
Crystal Structure of New 1-Phenyl-Substituted Tribenzsilatranes
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Single Crystal X-ray Diffraction
2.2. Modeling and Quantitative Analysis of Crystal Structures
2.3. Computational Details
2.4. Synthesis of the Compounds
3. Results
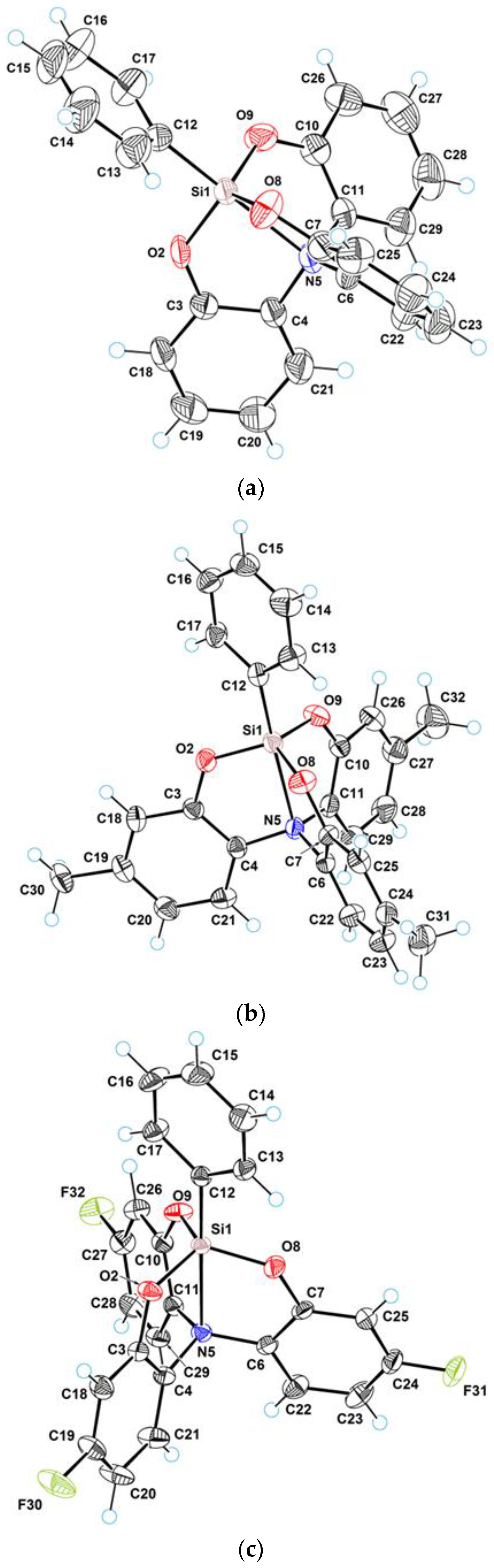
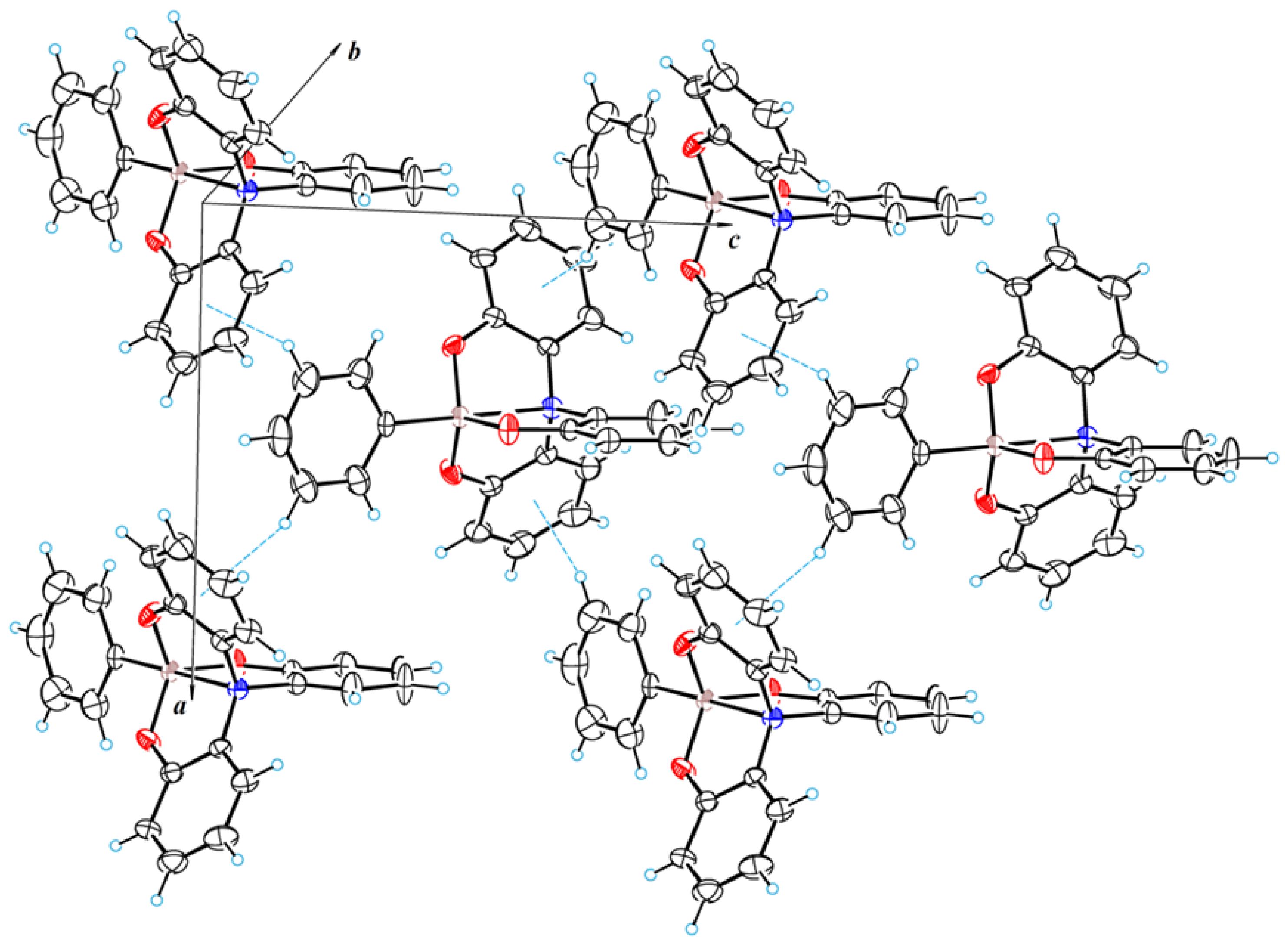
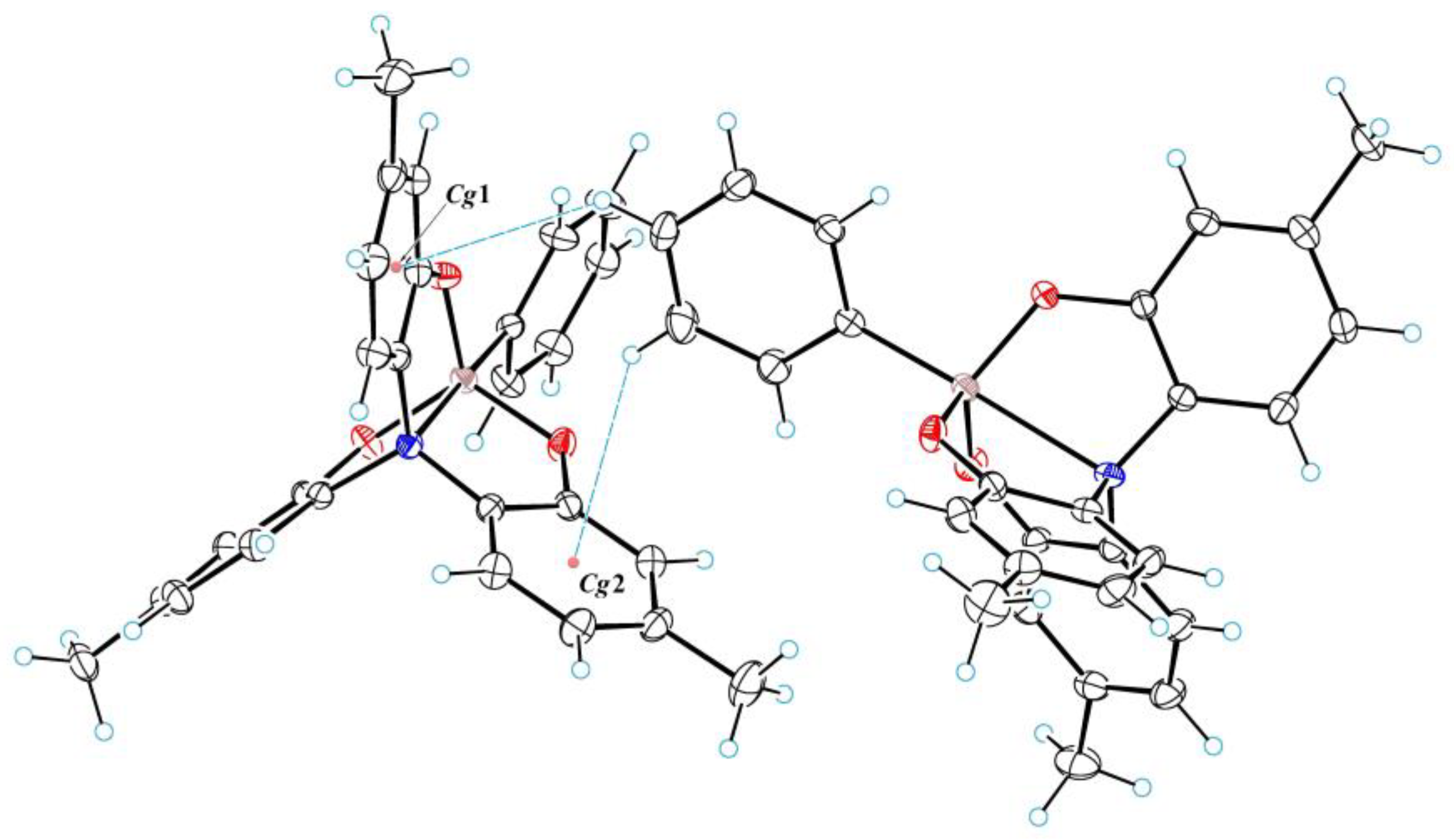
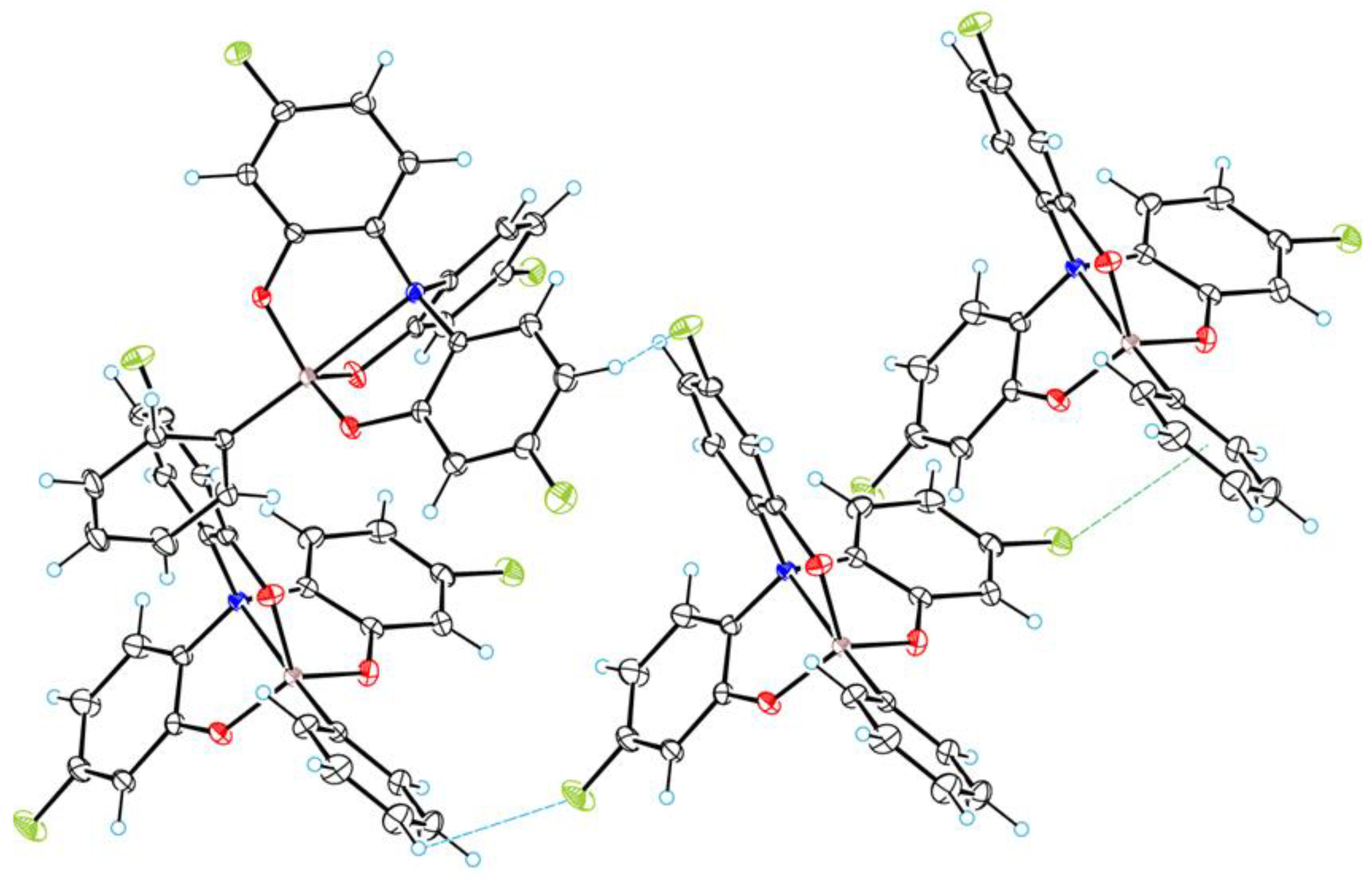
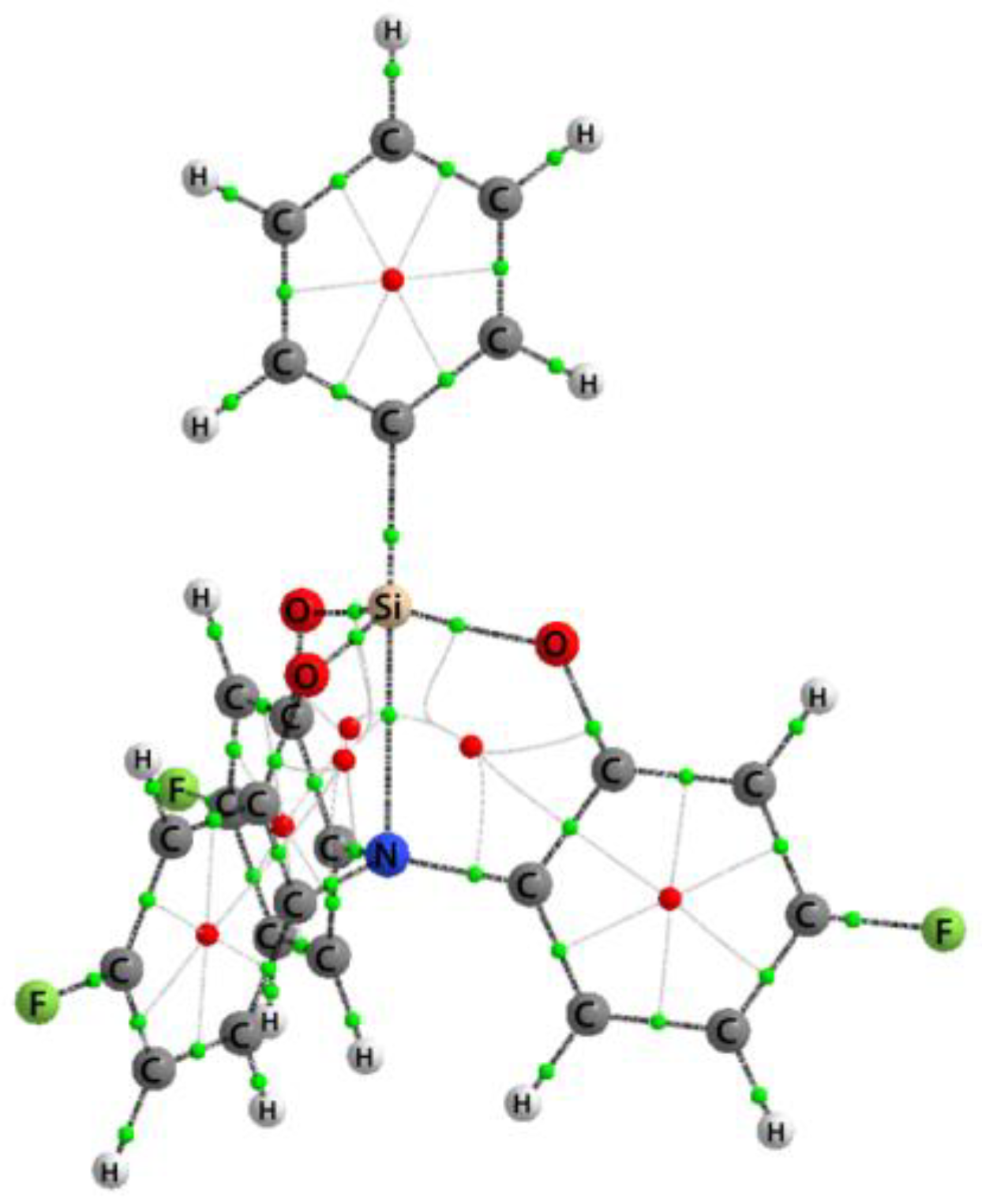
3.1. Crystal Structure of 2a–c
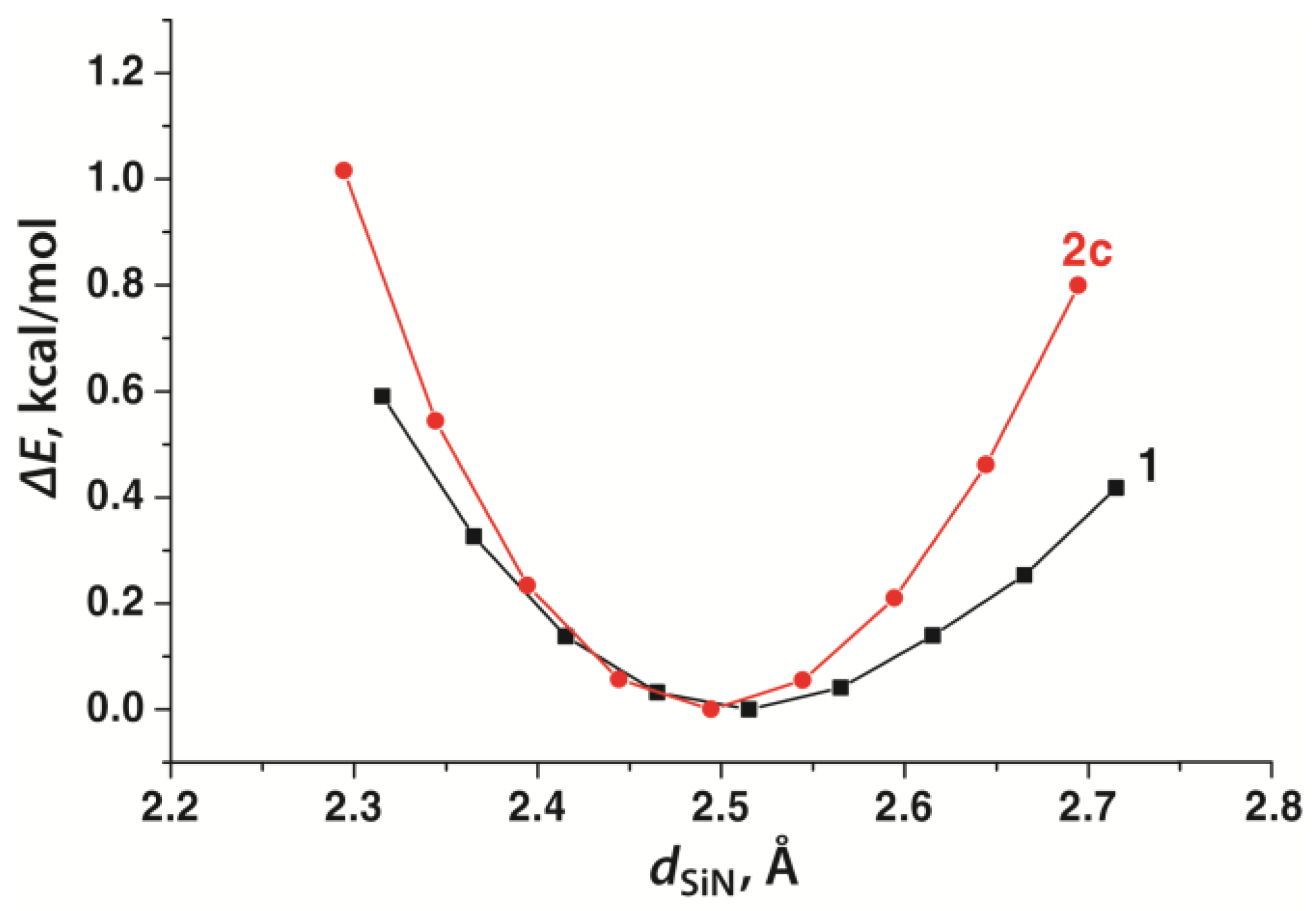
3.2. Geometry of Silatrane Cage in 2a–c
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verkade, J.G. Main group atranes: Chemical and structural features. Coord. Chem. Rev. 1994, 137, 233–295. [Google Scholar] [CrossRef]
- Pestunovich, V.; Kirpichenko, S.; Voronkov, M. The Chemistry of Organic Silicon Compounds; Rappoport, Z., Apeloig, Y., Eds.; Wiley: Chichester, UK, 1998; Volume 2, pp. 1447–1537. [Google Scholar]
- Voronkov, M.G.; Baryshok, V.P. Use of Silatranes for Medicine and Agriculture; Tolstikov, G.A., Ed.; Publishing House of the Siberian Branch of Russian Academy of Sciences: Novosibirsk, Russia, 2005; p. 258. [Google Scholar]
- D’yakov, V.M.; Voronkov, M.G.; Kazimirovskaya, V.B.; Loginov, S.V.; Rasulov, M.M. Organosilicon Chemistry VI: From Molecules to Materials; Auner, N., Weis, J., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2008; pp. 588–594. [Google Scholar]
- Puri, J.K.; Singh, R.; Chahal, V.K. Silatranes: A review on their synthesis, structure, reactivity and applications. Chem. Soc. Rev. 2011, 40, 1791–1840. [Google Scholar] [CrossRef] [PubMed]
- Sidorkin, V.F.; Belogolova, E.F.; Wang, Y.; Jouikov, V.; Doronina, E.P. Electrochemical Oxidation and Radical Cations of Structurally Non-rigid Hypervalent Silatranes: Theoretical and Experimental Studies. Chem. Eur. J. 2017, 8, 1910–1919. [Google Scholar] [CrossRef]
- Belogolova, E.F.; Sidorkin, V.F. Correlation among the Gas-Phase, Solution, and Solid-Phase Geometrical and NMR Parameters of Dative Bonds in the Pentacoordinate Silicon Compounds. 1-Substituted Silatranes. J. Phys. Chem. A 2013, 117/25, 5365–5376. [Google Scholar] [CrossRef] [PubMed]
- Belogolova, E.F.; Liu, G.; Doronina, E.P.; Ciborowski, S.M.; Sidorkin, V.F.; Bowen, K.H. “Outlaw” Dipole-Bound Anions of Intra-Molecular Complexes. J. Phys. Chem. Lett. 2018, 9, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Sidorkin, V.F.; Belogolova, E.F.; Doronina, E.P.; Liu, G.; Ciborowski, S.M.; Bowen, K.H. “Outlaw” Dipole-Bound Anions of Intra-Molecular Complexes. J. Am. Chem. Soc. 2020, 142, 2001–2011. [Google Scholar] [CrossRef]
- Belogolova, E.F.; Shlykov, S.A.; Eroshin, A.V.; Doronina, E.P.; Sidorkin, V.F. The hierarchy of ab initio and DFT methods for describing an intramolecular non-covalent Si···N contact in the silicon compounds using electron diffraction geometries. Phys. Chem. Chem. Phys. 2021, 23, 2762–2774. [Google Scholar] [CrossRef]
- Shen, Q.; Hilderbrandt, R.L. The structure of methyl silatrane (1-methyl-2,8,9-trioxa-5-aza-1-silabicyclo(3.3.3)undecane) as determined by gas phase electron diffraction. J. Mol. Struct. 1980, 64, 257–262. [Google Scholar] [CrossRef]
- Forgacs, G.; Kolonits, M.; Hargittai, I. The gas-phase molecular structure of 1-fluorosilatrane from electron diffraction. Struct. Chem. 1990, 1, 245–250. [Google Scholar] [CrossRef]
- Shishkov, I.F.; Khristenko, L.V.; Rudakov, F.M.; Golubinskii, A.V.; Vilkov, L.V.; Karlov, S.S.; Zaitseva, G.S.; Samdal, S. Molecular Structure of Silatrane Determined by Gas Electron Diffraction and Quantum-Mechanical Calculations. Struct. Chem. 2004, 15, 11–16. [Google Scholar] [CrossRef]
- Korlyukov, A.A.; Antipin, M.Y.; Bolgova, Y.I.; Trofimova, O.M.; Voronkov, M.G. Chemical bonding in the crystal structure of 1-hydrosilatrane. Russ. Chem. Bull. Int. Ed. 2009, 58, 25–30. [Google Scholar] [CrossRef]
- Boer, F.P.; Turley, J.W.; Flynn, J.J. Structural studies of pentacoordinate silicon. II. Phenyl(2′2′,2′′-nitrilotriphenoxy)silane. J. Am. Chem. Soc. 1968, 90, 5102–5105. [Google Scholar] [CrossRef]
- Chen, W.; Wu, G.L.; Luo, Y. Jiegou Huaxue (Chin.). Chin. J. Struct. Chem. 1987, 6, 165. [Google Scholar]
- Meshgi, M.A.; Zaitsev, K.V.; Vener, M.V.; Churakov, A.V.; Baumgartner, J.; Marschner, C. Hypercoordinated Oligosilanes Based on Aminotrisphenols. ACS Omega 2018, 3, 10317. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Section A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Section C 2015, 71, 3–8. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990; p. 458. [Google Scholar]
- Todd, A.; Keith, T.K. AIMAll (Version 19.02.13); Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Espinosa, E.; Lecomte, C.; Molins, E. Experimental electron density overlapping in hydrogen bonds: Topology vs. energetics. Chem. Phys. Lett. 1999, 300, 745–748. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E. Retrieving interaction potentials from the topology of the electron density distribution: The case of hydrogen bonds. J. Chem. Phys. 2000, 113, 5686–5694. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Rozas, I.; Elguero, J.; Molins, E. About the evaluation of the local kinetic, potential and total energy densities in closed-shell interactions. Chem. Phys. Lett. 2001, 336, 457–461. [Google Scholar] [CrossRef]
- Korlyukov, A.A. Coordination compounds of tetravalent silicon, germanium and tin: The structure, chemical bonding and intermolecular interactions in them. Russ. Chem. Rev. 2015, 84, 422–440. [Google Scholar] [CrossRef]
- Lyssenko, K.A.; Korlyukov, A.A.; Golovanov, D.G.; Ketkov, S.Y.; Antipin, M.Y. Estimation of the Barrier to Rotation of Benzene in the (η6-C6H6)2Cr Crystal via Topological Analysis of the Electron Density Distribution Function. J. Phys. Chem. A 2006, 110, 6545–6551. [Google Scholar] [CrossRef] [PubMed]
- Zhurova, E.A.; Stash, A.I.; Tsirelson, V.G.; Zhurov, V.V.; Bartashevich, E.V.; Potemkin, V.A.; Pinkerton, A.A. Atoms-in-Molecules Study of Intra- and Intermolecular Bonding in the Pentaerythritol Tetranitrate Crystal. J. Am. Chem. Soc. 2006, 128, 14728–14734. [Google Scholar] [CrossRef] [PubMed]
- Sidorkin, V.F.; Doronina, E.P.; Belogolova, E.F. A New Approach to the Design of Neutral 10-C-5 Trigonal-Bipyramidal Carbon Compounds: A “π-Electron Cap” Effect. Chem. Eur. J. 2013, 19, 10302–10311. [Google Scholar] [CrossRef]
- Sidorkin, V.F.; Doronina, E.P. Cage Silaphosphanes with a P→Si Dative Bond. Organometallics 2009, 28, 5305–5315. [Google Scholar] [CrossRef]
- Doronina, E.P.; Sidorkin, V.F.; Lazareva, N.F. (PO→Si) Chelates of Silylmethyl Derivatives of Phosphoric Acids R2P(O)ZCH2SiMe3−nHaln (n = 1−3; Z = O, NMe, CH2, S). Organometallics 2010, 29, 3327–3340. [Google Scholar] [CrossRef]
- Sidorkin, V.F.; Belogolova, E.F.; Doronina, E.P. Assignment of photoelectron spectra of silatranes: First ionization energies and the nature of the dative Si←N contact. Phys. Chem. Chem. Phys. 2015, 17, 26225–26237. [Google Scholar] [CrossRef]
- Doronina, E.P.; Sidorkin, V.F.; Belogolova, E.F.; Jouikov, V. Isomer-selective dative bond O→M (M = Si, Ge) for designing new photochromic hemi-indigo systems. J. Organomet. Chem. 2018, 858, 89–96. [Google Scholar] [CrossRef]
- Doronina, E.P.; Jouikov, V.V.; Sidorkin, V.F. Molecular Design of Silicon-Containing Diazenes: Absorbance of E and Z Isomers in the Near-Infrared Region. Chem. Eur. J. 2022, 28, e202201508. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Weinhold, F. NBO 5.G; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2004; Available online: https://nbo7.chem.wisc.edu/ (accessed on 20 March 2023).
- Granovsky, A.A. Firefly Version 8. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 20 March 2023).
- Tamao, K.; Hayashi, T.; Ito, Y.; Shiro, M. Pentacoordinate anionic bis(siliconates) containing a fluorine bridge between two silicon atoms. Synthesis, solid-state structures, and dynamic behavior in solution. Organometallics 1992, 11, 2099–2114. [Google Scholar] [CrossRef]
- Frye, C.L.; Vincent, G.A.; Hauschildt, G.L. Pentacoordinate Silicon Derivatives. III.1 2,2′2′-Nitrilotriphenol, a New Chelating Agent. J. Am. Chem. Soc. 1966, 88, 2727–2730. [Google Scholar] [CrossRef]
- Olsson, L.; Ottosson, C.-H.; Cremer, D. Properties of R3SiX Compounds and R3Si+ Ions: Do Silylium Ions Exist in Solution? J. Am. Chem. Soc. 1995, 117, 7460–7479. [Google Scholar] [CrossRef]
- Ottosson, C.-H.; Cremer, D. Intramolecularly Stabilized Phenylsilyl and Anthrylsilyl Cations. Organometallics 1996, 15, 5309–5320. [Google Scholar] [CrossRef]
- Turley, J.W.; Boer, F.P. Structural Studies of Pentacoordinate Silicon. I. Phenyl-(2,2′,2′′-nitrilotriethoxy)silane. J. Am. Chem. Soc. 1968, 90, 4026–4030. [Google Scholar] [CrossRef]
- Parkanyi, L.; Simon, K.; Nagy, J. Crystal and Molecular Structure of β-l-Phenylsilatrane, C12H17O3NSi. Acta Cryst. 1974, 30, 2328–2332. [Google Scholar] [CrossRef]
- Parkanyi, L.; Nagy, J.; Simon, K. Crystal and Molecular Structure of γ−l-Phenylsilatrane, Some Structural Features of Silatranes. J. Organomet. Chem. 1975, 101, 11–18. [Google Scholar] [CrossRef]
- Pestunovich, V.; Lukevics, E.; Pudova, O.; Sturkovich, R. Molecular Structure of Organosilicon Compounds; John Wiley: Chichester, NY, USA, 1989; p. 359. [Google Scholar]
- Greenberg, A.; Wu, G. Structural Relationships in Silatrane Molecules. Struct. Chem. 1990, 1, 79–85. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 2a | 2b | 2c |
|---|---|---|---|
| Empirical formula | C24H17NO3Si | C27H23NO3Si | C24H14F3NO3Si |
| Formula weight, Mr | 395.47 | 437.55 | 449.45 |
| Temperature (K) | 296(2) | 150(2) | 150(2) |
| Diffractometer | D8 VENTURE Bruker AXS | APEXII Bruker AXS | APEXII Bruker AXS |
| Crystal size (mm3) | 0.80 × 0.36 × 0.27 | 0.33 × 0.24 × 0.10 | 0.58 × 0.42 × 0.09 |
| Crystal system | orthorhombic | monoclinic | monoclinic |
| Space group | Cmc21 | P21/c | P21/n |
| a (Å) | 11.127(6) | 10.086(1) | 9.571(2) |
| b (Å) | 14.714(4) | 12.433(1) | 12.308(2) |
| c (Å) | 11.892(3) | 19.107(2) | 17.430(2) |
| β (°) | 90 | 104.594(6) | 91.533(5) |
| Unit cell volume (Å3) | 1946.9(13) | 2318.7(4) | 2052.5(5) |
| Molecular multiplicity | 4 | 4 | 4 |
| Absorption coefficient (mm−1) | 0.147 | 0.130 | 0.169 |
| F(000) | 824 | 920 | 920 |
| Calculated density (g/cm3) | 1.349 | 1.253 | 1.454 |
| 2θmax (°) | 55.0 | 51.0 | 55.0 |
| Reflections collected | 6266 | 15,749 | 11,432 |
| Independent reflections with I > 2σ(I) | 1901 | 2351 | 3348 |
| Number of refined parameters | 137 | 292 | 289 |
| R-factor | 0.1105 | 0.0636 | 0.0417 |
| CCDC number | 2247806 | 2247803 | 2247804 |
| Parameter | 2a | 2b | 2c | |||
|---|---|---|---|---|---|---|
| exp. | calc. | exp. | calc. | exp. | calc. | |
| Si1–O2 | 1.654(8) | 1.681 | 1.657(2) | 1.684 | 1.655(2) | 1.683 |
| Si1–C12 | 1.863(11) | 1.861 | 1.850(3) | 1.863 | 1.852(3) | 1.857 |
| O2–C3 | 1.342(14) | 1.353 | 1.370(4) | 1.353 | 1.369(4) | 1.349 |
| C3–C4 | 1.400(15) | 1.399 | 1.387(4) | 1.398 | 1.392(4) | 1.400 |
| N5–C4 | 1.443(13) | 1.438 | 1.446(4) | 1.440 | 1.444(4) | 1.437 |
| O2–Si1–O8 | 117.1(3) | 115.0 | 116.0(1) | 115.4 | 116.2(1) | 114.7 |
| O2–Si1–C12 | 100.3(4) | 102.1 | 98.9(1) | 102.2 | 102.5(1) | 103.2 |
| Si1–O2–C3 | 126.3(7) | 126.4 | 125.7(2) | 126.1 | 125.9(2) | 126.8 |
| O2–C3–C4 | 118.2(11) | 119.7 | 118.1(3) | 119.6 | 119.2(3) | 120.0 |
| C3–C4–N5 | 113.3(10) | 114.7 | 113.9(3) | 114.8 | 114.4(3) | 115.0 |
| C4–N5–C6 | 116.5(7) | 117.1 | 114.7(2) | 117.3 | 116.8(2) | 117.4 |
| N5–Si1–C12 | 179.8(4) | 179.6 | 178.2(1) | 179.6 | 178.0(1) | 179.5 |
| Si1–N5 | 2.329(9) | 2.463 | 2.358(3) | 2.451 | 2.411(3) | 2.494 |
| ΔN | 0.300(9) | 0.357 | 0.292(4) | 0.247 | 0.267(4) | 0.230 |
| ΔSi | 0.284(7) | 0.243 | 0.286(3) | 0.346 | 0.318(3) | 0.372 |
| ηe | 72.4 | 58.4 | 72.2 | 60.9 | 65.8 | 54.9 |
| Σ(O-Si-O) | 351.4(3) | 346.9 | 351.3(1) | 347.7 | 349.3(1) | 345.8 |
| Σ(C-N-C) | 347.4(7) | 351.6 | 348.2(2) | 351.3 | 350.0(2) | 352.5 |
| Compound | dSiN | ηe | ρ(r) | ∇2ρ(r) | E(rc) | ESiN | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|
| 1 | gas | 2.515 | 54 | 0.235 | 0.861 | −0.07 | 8.9 | ||
| crystal | α | 2.193 | 86 | 0.370 | 1.984 | −0.17 | 22.6 | [43] | |
| β | 2.156 | 87 | 0.391 | 2.554 | −0.18 | 25.1 | [44] | ||
| γ | 2.132 | 88 | 0.404 | 2.908 | −0.18 | 26.7 | [45] | ||
| 2a | gas | 2.463 | 58 | 0.256 | 0.737 | −0.08 | 10.2 | ||
| crystal | 2.344 | 72 | 0.299 | 0.866 | −0.13 | 14.6 | [15] | ||
| 2.329 | 72 | 0.311 | 0.823 | −0.14 | 15.5 | this work | |||
| 2b | gas | 2.451 | 61 | 0.261 | 0.701 | −0.09 | 10.5 | ||
| crystal | 2.358 | 72 | 0.296 | 0.745 | −0.12 | 13.9 | this work | ||
| 2c | gas | 2.494 | 55 | 0.244 | 0.786 | −0.07 | 9.4 | ||
| crystal | 2.411 | 66 | 0.274 | 0.749 | −0.10 | 11.9 | this work | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanovs, V.; Belyakov, S.; Doronina, E.; Sidorkin, V.; Roisnel, T.; Jouikov, V. Crystal Structure of New 1-Phenyl-Substituted Tribenzsilatranes. Crystals 2023, 13, 772. https://doi.org/10.3390/cryst13050772
Romanovs V, Belyakov S, Doronina E, Sidorkin V, Roisnel T, Jouikov V. Crystal Structure of New 1-Phenyl-Substituted Tribenzsilatranes. Crystals. 2023; 13(5):772. https://doi.org/10.3390/cryst13050772
Chicago/Turabian StyleRomanovs, Vitalijs, Sergey Belyakov, Evgeniya Doronina, Valery Sidorkin, Thierry Roisnel, and Viatcheslav Jouikov. 2023. "Crystal Structure of New 1-Phenyl-Substituted Tribenzsilatranes" Crystals 13, no. 5: 772. https://doi.org/10.3390/cryst13050772