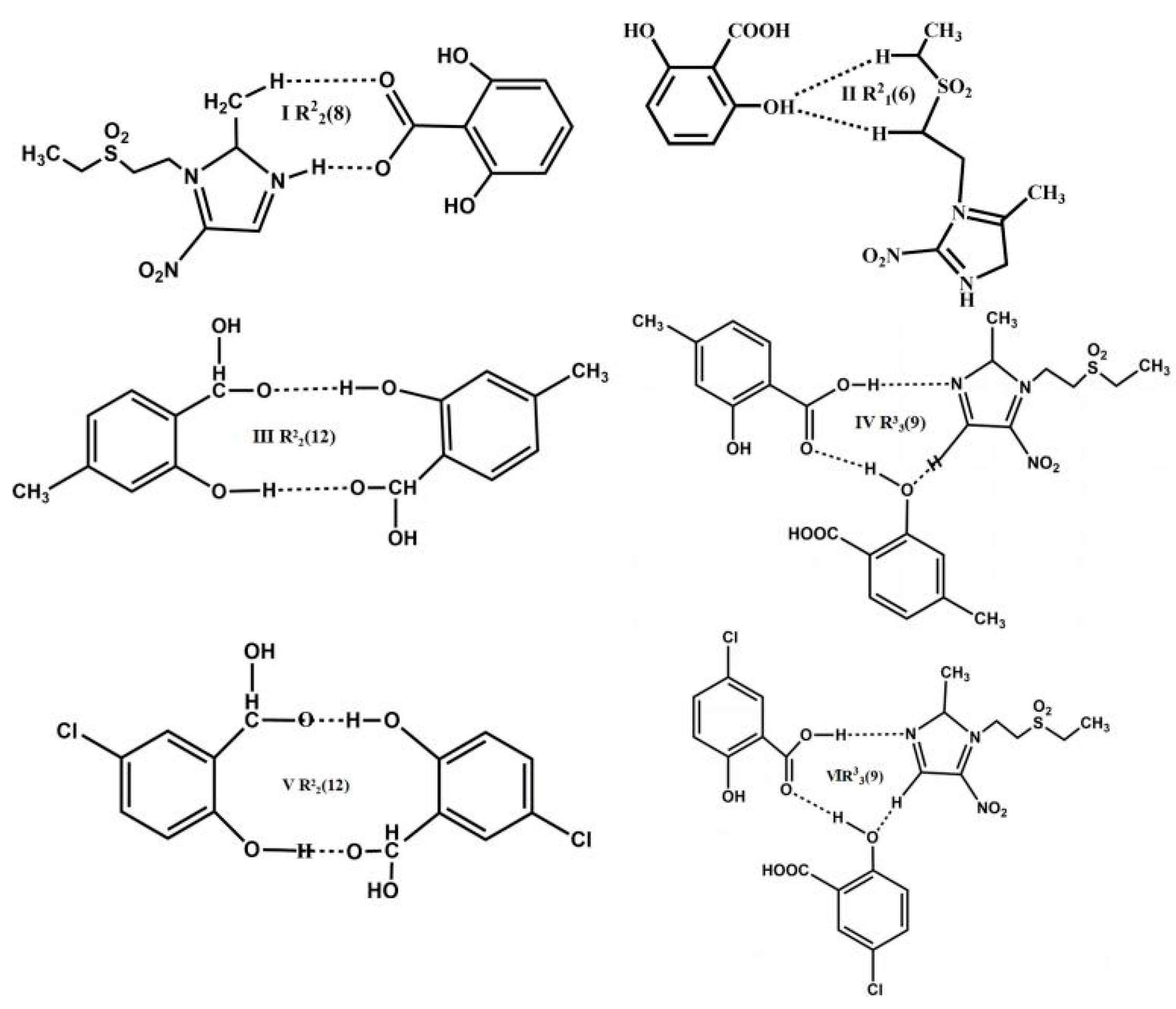
3. Results and Discussion
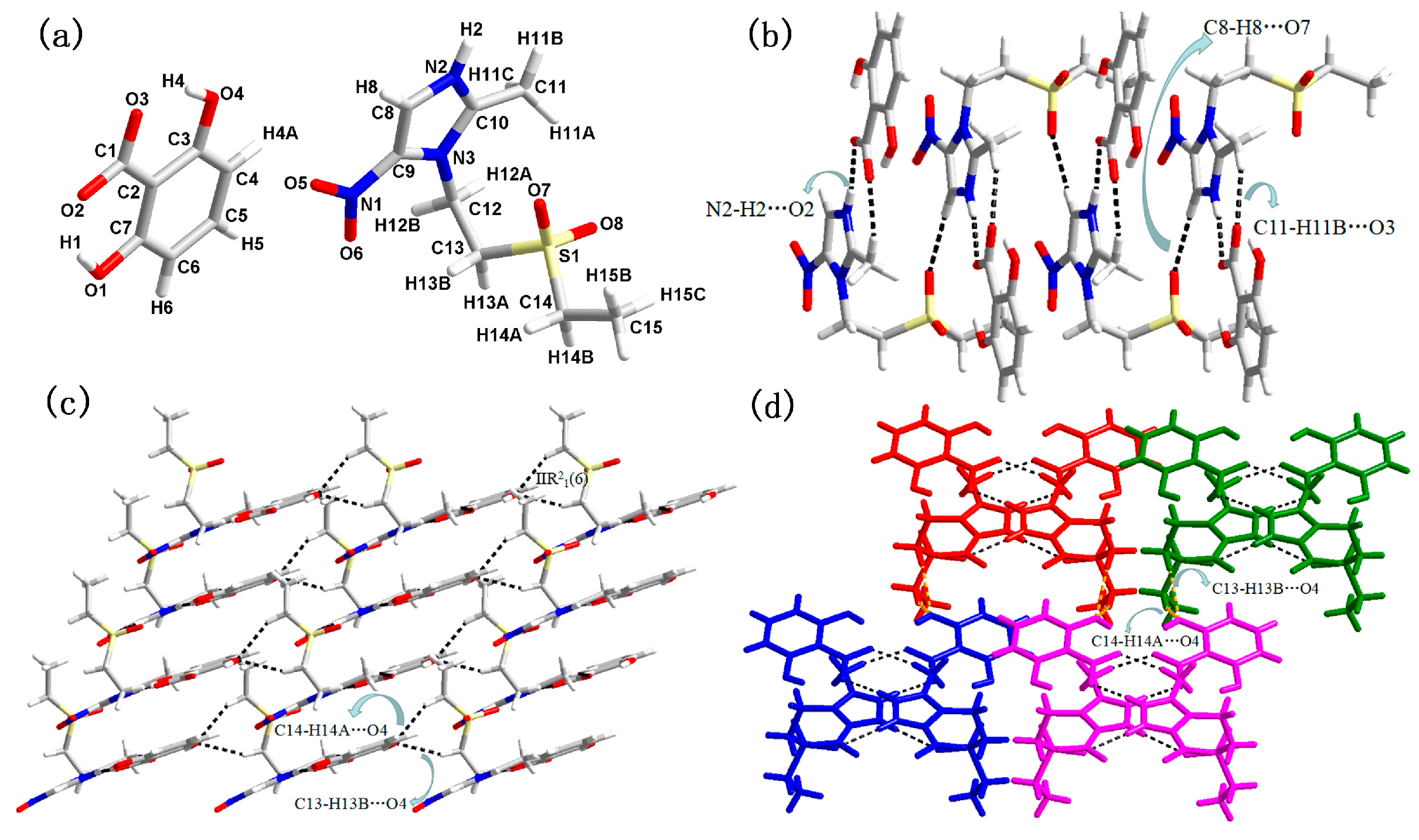
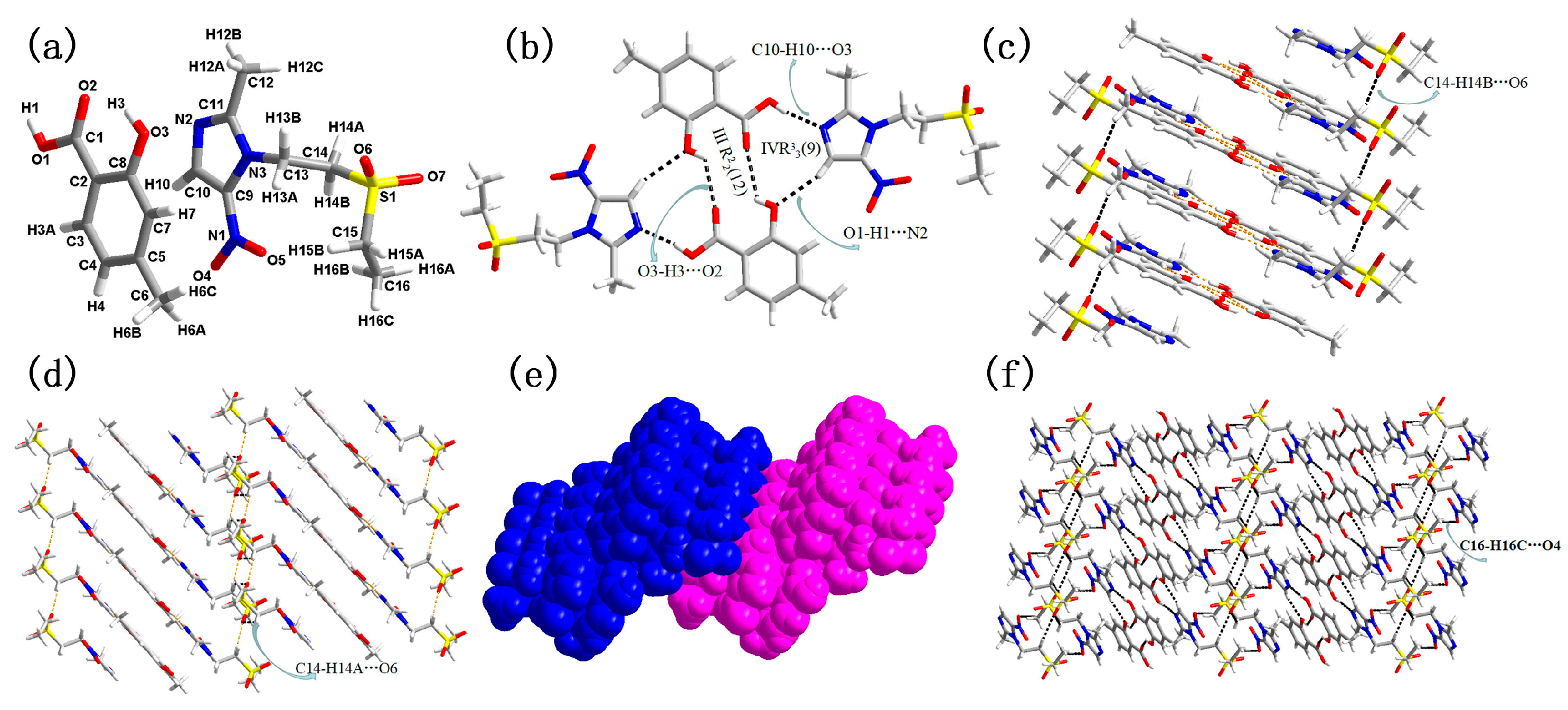
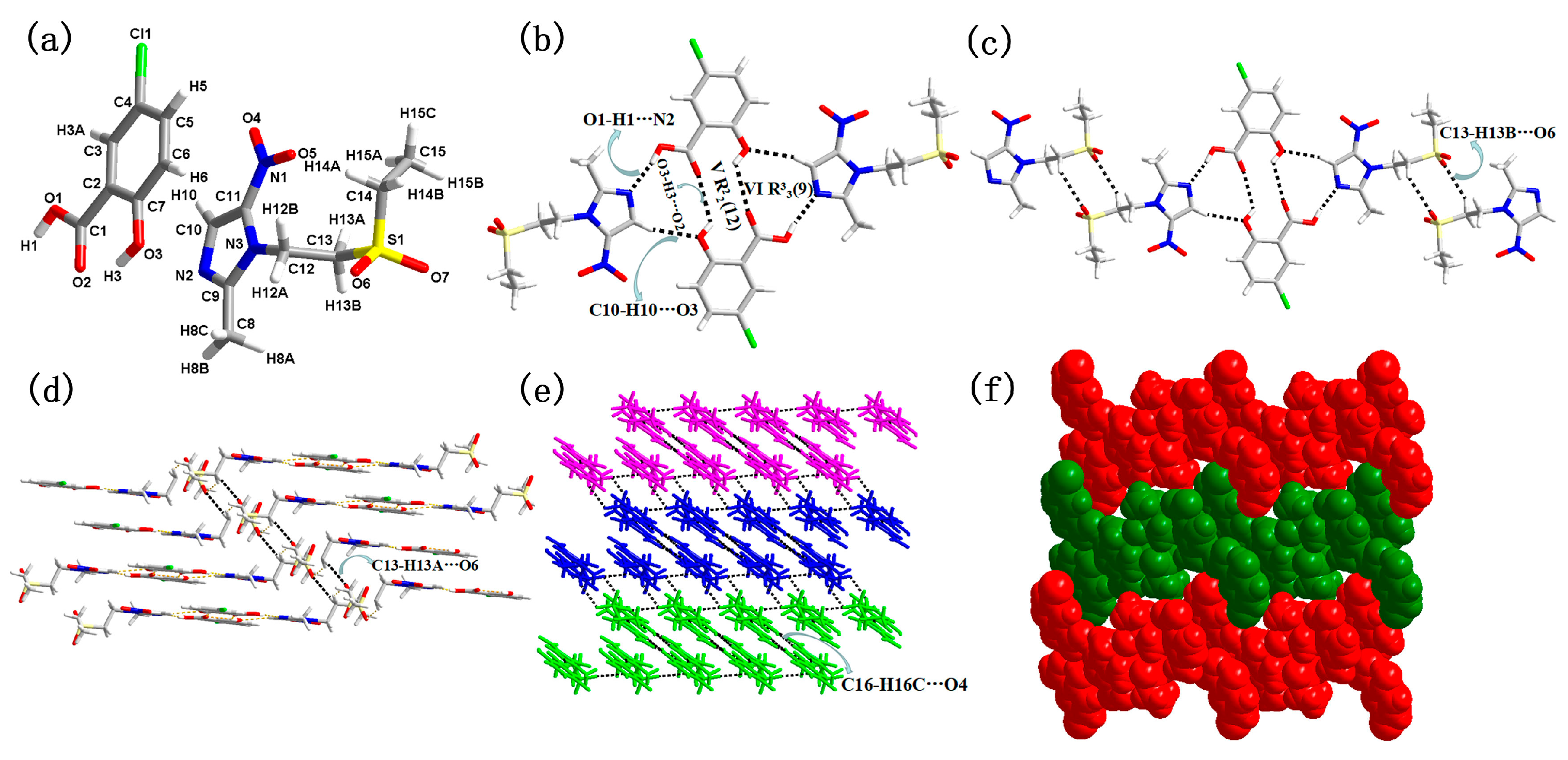
3.1. Molecular and Supramolecular Structural Descriptions of Compounds 1–3
3.2. Thermal Stability
3.3. IR Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bolla, G.; Sarma, B.; Nangia, A.K. Crystal Engineering of Pharmaceutical Cocrystals in the Discovery and Development of Improved Drugs. Chem. Rev. 2022, 122, 11514–11603. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Drozd, K.V.; Volkova, T.V.; Vasilev, N.; Batov, D.; Churakov, A.V.; Perlovich, G.L. Extending the Range of Nitrofurantoin Solid Forms: Effect of Molecular and Crystal Structure on Formation Thermodynamics and Physicochemical Properties. Cryst. Growth Des. 2022, 22, 2569–2586. [Google Scholar] [CrossRef]
- Beletskaya, I.; Tyurin, V.S.; Tsivadze, A.Y.; Guilard, R.; Stern, C. Supramolecular Chemistry of Metalloporphyrins. Chem. Rev. 2009, 109, 1659–1713. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, Y.; Bo, Y.; Chinnam, A.K.; Singh, J.; Staples, R.J.; He, X.; Wang, K.; Zhang, J.; Shreeve, J.M. Synthesis of a high-energy-density material through rapid replacement of crystal water of hydrates. Chem 2022, 8, 2678–2687. [Google Scholar] [CrossRef]
- Singh, M.; Liu, K.; Qu, S.; Ma, H.; Shi, H.; An, Z.; Huang, W. Recent Advances of Cocrystals with Room Temperature Phosphorescence. Adv. Opt. Mater. 2021, 9, 2002197. [Google Scholar] [CrossRef]
- Bennion, J.C.; Matzger, A.J. Development and Evolution of Energetic Cocrystals. Acc. Chem. Res. 2021, 54, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.Q.; Abbasi, N.M.; Smith, E.A.; Petrich, J.W.; Anderson, J.L. Characterizing the Solvation Characteristics of Deep Eutectic Solvents Composed of Active Pharmaceutical Ingredients as a Hydrogen Bond Donor and/or Acceptor. ACS Sustain. Chem. Eng. 2022, 10, 3066–3078. [Google Scholar] [CrossRef]
- Huang, Z.; Suzuki, H.; Ito, M.; Noguchi, S. Direct detection of the crystal form of an active pharmaceutical ingredient in tablets by X-ray absorption fine structure spectroscopy. Int. J. Pharmaceut. 2022, 625, 122057. [Google Scholar] [CrossRef] [PubMed]
- Sugden, I.J.; Braun, D.E.; Bowskill, D.H.; Adjiman, C.S.; Pantelides, C.C. Efficient Screening of Coformers for Active Pharmaceutical Ingredient Cocrystallization. Cryst. Growth Des. 2022, 22, 4513–4527. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yu, Y.; Bu, F.; Li, Y.; Yan, C.; Wu, Z. The First Cocrystallization of Milrinone with Nutraceuticals: The Adjusting Effects of Hydrophilicity/Hydrophobicity in Cavities on the In Vitro/In Vivo Properties of the Cocrystals. Cryst. Growth Des. 2022, 22, 1623–1637. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, F.; Zhao, X.; Wang, S.; Yang, Q.; Zhang, X. Crystal Structure, Solubility, and Pharmacokinetic Study on a Hesperetin Cocrystal with Piperine as Coformer. Pharmaceutics 2022, 14, 94. [Google Scholar] [CrossRef]
- Good, D.J.; Rodríguez-Hornedo, N. Solubility Advantage of Pharmaceutical Cocrystals. Cryst. Growth Des. 2009, 9, 2252–2264. [Google Scholar] [CrossRef]
- Liu, L.; Wang, J.; Mei, X. Enhancing the stability of active pharmaceutical ingredients by the cocrystal strategy. Cryst. Eng. Comm. 2022, 24, 2002–2022. [Google Scholar] [CrossRef]
- Nangia, A.K.; Desiraju, G.R. Heterosynthons, Solid Form Design and Enhanced Drug Bioavailability. Angew. Chem. Int. Ed. 2022, 61, e202207484. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ning, L. Pharmaceutical cocrystals of nomegestrol acetate with superior dissolution. CrystEngComm 2022, 24, 6385–6391. [Google Scholar] [CrossRef]
- Yang, Y.; Niu, H.; Xia, S.; Guo, Y.; Wu, X. Solubility and dissolution rate of progesterone cocrystals using 4-fluorobenzoic acid and 2-hydroxy-6-naphthoic acid as coformers. J. Cryst. Growth 2022, 585, 126601. [Google Scholar]
- Riley, D.D.; Nicolas, J.V.; Aaron, D.F.; Venkata, S.M.; Manish, A.M. The Effects of Humidity on Spontaneous Cocrystallization: A Survey of Diacid Cocrystals with Caffeine, Theophylline, and Nicotinamide. J. Chem. Crystallogr. 2022, 52, 479–484. [Google Scholar]
- Wang, L.; Luo, M.; Li, J.H.; Wang, J.M.; Zhang, H.L.; Deng, Z.W. Sweet Theophylline Cocrystal with Two Tautomers of Acesulfame. Cryst. Growth Des. 2015, 15, 2574–2578. [Google Scholar] [CrossRef]
- Yao, J.; Chen, J.M.; Xu, Y.B.; Lu, T.B. Enhancing the Solubility of 6-Mercaptopurine by Formation of Ionic Cocrystal with Zinc Trifluoromethanesulfonate: Single-Crystal-to-Single-Crystal Transformation. Cryst. Growth Des. 2014, 14, 5019–5025. [Google Scholar] [CrossRef]
- Nangia, A.; Rodriguez-Hornedo, N. Indo–U.S. Workshop on Pharmaceutical Cocrystals and Polymorphs. Cryst. Growth Des. 2009, 9, 3339–3341. [Google Scholar] [CrossRef]
- Childs, S.L.; Zaworotko, M.J. The Reemergence of Cocrystals: The Crystal Clear Writing Is on the Wall Introduction to Virtual Special Issue on Pharmaceutical Cocrystals. Cryst. Growth Des. 2009, 9, 4208–4211. [Google Scholar] [CrossRef]
- Zhang, X.M.; Tian, Y.Y.; Jia, J.T.; Zhang, T.T.; Zhu, G.S. Synthesis, characterization and dissolution of three pharmaceutical cocrystals based on deferiprone. J. Mole Struct. 2016, 1108, 560–566. [Google Scholar] [CrossRef]
- Bathori, N.B.; Lemmerer, A.; Venter, G.A.; Bourne, S.A.; Caira, M.R. Pharmaceutical Co-crystals with Isonicotinamide-Vitamin B3, Clofibric Acid, and Diclofenac—And Two Isonicotinamide Hydrates. Cryst. Growth Des. 2011, 11, 75–87. [Google Scholar] [CrossRef]
- Wang, C.H.; Wang, F.; Li, C.Y.; Xu, X.L.; Li, T.; Wang, C.F. Voltammetric sensor for tinidazole based on poly(carmine) film modified electrode and its application. Dyes Pigments 2007, 75, 213–217. [Google Scholar] [CrossRef]
- Rams, T.E.; Sautter, J.D.; van Winkelhoff, A.J. Comparative In Vitro Resistance of Human Periodontal Bacterial Pathogens to Tinidazole and Four Other Antibiotics. Antibiotics 2020, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Pandiyan, R.; Vinothkumar, V.; Chen, T.; Chen, S.; Abinaya, M.; Rwei, S.; Hsu, H.; Huang, C.; Yu, M. Synthesis of Ag@ZrO2 nanoparticles: A sensitive electrochemical sensor for determination of antibiotic drug tinidazole. Int. J. Electrochem. Sci. 2022, 17, 220414. [Google Scholar] [CrossRef]
- Smirnov, A.S.; Mikherdov, A.S.; Rozhkov, A.V.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y.; Bokach, N.A. Halogen Bond-Involving Supramolecular Assembly Utilizing Carbon as a Nucleophilic Partner of I···C Non-covalent Interaction. Chem.-Asian J. 2023, 18, e202300037. [Google Scholar] [CrossRef] [PubMed]
- Bosch, E.; Ferrence, G.M.; Powell, C.J.; Unruh, D.K.; Krueger, H.R., Jr.; Groeneman, R.H. Cooperative non-covalent interactions and synthetic feed as driving forces to structural diversity within organic co-crystals containing isosteric perhalobenzenes. CrystEngComm 2022, 24, 3841–3845. [Google Scholar] [CrossRef]
- Singh, J.; Kim, H.; Chi, K. Non-Covalent Interaction-Directed Coordination-Driven Self-Assembly of Non-Trivial Supramolecular Topologies. Chem. Rec. 2021, 21, 574–593. [Google Scholar] [CrossRef]
- Novikov, A.S. Non-Covalent Interactions in Organic, Organometallic, and Inorganic Supramolecular Systems Relevant for Medicine, Materials Science, and Catalysis. Crystals 2022, 12, 246. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Non-covalent interactions in the synthesis of coordination compounds: Recent advances. Coordin. Chem. Rev. 2017, 345, 54–72. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, J.; Jin, S.; Liu, B.; Shi, C.; Wang, D. Preparation and structure analysis of non-covalent interactions mediated 2D-3D supramolecular adducts from 6-methylnicotinamide and carboxylic acids. J. Mol. Struct. 2022, 1264, 133135. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Vasilev, N.A.; Ilyukhin, A.B.; Perlovich, G.L. Novel cocrystals of the potent 1,2,4-thiadiazole-based neuroprotector with carboxylic acids: Virtual screening, crystal structures and solubility performance. New J. Chem. 2021, 45, 3034–3047. [Google Scholar] [CrossRef]
- SMART and SAINT+ for Windows NT, Version 6.02a; Bruker Analytical X-ray Instruments Inc.: Madison, WI, USA, 1998.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H.J. OLEX2: A complete structure solution, refinement and analysis program. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Tobón Zapata, G.E.; Martínez Carmona, D.M.; Echeverría, G.A.; Piro, O.E. Molecular structures of two copper complexes with the pharmaceuticals norfloxacin and tinidazole, when powder X-ray diffraction assists multi-domain single-crystal X-ray diffraction. Acta Cryst. B 2022, 78, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Fayaz, T.K.S.; Palanisamy, V.; Sanphui, P.; Chernyshev, V. Multicomponent solid forms of antibiotic cephalexin towards improved chemical stability. CrystEngComm 2023, 25, 1252–1262. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| Empirical formula | C15H19N3O8S | C16H21N3O7S | C15H18ClN3O7S |
| Formula weight | 401.39 | 399.42 | 419.83 |
| T/K | 293(2) | 293(2) | 293(2) |
| Crystal system | monoclinic | triclinic | triclinic |
| Space group | Cc | P−1 | P−1 |
| a/Å | 19.2461(9) | 5.7197(3) | 5.8506(4) |
| b/Å | 11.1923(5) | 11.0515(13) | 11.0083(5) |
| c/Å | 8.9518(4) | 15.4804(12) | 15.0294(9) |
| α/° | 90 | 89.780(8) | 92.248(4) |
| β/° | 115.967(4) | 79.433(6) | 101.203(5) |
| γ/° | 90 | 77.183(7) | 103.511(5) |
| V/Å3 | 1733.62(15) | 937.28(14) | 919.61(10) |
| Z | 4 | 2 | 2 |
| Dc/g cm−3 | 1.538 | 1.415 | 1.516 |
| μ/mm−1 | 0.239 | 0.217 | 0.365 |
| F(000) | 840 | 420 | 436 |
| Reflns collected | 3679 | 6238 | 6058 |
| Data/restraints/parameters | 2090/2/248 | 3306/0/249 | 3229/0/248 |
| S(GOF on F2) | 1.059 | 1.095 | 1.069 |
| h, k, lmax | 18, 13, 10 | 6, 13, 13 | 5, 13, 17 |
| R (I > 2σ (I)) | a R1 = 0.0285, b wR2 = 0.0617 | a R1 = 0.0511, b wR2 = 0.1127 | a R1 = 0.0366, b wR2 = 0.0922 |
| R (all data) | a R1 = 0.0310, b wR2 = 0.0634 | a R1 = 0.0859, b wR2 = 0.1375 | a R1 = 0.0474, b wR2 = 0.0998 |
| D–H···A (Å) | D-H(Å) | H···A(Å) | D···A(Å) | D-H···A (deg) | |
|---|---|---|---|---|---|
| 1 | O1–H1···O2 (x, y, z) | 0.820(1) | 1.874(1) | 2.598(1) | 146.515(2) |
| O4–H4···O3 (x, y, z) | 0.820(1) | 1.746(1) | 2.480(1) | 148.086(1) | |
| N2–H2···O2 (x, −1 − y, 0.5 + z) | 0.860(1) | 1.832(1) | 2.681(1) | 168.669(3) | |
| C11–H11B···O30 (x, −1 − y, 0.5 + z) | 0.960(1) | 2.146(1) | 3.068(1) | 160.688(2) | |
| C8–H8···O7 (x, −1 − y, 0.5 + z) | 0.930(1) | 2.396(1) | 3.325(1) | 176.673(2) | |
| C13–H13B···O4 (0.5 + x, −0.5 − y, 0.5 + z) | 0.970(1) | 2.417(1) | 3.321(2) | 154.897(4) | |
| O14A–H14A···O4 (0.5 + x, −0.5 − y, 0.5 + z) | 0.970(1) | 2.487(1) | 3.384(1) | 153.754(4) | |
| 2 | O3–H3···O2 (x, y, z) | 0.820(1) | 1.914(2) | 2.629(3) | 145.206(9) |
| O1–H1···N2 (−1 − x, 1 − y, 2 − z) | 0.820(1) | 1.886(2) | 2.695(3) | 168.959(1) | |
| C10–H10···O3 (1 + x, y, z) | 0.930(1) | 2.366(2) | 3.260(2) | 161.053(8) | |
| O3–H3···O2 (−x, 1 − y, 2 − z) | 0.820(1) | 2.441(3) | 2.996(3) | 125.84(12) | |
| C6–H6A···O3 (x, 1 + y, z) | 0.975(0) | 2.407(1) | 3.286(2) | 149.764(3) | |
| C14–H14B···O6 (1 + x, y, z) | 0.970(1) | 2.475(1) | 3.403(2) | 159.988(8) | |
| C14–H14A···O6 (x, y, z) | 0.970(1) | 2.578(3) | 3.405(4) | 143.210(1) | |
| C16–H16C···O4 (2 + x, y, z) | 0.960(1) | 2.586(3) | 3.469(4) | 153.025(10) | |
| 3 | O3–H3···O2 (x, y, z) | 0.820(1) | 1.897(1) | 2.611(1) | 145.061(7) |
| O1–H1···N2 (1 − x, 1 − y, −z) | 0.820(1) | 1.866(1) | 2.677(1) | 169.912(7) | |
| C10–H10···O3 (−1 + x, y, z) | 0.930(1) | 2.371(1) | 3.270(2) | 162.369(6) | |
| O3–H1···O2 (−x, 1 − y,−z) | 0.820(1) | 2.491(2) | 3.039(2) | 125.248(7) | |
| C13–H13B···O6 (x, y, 1 + z) | 0.970(1) | 2.561(2) | 3.385(2) | 142.839(6) | |
| C13–H13A···O6 (x, y, z) | 0.970(1) | 2.539(2) | 3.452(3) | 156.959(5) | |
| C16–H16C···O4 (x, y, 1 + z) | 0.960(1) | 2.620(2) | 3.424(2) | 141.561(6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Chen, Y.; Chen, R.; Zhang, M.; Wu, T.; Liu, K. Synthesis and Structure Characterization of Three Pharmaceutical Compounds Based on Tinidazole. Crystals 2023, 13, 947. https://doi.org/10.3390/cryst13060947
Li N, Chen Y, Chen R, Zhang M, Wu T, Liu K. Synthesis and Structure Characterization of Three Pharmaceutical Compounds Based on Tinidazole. Crystals. 2023; 13(6):947. https://doi.org/10.3390/cryst13060947
Chicago/Turabian StyleLi, Na, Yuting Chen, Ruixin Chen, Mingjuan Zhang, Tingting Wu, and Kang Liu. 2023. "Synthesis and Structure Characterization of Three Pharmaceutical Compounds Based on Tinidazole" Crystals 13, no. 6: 947. https://doi.org/10.3390/cryst13060947