
A New Anti-Alias Model of Ab Initio Calculations of the Generalized Stacking Fault Energy in Face-Centered Cubic Crystals

Abstract
:1. Introduction
2. Computational Approach
3. Results and Discussion

{kind=link}
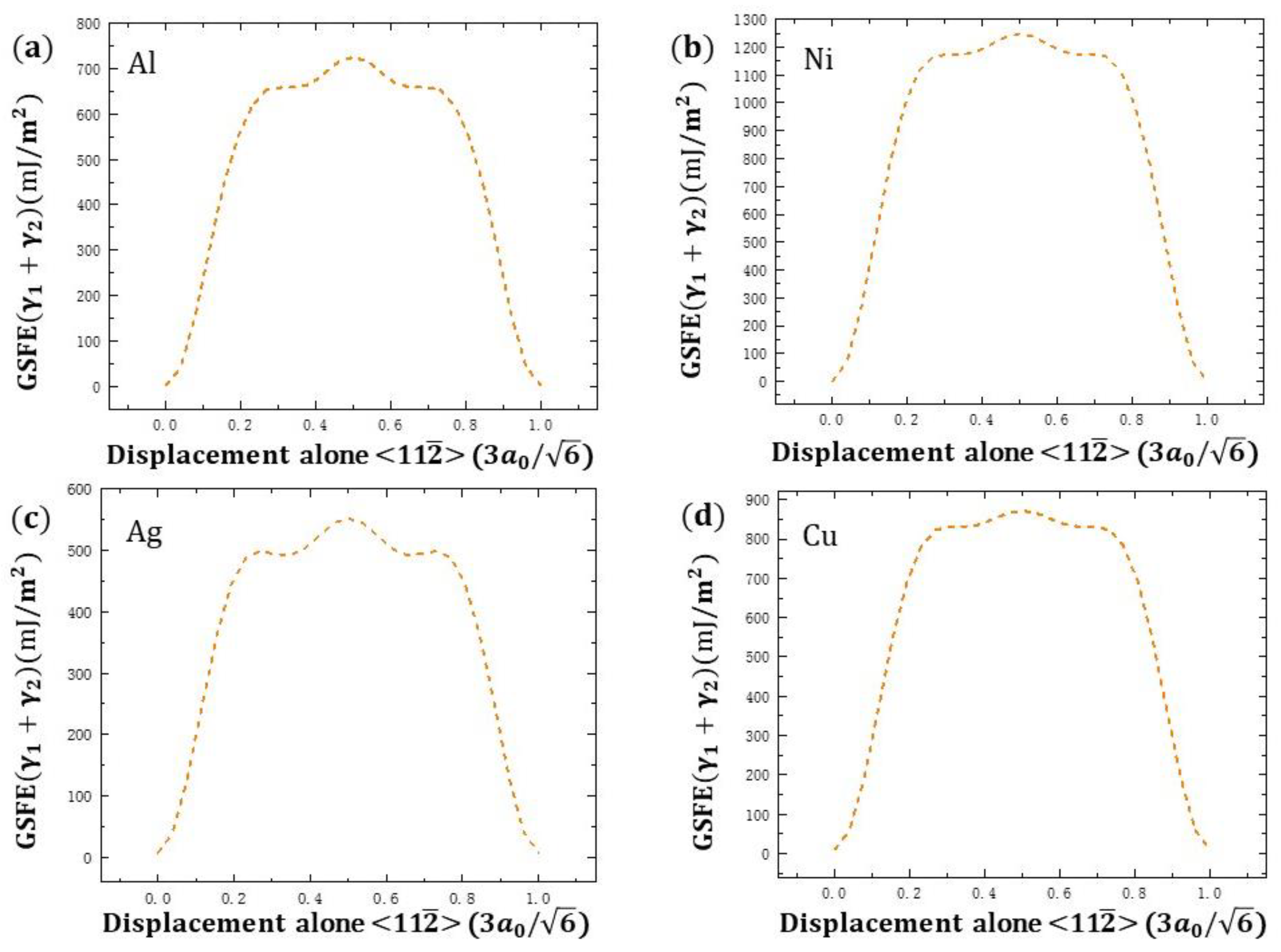{kind=link}
{kind=link}
| Model | |||
|---|---|---|---|
| Al | 133 | 189 | 0.7 |
| 130 [41], 146 [42], 158 [43] | 162 [41], 178 [42], 175 [43] | ||
| Ni | 129 | 280 | 0.46 |
| 110 [44], 120–130 [45], 132 [33] | 305 [33], 278 [42], 273 [41] | ||
| Ag | 26 | 119 | 0.21 |
| 17 [37], 25.9 [4], 18 [41] | 93 [24], 111 [37], 190 [46] | ||
| Cu | 42 | 171 | 0.24 |
| 43 [37], 51 [47], 40.5 [9] | 158 [31], 161 [9], 175 [37] |
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cheng, Z.; Zhou, H.F.; Lu, Q.H.; Gao, H.J.; Lu, L. Extra strengthening and work hardening in gradient nanotwinned metals. Science 2018, 362, 559. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Wang, L.; Fan, D.; Bie, B.X.; Zhou, X.M.; Suo, T.; Li, Y.L.; Chen, M.W.; Liu, C.L.; Qi, M.L.; et al. Macrode formation Twins in Single-Crystal Aluminum. Phys. Rev. Lett. 2016, 116, 075501. [Google Scholar] [CrossRef] [Green Version]
- Edalati, K.; Akama, D.; Nishio, A.; Lee, S.; Yonenaga, Y.; Cubero-Sesin, J.M.; Horita, Z.J. Influence of dislocation–solute atom interactions and stacking fault energy on grain size of single-phase alloys after severe plastic deformation using high-pressure torsio. Acta Mater. 2014, 69, 68–77. [Google Scholar] [CrossRef]
- Cai, T.; Zhang, Z.J.; Zhang, P.; Yang, J.B.; Zhang, Z.F. Competition between slip and twinning in face-centered cubic metals. J. Appl. Phys. 2014, 116, 163512. [Google Scholar] [CrossRef]
- Chowdhury, P.B.; Sehitoglu, H.; Rateick, R.G. Predicting fatigue resistance of nano-twinned materials: Part I—Role of cyclic slip irreversibility and Peierls stress. Int. J. Fatigue 2014, 68, 277–291. [Google Scholar] [CrossRef]
- Meyers, M.A.; Benson, D.J.; Vöhringer, O.; Kad, B.K.; Xue, Q.; Fu, H.H. Constitutive description of dynamic deformation: Physically-based mechanisms. Mater. Sci. Eng. A 2002, 322, 194–216. [Google Scholar] [CrossRef]
- Nishikawa, O.; Walko, R.J. Field ion microscopical observation of twinning in iridium induced by a mechanical contact. Acta Metall. 1971, 19, 1163–1168. [Google Scholar] [CrossRef]
- Vítek, V. Intrinsic stacking faults in body-centred cubic crystals. Philos. Mag. 1968, 18, 773–786. [Google Scholar] [CrossRef]
- Zhao, D.D.; Løvvik, O.M.; Marthinsen, K.; Li, Y.J. Impurity effect of Mg on the generalized planar fault energy of Al. J. Mater. Sci. 2016, 51, 6552–6568. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.P.; Xiao, H.; Ma, L.; Fan, T.W.; Tang, P.Y. Effects of rare-earth elements on twinning deformation of Al alloys from first-principles calculations. Solid State Commun. 2022, 356, 114946. [Google Scholar] [CrossRef]
- Liu, L.H.; Chen, J.H.; Fan, T.W.; Shang, S.L.; Shao, Q.Q.; Yuan, D.W.; Dai, Y. The stability of deformation twins in aluminum enhanced by alloying elements. J. Mater. Sci. Technol. 2019, 35, 2625–2629. [Google Scholar] [CrossRef]
- Schönecker, S.; Li, W.; Vitos, L.; Li, X.Q. Effect of strain on generalized stacking fault energies and plastic deformation modes in fcc-hcp polymorphic high-entropy alloys: A first-principles investigation. Phys. Rev. Mater. 2021, 5, 075004. [Google Scholar] [CrossRef]
- Janković, B.; Joksimović, T.; Stare, J.; Losev, E.; Zemtsova, V.; Srčič, S.; Boldyrev, E. Quantification and modeling of nanomechanical properties of chlorpropamide α, β, and γ conformational polymorphs. Eur. J. Pharm. Sci. 2017, 110, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Arkhipov, S.G.; Sherin, P.S.; Kiryutin, A.S.; Lazarenko, V.A.; Tantardini, C. The Role of S-bond in Tenoxicam Keto-Enolic Tautomerization. CrystEngComm 2019, 36, 5392–5401. [Google Scholar] [CrossRef]
- Arkhipov, S.G.; Rychkov, D.A.; Pugachevc, A.M.; Boldyrevaa, E.V. New hydrophobic L-amino acid salts: Maleates of L-leucine, L-isoleucine and L-norvaline. Acta Crystallogr. C Struct. Chem. 2015, 71, 584–592. [Google Scholar] [CrossRef]
- Asanbaeva, N.B.; Rychkov, D.A.; Tyapkin, P.Y.; Arkhipov, S.G.; Uvarov, N.F. The unique structure of [(C4H9)4N]3[Pb(NO3)5]—One step forward in understanding transport properties in tetra-n-butylammonium-based solid electrolytes. Struct. Chem. 2021, 32, 1261–1267. [Google Scholar] [CrossRef]
- Desta, I.T.; Chizhik, S.A.; Sidelnikov, A.A.; Karothu, D.P.; Boldyreva, E.V.; Naumov, P. Mechanically Responsive Crystals: Analysis of Macroscopic Strain Reveals “Hidden” Processes. J. Phys. Chem. A 2020, 124, 300–310. [Google Scholar] [CrossRef]
- Tantardini, C.; Arkhipov, S.G.; Cherkashina, K.A.; Kil’met’eva, A.S.; Boldyreva, E.A. Crystal structure of a 2:1 co-crystal of meloxicam with acetylendicarboxylic acid. Acta Crystallogr. 2016, 72, 1856–1859. [Google Scholar] [CrossRef]
- Tantardini, C.; Arkipov, S.G.; Cherkashina, K.A.; Kil’met’ev, A.S.; Boldyreva, E.V. Synthesis and crystal structure of a meloxicam co-crystal with benzoic acid. Struct. Chem. 2018, 29, 1867–1874. [Google Scholar] [CrossRef]
- Vaid, A.; Wei, D.; Bitzek, E.; Nasiri, S.; Zaiser, M. Pinning of extended dislocations in atomically disordered crystals. Acta Mater. 2022, 236, 118095. [Google Scholar] [CrossRef]
- Shih, M.; Miao, J.S.; Mills, M.; Ghazisaeidi, M. Stacking fault energy in concentrated alloys. Nat. Commun. 2021, 12, 3590. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Zheng, G.P.; Jiao, Z.B. Alloying effects on phase stability, mechanical properties, and deformation behavior of CoCrNi-based medium-entropy alloys at low temperatures. Intermetallics 2022, 140, 107399. [Google Scholar] [CrossRef]
- Van Swygenhoven, H.; Derlet, P.M.; Froseth, A.G. Stacking fault energies and slip in nanocrystalline metals. Nat. Mater. 2004, 3, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Tadmor, E.B.; Bernstein, N. A First-Principles Measure for the Twinnability of FCC Metals. J. Mech. Phys. Solids 2004, 52, 2507–2519. [Google Scholar] [CrossRef]
- Achmad, T.L.; Fu, W.X.; Chen, H.; Zhang, C.; Yang, Z.G. First-principles calculations of generalized-stacking-fault-energy of Co-based alloys. Comput. Mater. Sci. 2016, 121, 86–96. [Google Scholar] [CrossRef]
- Liu, L.L.; Wang, R.; Wu, X.Z.; Gan, L.Y.; Wei, Q.Y. Temperature effects on the generalized planar fault energies and twinnabilities of Al, Ni and Cu: First principles calculations. Comput. Mater. Sci. 2014, 88, 124–130. [Google Scholar] [CrossRef]
- Asaro, R.J.; Suresh, S. Mechanistic models for the activation volume and rate sensitivity in metals with nanocrystalline grains and nano-scale twins. Acta Mater. 2005, 53, 3369–3382. [Google Scholar] [CrossRef]
- Aitken, Z.H.; Sorkin, V.; Yu, Z.G.; Chen, S.; Wu, Z.X.; Zhang, Y.W. Modified embedded-atom method potentials for the plasticity and fracture behaviors of unary fcc metals. Phys. Rev. B 2021, 103, 094116. [Google Scholar] [CrossRef]
- Xu, L.B.; Casillas-Trujillo, L.; Gao, Y.F.; Xu, H.X. Compositional effects on stacking fault energies in Ni-based alloys using first-principles and atomistic simulations. Comput. Mater. Sci. 2021, 197, 110618. [Google Scholar] [CrossRef]
- Chang, L.; Liu, X.R.; Zhao, J.L.; Zhou, C.Y. Effect of interatomic potential on modelling fracture behavior in hcp titanium: A molecular dynamics study. J. Mater. Res. Technol. 2022, 17, 2118–2133. [Google Scholar] [CrossRef]
- Qing, D.; Luo, Z.; Zhu, H.; Wang, L.Y.; Ying, T.; Jin, Z.H.; Li, D.J.; Ding, W.J.; Zeng, X.Q. Basal-plane stacking-fault energies of Mg alloys: A first-principles study of metallic alloying effects. J. Mater. Sci. Technol. 2018, 34, 1773–1780. [Google Scholar]
- Zhao, S.J.; Malcolm Stocks, G.; Zhang, Y.W. Stacking fault energies of face-centered cubic concentrated solid solution alloys. Acta Mater. 2017, 134, 334–345. [Google Scholar] [CrossRef]
- Shang, S.L.; Wang, W.Y.; Zhou, B.C.; Wang, Y.; Darling, K.A.; Kecskes, L.J.; Mathaudhu, S.N.; Liu, Z.K. Generalized stacking fault energy, ideal strength and twinnability of dilute Mg-based alloys: A first-principles study of shear deformation. Acta Mater. 2014, 67, 168–180. [Google Scholar] [CrossRef]
- Liu, L.H.; He, M.D.; Li, J.B.; Peng, X.F.; Chen, H.; Wang, Z.P.; Fan, T.W. A novel simplified model for measuring the twinnability of face-centred-cubic metals. Philos. Mag. Lett. 2020, 100, 63–73. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Arshad Javid, M.; Khan, Z.U.; Mehmood, Z.; Nabi, A.; Hussain, F.; Imran, M.; Nadeem, N.; Anjum, N. Structural, electronic and optical properties of LiNbO3 using GGA-PBE and TB-mBJ functionals: A DFT study. Int. J. Mod. Phys. B 2018, 32, 1850168. [Google Scholar] [CrossRef]
- Wu, X.Z.; Wang, R.; Wang, S.F.; Wei, Q.Y. Ab initio calculations of generalized-stacking-fault energy surfaces and surface energies for FCC metals. Appl. Surf. Sci. 2010, 256, 6345–6349. [Google Scholar] [CrossRef]
- Kibey, S.; Liu, J.B.; Curtis, M.J.; Johnson, D.D.; Sehitoglu, H. Effect of nitrogen on generalized stacking fault energy and stacking fault widths in high nitrogen steels. Acta Mater. 2006, 54, 2991–3001. [Google Scholar] [CrossRef]
- Rice, J.R. Dislocation Nucleation from a Crack Tip: An Analysis Based on the Peierls Concept. J. Mech. Phys. Solids 1992, 40, 239–271. [Google Scholar] [CrossRef]
- Schiøtz, J.; Jacobsen, K.W. A Maximum in the Strength of Nanocrystalline Copper. Science 2003, 301, 1357. [Google Scholar] [CrossRef] [Green Version]
- Kibey, S.; Liu, J.B.; Johnson, D.D.; Sehitoglu, H. Predicting twinning stress in fcc metals: Linking twin-energy pathways to twin nucleation. Acta Mater. 2007, 55, 6843–6851. [Google Scholar] [CrossRef]
- Brandl, C.; Derlet, P.M.; Van Swygenhoven, H. General-stacking-fault energies in highly strained metallic environments: Ab initio calculations. Phys. Rev. B 2007, 76, 054124. [Google Scholar] [CrossRef] [Green Version]
- Ogata, s.; Li, j.; Yip, S. Ideal pure shear strength of aluminum and copper. Science 2002, 298, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Donald, J. Siegel. Generalized stacking fault energies, ductilities, and twinnabilities of Ni and selected Ni alloys. Appl. Phys. Lett. 2005, 87, 121901. [Google Scholar]
- Carter, C.B.; Holmes, S. The stacking-fault energy of nickel. Philos. Mag. 1977, 35, 1161. [Google Scholar] [CrossRef]
- Kioussis, N.; Herbranson, M.; Collins, E.; Eberhart, M.E. Topology of Electronic Charge Density and Energetics of Planar Faults in fcc Metals. Phys. Rev. Lett. 2002, 88, 125501. [Google Scholar] [CrossRef] [Green Version]
- Hartford, J.; von Sydow, B.; Wahnström, G.; Lundqvist, B.I. Peierls barriers and stresses for edge dislocations in Pd and Al calculated from first principles. Phys. Rev. B 1998, 58, 2487. [Google Scholar] [CrossRef]



| Al | 246.0 | −63.0 | −17.7 | −60.6 |
| Ni | 479.8 | −130.3 | −26.1 | 177.0 |
| Ag | 208.7 | −53.8 | −14.5 | 85.1 |
| Cu | 329.2 | −91.2 | −16.8 | 144.5 |
| Sliding Displacement | (mJ/m2) | (mJ/m2) | (mJ/m2) | (mJ/m2) |
|---|---|---|---|---|
| 1/26 | 20 | 19 | 39 | 41 |
| 2/26 | 82 | 67 | 149 | 151 |
| 3/26 | 174 | 125 | 299 | 300 |
| 4/26 | 277 | 170 | 447 | 444 |
| 5/26 | 372 | 189 | 561 | 550 |
| 6/26 | 448 | 183 | 630 | 620 |
| 7/26 | 498 | 159 | 657 | 654 |
| 8/26 | 523 | 137 | 660 | 659 |
| 9/26 | 526 | 133 | 659 | 659 |
| 10/26 | 511 | 158 | 669 | 667 |
| 11/26 | 477 | 210 | 687 | 690 |
| 12/26 | 425 | 281 | 705 | 716 |
| 13/26 | 357 | 357 | 713 | 726 |
| 14/26 | 281 | 425 | 705 | 716 |
| 15/26 | 210 | 477 | 687 | 690 |
| 16/26 | 158 | 511 | 669 | 667 |
| 17/26 | 133 | 526 | 659 | 659 |
| 18/26 | 137 | 523 | 660 | 659 |
| 19/26 | 159 | 498 | 657 | 654 |
| 20/26 | 183 | 448 | 630 | 620 |
| 21/26 | 189 | 372 | 561 | 550 |
| 22/26 | 170 | 277 | 447 | 444 |
| 23/26 | 125 | 174 | 299 | 300 |
| 24/26 | 67 | 82 | 149 | 151 |
| 25/26 | 19 | 20 | 39 | 41 |
| 1 | 0 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, D.; Zhang, Q.; Fan, T.; He, M.; Liu, L. A New Anti-Alias Model of Ab Initio Calculations of the Generalized Stacking Fault Energy in Face-Centered Cubic Crystals. Crystals 2023, 13, 461. https://doi.org/10.3390/cryst13030461
Fan D, Zhang Q, Fan T, He M, Liu L. A New Anti-Alias Model of Ab Initio Calculations of the Generalized Stacking Fault Energy in Face-Centered Cubic Crystals. Crystals. 2023; 13(3):461. https://doi.org/10.3390/cryst13030461
Chicago/Turabian StyleFan, Dawei, Qingzhou Zhang, Touwen Fan, Mengdong He, and Linghong Liu. 2023. "A New Anti-Alias Model of Ab Initio Calculations of the Generalized Stacking Fault Energy in Face-Centered Cubic Crystals" Crystals 13, no. 3: 461. https://doi.org/10.3390/cryst13030461