

Computational Discovery of New Feasible Crystal Structures in Ce3O3N
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Candidates Generated via Global Exploration of the Energy Landscape at the Empirical Level
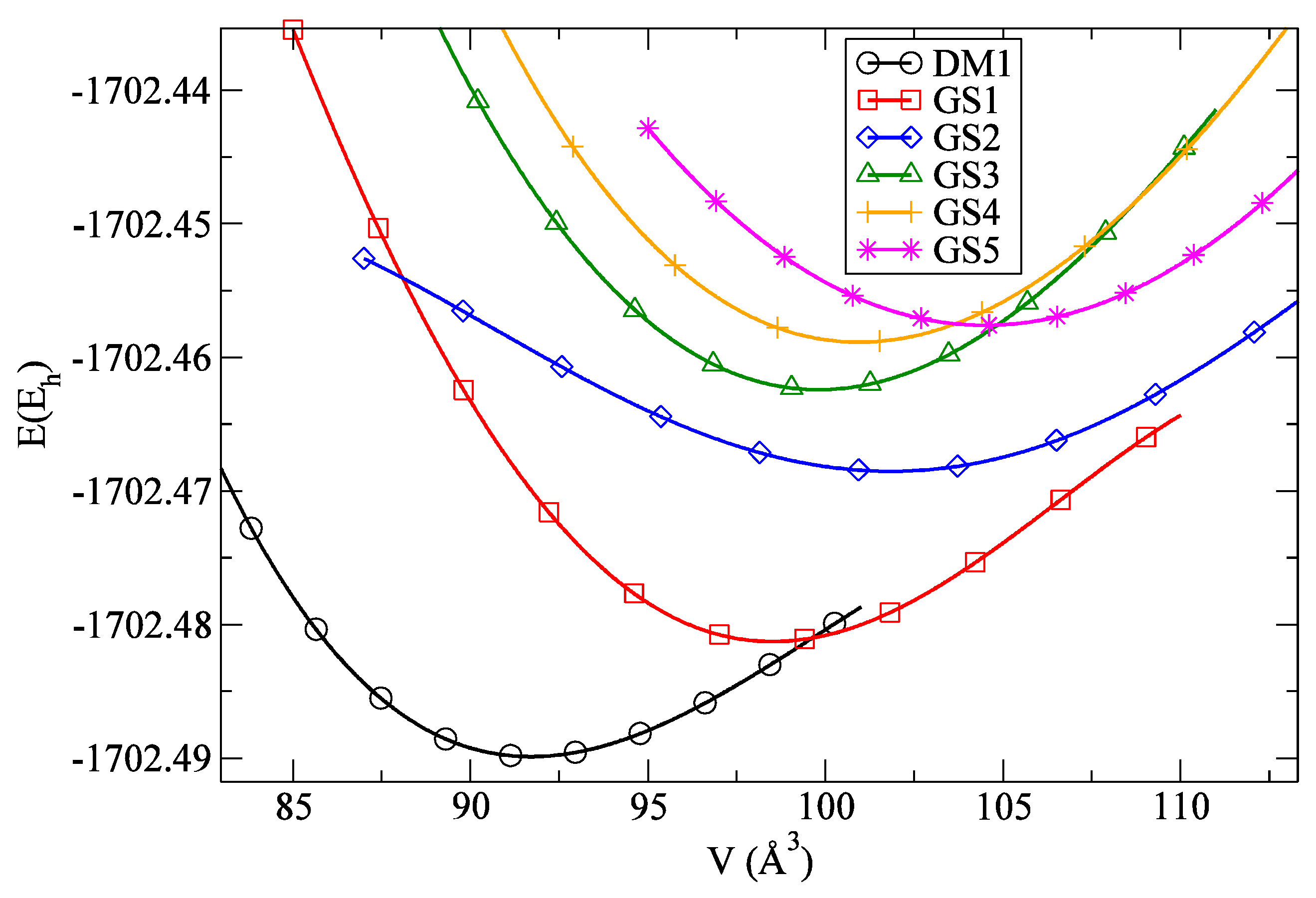
3.2. Candidates Generated via Data-Mining-Based Searches
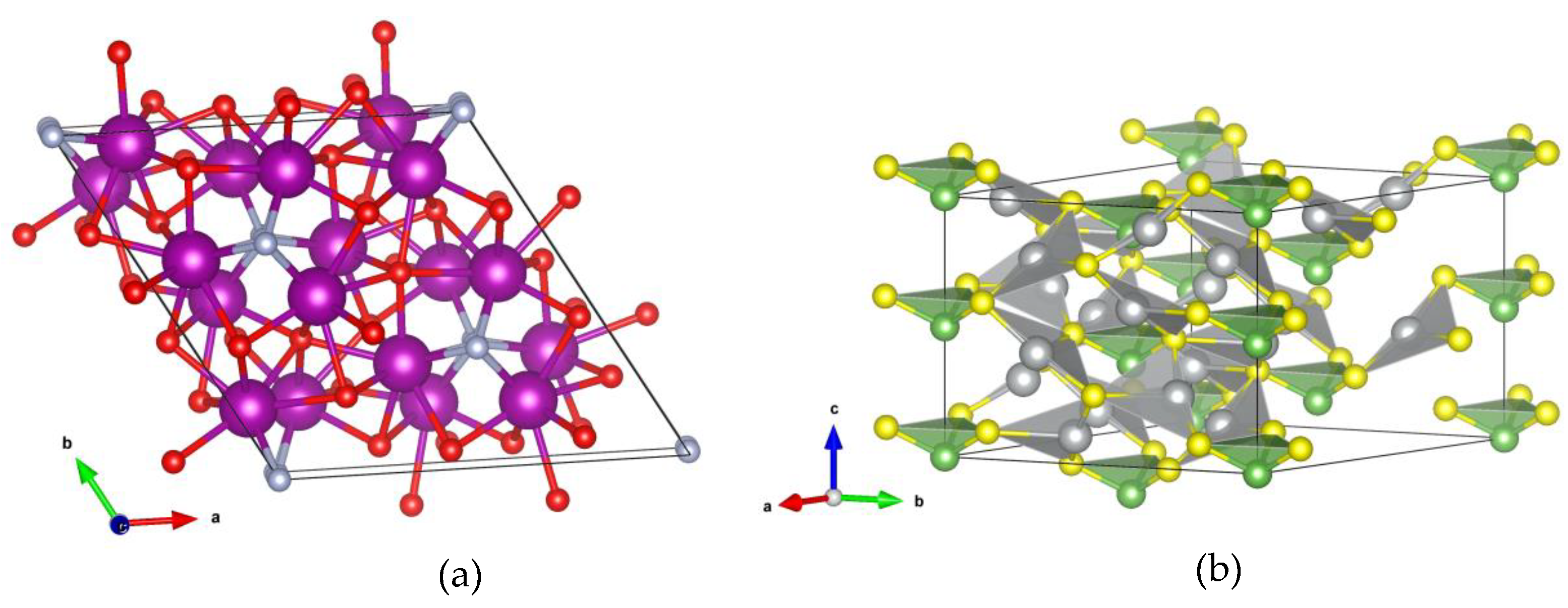
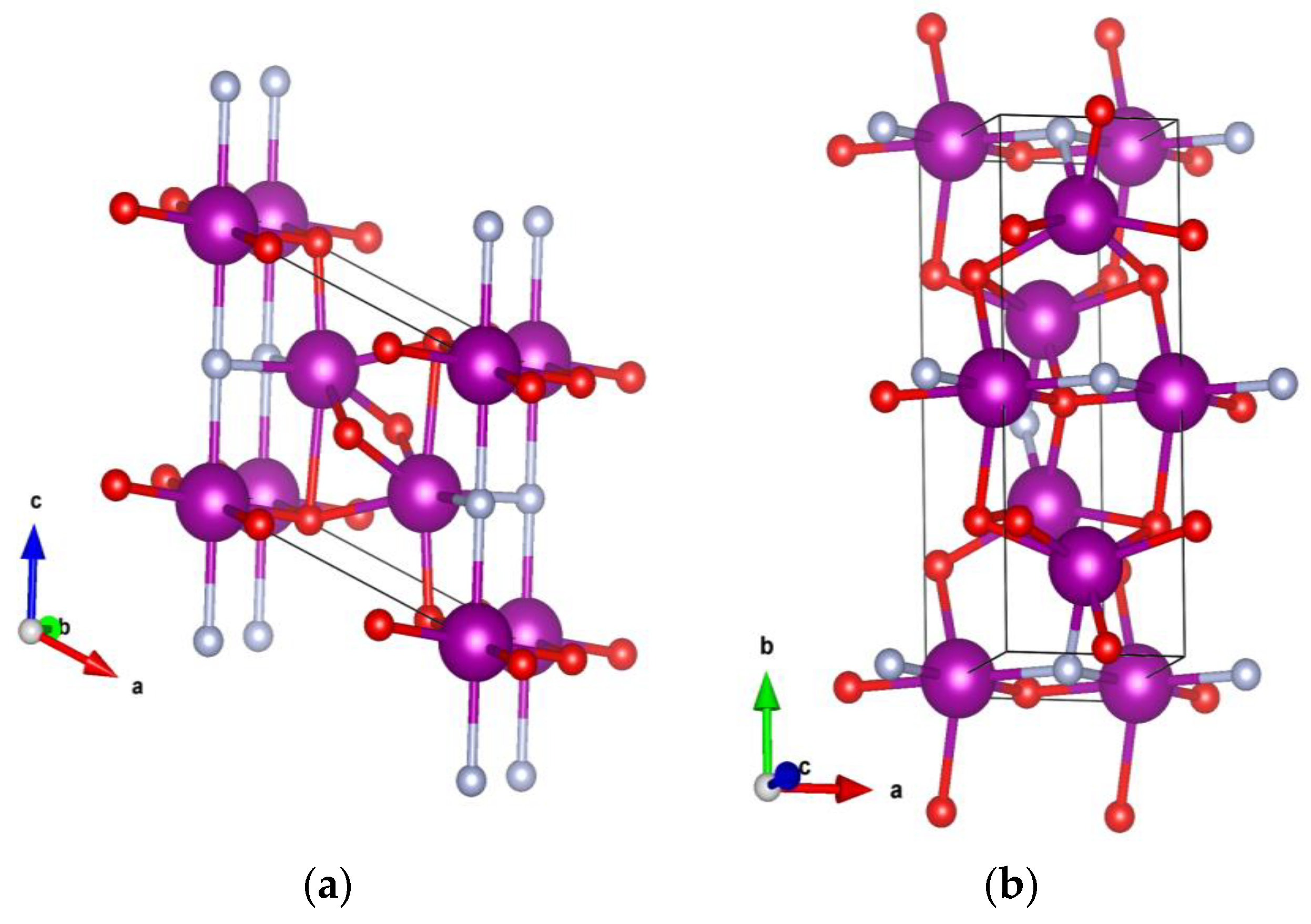
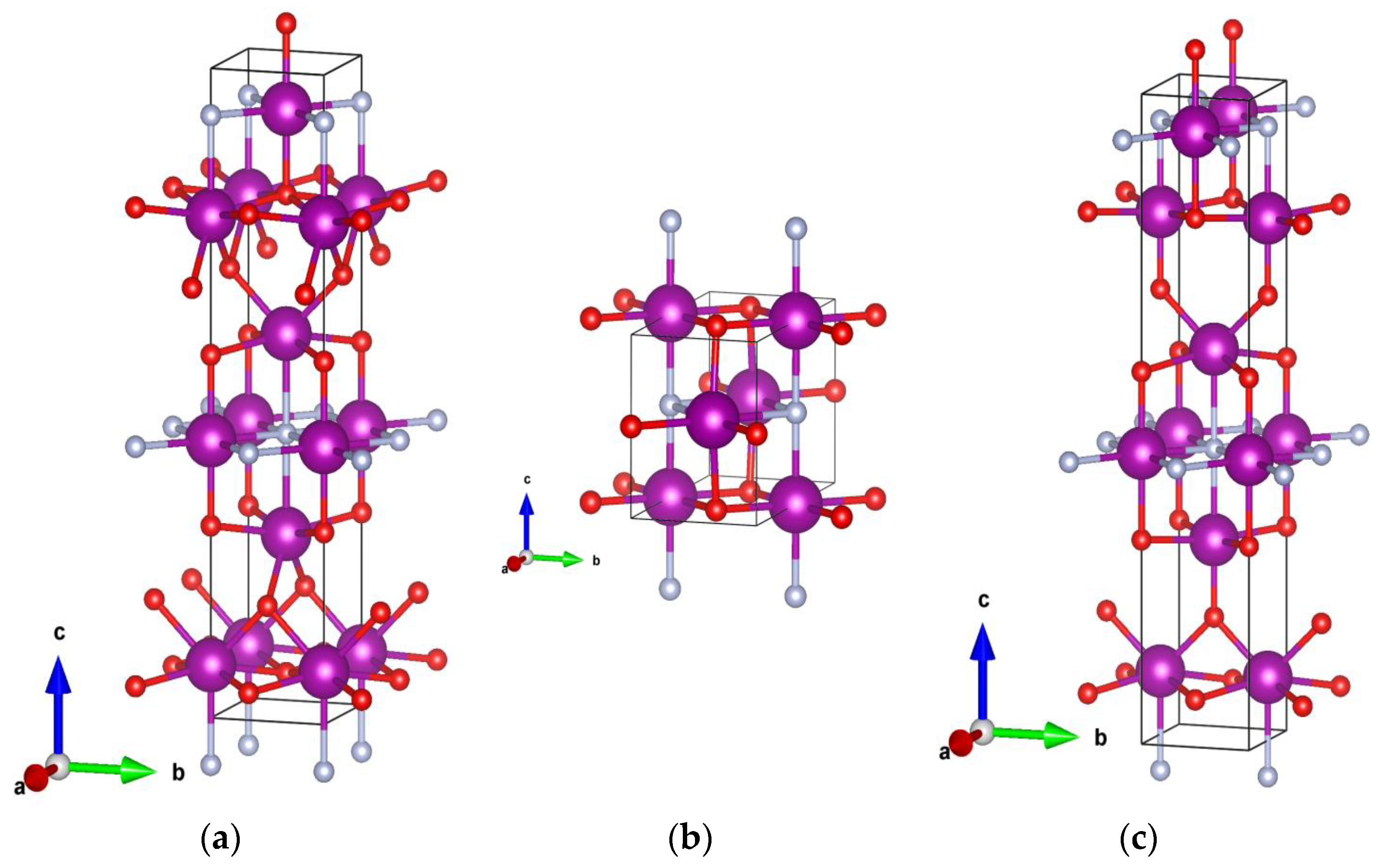
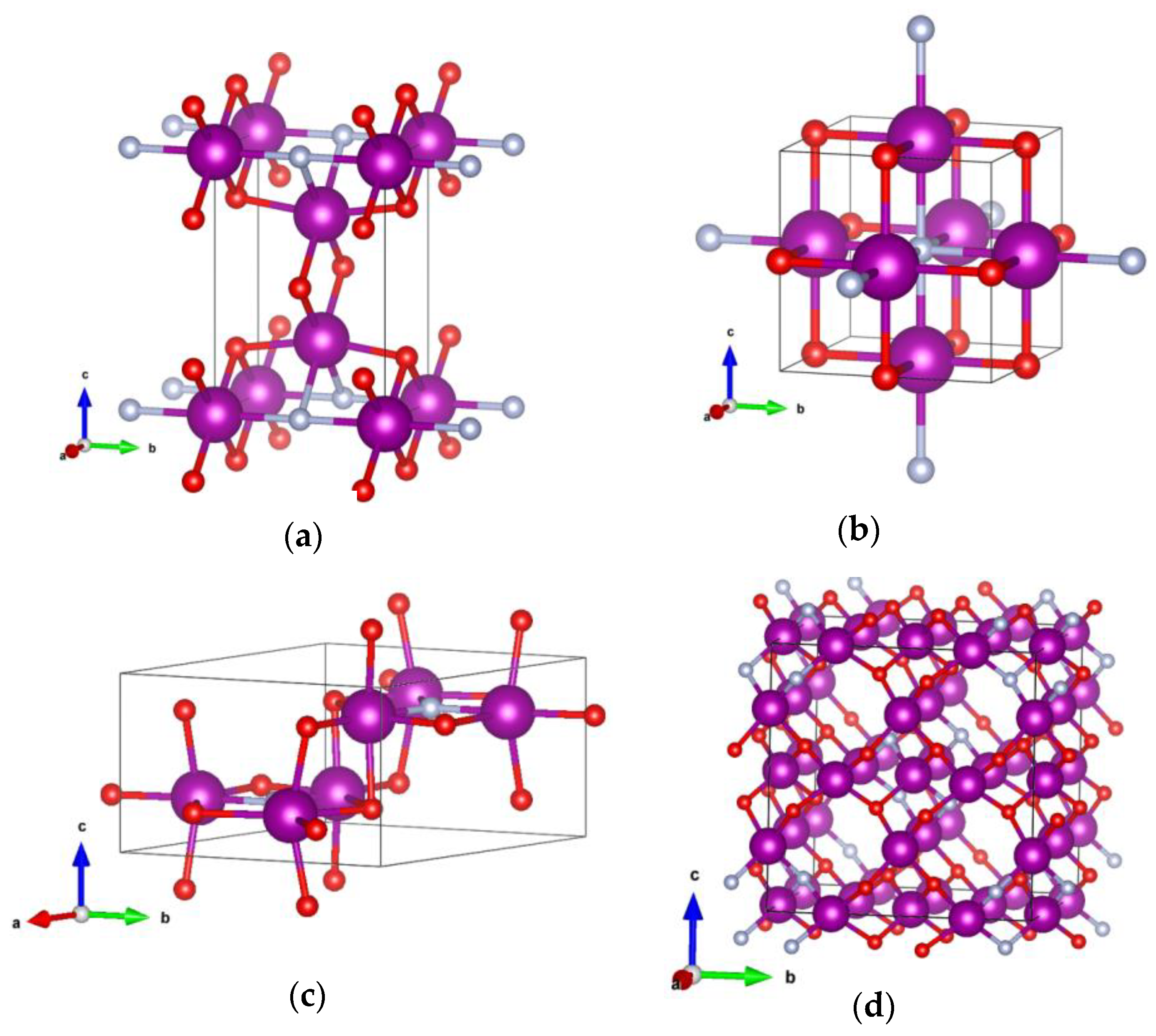
4. Crystal Structure Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Higashi, M.; Abe, R.; Takata, T.; Domen, K. Photocatalytic Overall Water Splitting under Visible Light Using ATaO2N (A = Ca, Sr, Ba) and WO3 in a IO3−/I− Shuttle Redox Mediated System. Chem. Mater. 2009, 21, 1543–1549. [Google Scholar] [CrossRef]
- Yang, M.; Oró-Solé, J.; Kusmartseva, A.; Fuertes, A.; Attfield, J.P. Electronic Tuning of Two Metals and Colossal Magnetoresistances in EuWO1+xN2−x Perovskites. J. Am. Chem. Soc. 2010, 132, 4822–4829. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.B.; Oró-Solé, J.; Bea, A.M.; Mufti, N.; Palstra, T.T.M.; Rodgers, J.A.; Attfield, J.P.; Fuertes, A. Large Coupled Magnetoresponses in EuNbO2N. J. Am. Chem. Soc. 2008, 130, 12572–12573. [Google Scholar] [CrossRef]
- Kim, Y.-I.; Woodward, P.M.; Baba-Kishi, K.Z.; Tai, C.W. Characterization of the Structural, Optical, and Dielectric Properties of Oxynitride Perovskites AMO2N (A = Ba, Sr, Ca; M = Ta, Nb). Chem. Mater. 2004, 16, 1267–1276. [Google Scholar] [CrossRef]
- Jansen, M.; Letschert, H.P. Inorganic yellow-red pigments without toxic metals. Nature 2000, 404, 980–982. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.B.; Fraxedas, J.; Cantarero, A.; Williams, A.J.; Rodgers, J.; Attfield, J.P.; Fuertes, A. Nitrogen Doping of Ceria. Chem. Mater. 2008, 20, 1682–1684. [Google Scholar] [CrossRef]
- Lee, J.-S.; Lerch, M.; Maier, J. Nitrogen-doped zirconia: A comparison with cation stabilized zirconia. J. Solid State Chem. 2006, 179, 270–277. [Google Scholar] [CrossRef]
- Sun, Y.; Lin, S.; Li, W.; Cheng, S.; Zhang, Y.; Liu, Y.; Liu, W. Review on Alkali Element Doping in Cu(In,Ga)Se2 Thin Films and Solar Cells. Engineering 2017, 3, 452–459. [Google Scholar]
- Kageyama, H.; Hayashi, K.; Maeda, K.; Attfield, J.P.; Hiroi, Z.; Rondinelli, J.M.; Poeppelmeier, K.R. Expanding frontiers in materials chemistry and physics with multiple anions. Nat. Commun. 2018, 9, 772. [Google Scholar] [CrossRef]
- Wu, Y.; Lazic, P.; Hautier, G.; Persson, K.; Ceder, G. First principles high throughput screening of oxynitrides for water-splitting photocatalysts. Energy Environ. Sci. 2013, 6, 157–168. [Google Scholar] [CrossRef]
- Sawada, K.; Nakajima, T. High-throughput screening of perovskite oxynitride and oxide materials for visible-light photocatalysis. APL Mater. 2018, 6, 101103. [Google Scholar]
- Castelli, I.E.; Olsen, T.; Datta, S.; Landis, D.D.; Dahl, S.; Thygesen, K.S.; Jacobsen, K.W. Computational screening of perovskite metal oxides for optimal solar light capture. Energy Environ. Sci. 2012, 5, 5814–5819. [Google Scholar]
- Sharan, A.; Lany, S. Computational discovery of stable and metastable ternary oxynitrides. J. Chem. Phys. 2021, 154, 234706. [Google Scholar] [CrossRef]
- Prabhakaran, V.; Ramani, V. Structurally-Tuned Nitrogen-Doped Cerium Oxide Exhibits Exceptional Regenerative Free Radical Scavenging Activity in Polymer Electrolytes. J. Electrochem. Soc. 2013, 161, F1–F9. [Google Scholar]
- Zhang, Y.C.; Liu, Y.K.; Zhang, L.; Xiu-tian-feng, E.; Pan, L.; Zhang, X.; Zou, D.R.; Liu, S.H.; Zou, J.J. DFT study on water oxidation on nitrogen-doped ceria oxide. Appl. Surf. Sci. 2018, 452, 423–428. [Google Scholar]
- Mao, C.; Zhao, Y.; Qiu, X.; Zhu, J.; Burda, C. Synthesis, characterization and computational study of nitrogen-doped CeO2 nanoparticles with visible-light activity. Phys. Chem. Chem. Phys. 2008, 10, 5633–5638. [Google Scholar] [CrossRef]
- Shi, H.; Hussain, T.; Ahuja, R.; Kang, T.W.; Luo, W. Role of vacancies, light elements and rare-earth metals doping in CeO2. Sci. Rep. 2016, 6, 31345. [Google Scholar]
- Matović, B.; Dukić, J.; Babić, B.; Bučevac, D.; Dohčević-Mitrović, Z.; Radović, M.; Bošković, S. Synthesis, calcination and characterization of Nanosized ceria powders by self-propagating room temperature method. Ceram. Int. 2013, 39, 5007–5012. [Google Scholar] [CrossRef]
- Dmitrović, S.; Nikolić, M.G.; Jelenković, B.; Prekajski, M.; Rabasović, M.; Zarubica, A.; Branković, G.; Matović, B. Photoluminescent properties of spider silk coated with Eu-doped nanoceria. J. Nanoparticle Res. 2017, 19, 1–11. [Google Scholar]
- Mićović, D.; Pagnacco, M.C.; Banković, P.; Maletaškić, J.; Matović, B.; Djokić, V.R.; Stojmenović, M. The influence of short thermal treatment on structure, morphology and optical properties of Er and Pr doped ceria pigments: Comparative study. Process. Appl. Ceram. 2019, 13, 310–321. [Google Scholar] [CrossRef]
- WoŁcyrz, M.; Kepinski, L. Rietveld refinement of the structure of CeOCI formed in Pd/CeO2 catalyst: Notes on the existence of a stabilized tetragonal phase of La2O3 in La-Pd-O system. J. Solid State Chem. Fr. 1992, 99, 409–413. [Google Scholar] [CrossRef]
- Coduri, M.; Scavini, M.; Allieta, M.; Brunelli, M.; Ferrero, C. Defect Structure of Y-Doped Ceria on Different Length Scales. Chem. Mater. 2013, 25, 4278–4289. [Google Scholar] [CrossRef]
- Mamontov, E.; Egami, T.; Brezny, R.; Koranne, M.; Tyagi, S. Lattice Defects and Oxygen Storage Capacity of Nanocrystalline Ceria and Ceria-Zirconia. J. Phys. Chem. B 2000, 104, 11110–11116. [Google Scholar] [CrossRef]
- Skorodumova, N.V.; Ahuja, R.; Simak, S.I.; Abrikosov, I.A.; Johansson, B.; Lundqvist, B.I. Electronic, bonding, and optical properties of CeO2 and Ce2O3 from first principles. Phys. Rev. B 2001, 64, 115108. [Google Scholar] [CrossRef]
- Zagorac, J.; Schön, J.C.; Matović, B.; Škundrić, T.; Zagorac, D. Predicting Feasible Modifications of Ce2ON2 Using a Combination of Global Optimization and Data Mining. J. Phase Equilibria Diffus. 2020, 41, 538–549. [Google Scholar] [CrossRef]
- Čebela, M.; Zagorac, D.; Batalović, K.; Radaković, J.; Stojadinović, B.; Spasojević, V.; Hercigonja, R. BiFeO3 perovskites: A multidisciplinary approach to multiferroics. Ceram. Int. 2017, 43, 1256–1264. [Google Scholar] [CrossRef]
- Zagorac, J.; Zagorac, D.; Rosić, M.; Schön, J.C.; Matović, B. Structure prediction of aluminum nitride combining data mining and quantum mechanics. CrystEngComm 2017, 19, 5259–5268. [Google Scholar] [CrossRef]
- Zagorac, D.; Schön, J.C.; Rosić, M.; Zagorac, J.; Jordanov, D.; Luković, J.; Matović, B. Theoretical and Experimental Study of Structural Phases in CoMoO4. Cryst. Res. Technol. 2017, 52, 1700069. [Google Scholar] [CrossRef]
- Kirkpatrick, S.; Gelatt, C.D.; Vecchi, M.P. Optimization by Simulated Annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef]
- Schön, J.C. Nanomaterials—What energy landscapes can tell us. Process. Appl. Ceram. 2015, 9, 157–168. [Google Scholar] [CrossRef]
- Schön, J.C.; Jansen, M. Determination of candidate structures for simple ionic compounds through cell optimisation. Comput. Mater. Sci. 1995, 4, 43–58. [Google Scholar] [CrossRef]
- Bergerhoff, G.; Brown, I.D. Crystallographic Databases; International Union of Crystallography: Chester, UK, 1987. [Google Scholar]
- Zagorac, D.; Muller, H.; Ruehl, S.; Zagorac, J.; Rehme, S. Recent developments in the Inorganic Crystal Structure Database: Theoretical crystal structure data and related features. J. Appl. Crystallogr. 2019, 52, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Sokol, A.A.; Catlow, C.R.A.; Miskufova, M.; Shevlin, S.A.; Al-Sunaidi, A.A.; Walsh, A.; Woodley, S.M. On the problem of cluster structure diversity and the value of data mining. Phys. Chem. Chem. Phys. 2010, 12, 8438–8445. [Google Scholar] [CrossRef] [PubMed]
- Ceder, G.; Morgan, D.; Fischer, C.; Tibbetts, K.; Curtarolo, S. Data-Mining-Driven Quantum Mechanics for the Prediction of Structure. MRS Bull. 2011, 31, 981–985. [Google Scholar] [CrossRef]
- Dovesi, R.; Orlando, R.; Civalleri, B.; Roetti, C.; Saunders Victor, R.; Zicovich-Wilson Claudio, M. CRYSTAL: A computational tool for the ab initio study of the electronic properties of crystals. Z. Für Krist. Cryst. Mater. 2005, 220, 571. [Google Scholar] [CrossRef]
- Doll, K.; Dovesi, R.; Orlando, R. Analytical Hartree–Fock gradients with respect to the cell parameter for systems periodic in three dimensions. Theor. Chem. Acc. 2004, 112, 394–402. [Google Scholar] [CrossRef]
- Doll, K.; Saunders, V.R.; Harrison, N.M. Analytical Hartree–Fock gradients for periodic systems. Int. J. Quantum Chem. 2001, 82, 1–13. [Google Scholar] [CrossRef]
- Graciani, J.; Márquez, A.M.; Plata, J.J.; Ortega, Y.; Hernández, N.C.; Meyer, A.; Zicovich-Wilson, C.M.; Sanz, J.F. Comparative Study on the Performance of Hybrid DFT Functionals in Highly Correlated Oxides: The Case of CeO2 and Ce2O3. J. Chem. Theory Comput. 2011, 7, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Towler, M.D.; Allan, N.L.; Harrison, N.M.; Saunders, V.R.; Mackrodt, W.C.; Aprà, E. Ab initio study of MnO and NiO. Phys. Rev. B 1994, 50, 5041–5054. [Google Scholar] [CrossRef]
- Zagorac, D.; Schön, J.C.; Zagorac, J.; Jansen, M. Prediction of structure candidates for zinc oxide as a function of pressure and investigation of their electronic properties. Phys. Rev. B 2014, 89, 075201. [Google Scholar] [CrossRef]
- Dovesi, R.; Causa’, M.; Orlando, R.; Roetti, C.; Saunders, V.R. Ab initio approach to molecular crystals: A periodic Hartree–Fock study of crystalline urea. J. Chem. Phys. 1990, 92, 7402–7411. [Google Scholar] [CrossRef]
- Zagorac, D.; Zagorac, J.; Djukic, M.B.; Jordanov, D.; Matović, B. Theoretical study of AlN mechanical behaviour under high pressure regime. Theor. Appl. Fract. Mech. 2019, 103, 102289. [Google Scholar] [CrossRef]
- Hundt, R. KPLOT, A Program for Plotting and Analyzing Crystal Structures; Technicum Scientific Publishing: Stuttgart, Germany, 2016. [Google Scholar]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Hundt, R.; Schon, J.C.; Hannemann, A.; Jansen, M. Determination of symmetries and idealized cell parameters for simulated structures. J. Appl. Crystallogr. 1999, 32, 413–416. [Google Scholar] [CrossRef]
- Hannemann, A.; Hundt, R.; Schön, J.C.; Jansen, M. A New Algorithm for Space-Group Determination. J. Appl. Crystallogr. 1998, 31, 922–928. [Google Scholar] [CrossRef]
- Hundt, R.; Schon, J.C.; Jansen, M. CMPZ—An algorithm for the efficient comparison of periodic structures. J. Appl. Crystallogr. 2006, 39, 6–16. [Google Scholar] [CrossRef]
- Tsokol, A.O.; Bodak, O.I.; Marusin, E.P.; Baivelman, M.G. Crystal structure of the compound ScAl3C3. Sov. Phys. Crystallogr. (=Kristalogr.) 1986, 31, 467–468. [Google Scholar]
- Rabenau, A.; Kniep, R.; Höhn, P. Ba3[FeN3]: Ein neues Nitridoferrat(III) mit [CO3]2--isosteren Anionen [FeN3]6. Z. Für Krist. 1991, 196, 153–158. [Google Scholar] [CrossRef]
- Lang, J.; Hamon, C.; Marchand, R.; Laurent, Y. Étude d’halogénopnictures. III. Structure de Ca2PI et Ca3PI3. Surstructures de type NaCl. Bull. De Minéralogie 1974, 97, 6–12. [Google Scholar]
- Cordier, G.; Schaefer, H.; Stelter, M. Neue Zintlphasen: Ba3GaSb3, Ca3GaAs3 und Ca3InP3. Z. Fuer Nat. Teil B Anorg. Chem. Org. Chem. 1985, 40, 1100–1104. [Google Scholar]
- Machatsehki, F. XII. Präzisionsmessungen der Gitterkonstanten verschiedener Fahlerze. Form. Und Struktur Derselben 1928, 68, 204–222. [Google Scholar] [CrossRef]
- Klepp, K.; Boller, H. Die Kristallstruktur von TlFe3Te3. Mon. Für Chem. Chem. Mon. 1979, 110, 677–684. [Google Scholar] [CrossRef]
- Pollock, C.B.; Stadelmaier, H.H. The eta carbides in the Fe−W−C and Co−W−C systems. Metall. Trans. 1970, 1, 767–770. [Google Scholar] [CrossRef]
- Ebihara, M.; Martin, J.D.; Corbett, J.D. Novel Chain and Oligomeric Condensed Cluster Phases for Gadolinium Iodides with Manganese Interstitials. Inorg. Chem. 1994, 33, 2079–2084. [Google Scholar] [CrossRef]
- Crystal structure of hexapotassium di-μ-selenido-bis(diselenidoaluminate), K6Al2Se6. Z. Für Krist. 1991, 197, 173–174. [CrossRef]
- Kuchinke, J.; Jansen, C.; Lindemann, A.; Krebs, B. Syntheses and Crystal Structures of the Novel Ternary Thioborates Na3BS3, K3BS3, and Rb3BS3. Z. Anorg. Allg. Chem. 2001, 627, 896–902. [Google Scholar] [CrossRef]
- Dittmar, G. Die KristallStrukturen von K6[Ge2Te6] und K6[Sn2Te6] und ihre kristall-chemische Beziehung zum K6[Si2Te6]-Typ. Z. Anorg. Allg. Chem. 1979, 453, 68–78. [Google Scholar] [CrossRef]
- Kuznetsov, I.Y.; Vinitskii, D.M.; Solntsev, K.A.; Kuznetsov, N.T.; Butman, L.A. The crystal structure of K2B6H6 and Cs2B6H6. Zhurnal Neorgnicheskoi Khimii 1987, 32, 3112–3114. [Google Scholar]
- Palazzi, M. Structure cristalline de l’orthotrithioarsenite trisodique Na3AsS3. Acta Crystallogr. Sect. B 1976, 32, 3175–3177. [Google Scholar] [CrossRef]
- Mruz, O.Y.; Pecharskii, V.K.; Sobolev, A.N.; Bodak, O.I. Crystal structure of SmNi3Ge3. Kristallografiya 1990, 35, 202–204. [Google Scholar]
- Kotur, B.Y.; Gladyshevskii, E.I. Crystal structure of scandium-nickel silicide (Sc3NiSi3). Kristallografiya 1983, 28, 461–464. [Google Scholar]
- Harker, D. The Application of the Three-Dimensional Patterson Method and the Crystal Structures of Proustite, Ag3AsS3, and Pyrargyrite, Ag3SbS3. J. Chem. Phys. 1936, 4, 381–390. [Google Scholar] [CrossRef]
- Wei, C.H. Structural analyses of tetracobalt dodecacarbonyl and tetrarhodium dodecacarbonyl. Crystallographic treatments of a disordered structure and a twinned composite. Inorg. Chem. 1969, 8, 2384–2397. [Google Scholar] [CrossRef]
- Hong, H.Y.P.; Mikkelsen, J.C.; Roland, G.W. Crystal structure of Tl3AsSe3. Mater. Res. Bull. 1974, 9, 365–369. [Google Scholar] [CrossRef]
- Engel, P.; Nowacki, W. Die Kristallstruktur von Xanthokon, Ag3AsS3. Acta Crystallogr. Sect. B 1968, 24, 77–81. [Google Scholar] [CrossRef]
- Fischer, D.; Andriyevsky, B.; Schön, C. Systematics of the allotrope formation in elemental gallium films. Mater. Res. Express 2019, 6, 116401. [Google Scholar] [CrossRef]
- Zagorac, D.; Zagorac, J.; Schön, J.C.; Stojanovic, N.; Matovic, B. ZnO/ZnS (hetero)structures: Ab initio investigations of polytypic behavior of mixed ZnO and ZnS compounds. Acta Crystallogr. B 2018, 74, 628–642. [Google Scholar] [CrossRef]
- Zagorac, D.; Zagorac, J.; Pejić, M.; Matović, B.; Schön, J.C. Band Gap Engineering of Newly Discovered ZnO/ZnS Polytypic Nanomaterials. Nanomaterials 2022, 12, 1595. [Google Scholar] [CrossRef]
- Pejić, M.; Zagorac, D.; Zagorac, J.; Matović, B.; Schön, J.C. Structure prediction via global energy landscape exploration of the ternary rare-earth compound LaOI. Z. Anorg. Allg. Chem. 2022, 648, e202200308. [Google Scholar] [CrossRef]
- Schön, J.C. Energy landscapes in inorganic chemistry. In Comprehensive Inorganic Chemistry III; Poeppelmeier, K., Reedijk, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 262–392. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pressure (GPa) | Space Group No. | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 5 | 6 | 8 | 10 | 16 | 25 | 38 | 44 | 47 | 99 | 115 | 146 | 155 | 160 | 207 | 221 | |
| 0 | 379 | - | - | 32 | 11 | - | - | 7 | - | 2 | - | 1 | - | - | - | - | - | - |
| 0.016 | 376 | 2 | 1 | 27 | 7 | - | - | 10 | 2 | - | - | 1 | 1 | - | 1 | - | 1 | 3 |
| 0.16 | 368 | - | - | 38 | 3 | 1 | 1 | 5 | - | 1 | 1 | 1 | - | - | - | - | 3 | 10 |
| 1.6 | 373 | 1 | 2 | 26 | 9 | - | - | 7 | 3 | - | - | 1 | - | - | 1 | - | 2 | 7 |
| 16 | 370 | 2 | 3 | 25 | 10 | - | - | - | 7 | 2 | - | - | - | - | - | - | 6 | 7 |
| 160 | 332 | - | 3 | 36 | 20 | 1 | 1 | 5 | 16 | 1 | 11 | - | - | 4 | - | 2 | - | - |
| Σ | 2198 | 5 | 9 | 184 | 60 | 2 | 2 | 34 | 28 | 6 | 12 | 4 | 1 | 4 | 2 | 2 | 12 | 27 |
| Modification | Space Group (No.) | Total Energy (Eh) | Relative Energy in Eh (kcal/mol) |
|---|---|---|---|
| Ce3O3N-DM1 | R3c (161) | −1702.4875 | 0 |
| Ce3O3N-GS1 | P2/m (10) | −1702.4813 | 0.0062 (3.89) |
| Ce3O3N-GS2 | Amm2 (38) | −1702.4685 | 0.0190 (11.92) |
| Ce3O3N-GS3 | Imm2 (44) | −1702.4627 | 0.0248 (15.56) |
| Ce3O3N-GS4 | Pmmm (47) | −1702.4588 | 0.0287 (18.01) |
| Ce3O3N-GS5 | Amm2 (38) | −1702.4576 | 0.0299 (18.76) |
| Ce3O3N-GS6 | Pmmm (47) | −1702.4509 | 0.0366 (22.97) |
| Ce3O3N-GS7 | Pm-3m (221) | −1702.4394 | 0.0481 (30.18) |
| Ce3O3N-DM2 | P63/m (176) | −1702.4007 | 0.0868 (54.47) |
| Ce3O3N-DM3 | I-43m (217) | −1702.2837 | 0.2038 (127.89) |
| Modification | Space Group (No.) | Cell Parameters (Å) and Fractional Coordinates |
|---|---|---|
| Ce3O3N-DM1 | R3c (161) | a = 10.17; c = 6.15 Ce (−0.1996 −0.0378 0.2416) O (−0.0656 0.2200 0.3762) N (0 0 0.0066) |
| Ce3O3N-GS1 | P2/m (10) | a = 5.89; b = 3.62; c = 5.04; β = 113.2° Ce (0 0 0) Ce (0.3017 1/2 0.6238) O (0.2702 1/2 0.0652) O (1/2 0/2) N (0 0 1/2) |
| Ce3O3N-GS2 | Amm2 (38) | a = 3.59; b = 9.83; c = 5.78 Ce (1/2 0.1729 0.8911) Ce (0 0 0.3964) Ce (1/2 0 0.8038) O (0.5 0 0.1402) O (0 0.7712 0.1681) N (1/2 0 0.6528) |
| Ce3O3N-GS3 | Imm2 (44) | a = 3.38; b = 3.39; c = 17.39 Ce (0 0 0.7735) Ce (0 0 0.4276) Ce (0 0 0.0814) O (0 0 0.5589) O (0 0 0.2961) O (0 1/2 0.1775) N (0 0 0.9277) |
| Ce3O3N-GS4 | Pmmm (47) | a = 6.86; b = 3.53; c = 4.62 Ce (1/2 0 0) Ce (0.7607 1/2 1/2) O (0.2869 1/2 0) O (0 0 1/2) N (1/2 0 1/2) |
| Ce3O3N-GS5 | Amm2 (38) | a = 3.49; b = 3.32; c = 17.93 Ce (1/2 0 0.0970) Ce (0 0 0.4475) Ce (1/2 0 0.8038) O (0 0 0.5730) O (0 0 0.3143) O (1/2 0 0.6850) N (1/2 0 0.9444) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zagorac, J.; Schön, J.C.; Matović, B.; Pejić, M.; Prekajski Đorđević, M.; Zagorac, D. Computational Discovery of New Feasible Crystal Structures in Ce3O3N. Crystals 2023, 13, 774. https://doi.org/10.3390/cryst13050774
Zagorac J, Schön JC, Matović B, Pejić M, Prekajski Đorđević M, Zagorac D. Computational Discovery of New Feasible Crystal Structures in Ce3O3N. Crystals. 2023; 13(5):774. https://doi.org/10.3390/cryst13050774
Chicago/Turabian StyleZagorac, Jelena, Johann Christian Schön, Branko Matović, Milan Pejić, Marija Prekajski Đorđević, and Dejan Zagorac. 2023. "Computational Discovery of New Feasible Crystal Structures in Ce3O3N" Crystals 13, no. 5: 774. https://doi.org/10.3390/cryst13050774