
Investigation of Solid-State Hydrate-Anhydrous Phase Transformations of Dabigatran Etexilate Hemi-Edisylate

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Production of DBE through Reaction Crystallization
2.3. Single Crystal X-ray Diffraction (SXD)
2.4. Differential Scanning Calorimetry (DSC)
2.5. Thermogravimetric Analysis (TGA)
2.6. Powder X-ray Diffraction (PXRD)
2.7. Solution-State Nuclear Magnetic Resonance Spectroscopy (Solution-State NMR)
2.8. Solid-State Nuclear Magnetic Resonance Spectroscopy (Solid-STATE CP/MAS 13C-NMR)
2.9. Hot-Stage Microscopy (HSM)
2.10. Hygroscopic Test
3. Results and Discussion
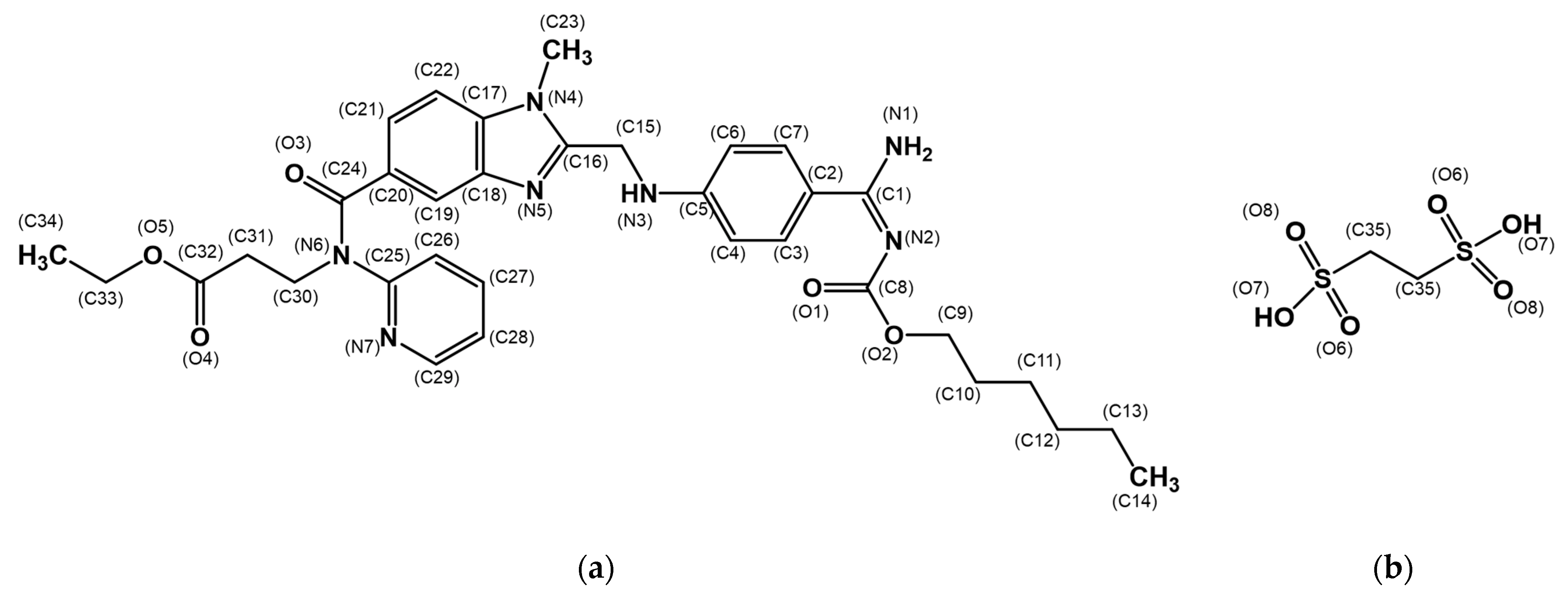
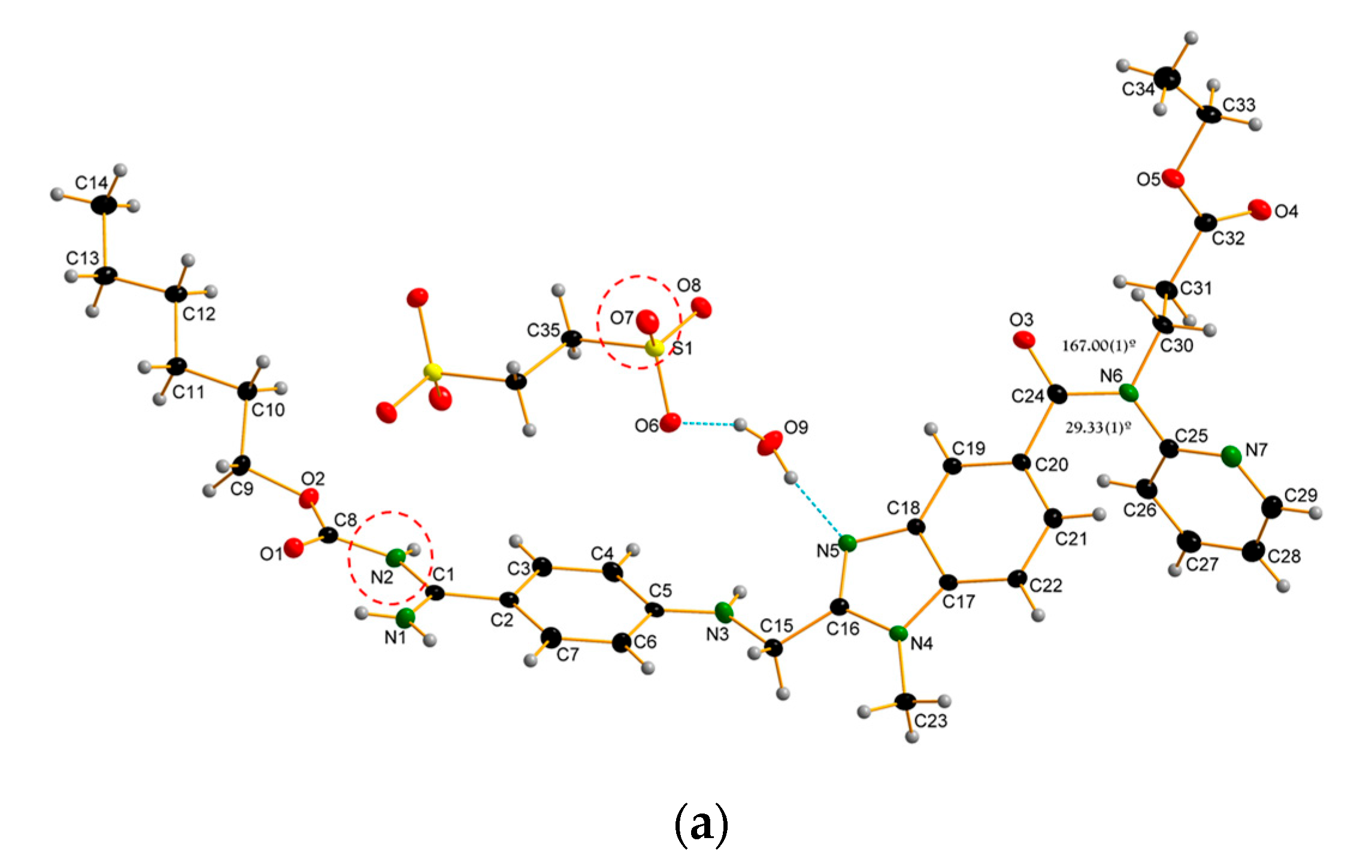
3.1. Crystal Structure of the Dabigatran Etexilate Hemi-Edisylate (DBE) Monohydrate (I)
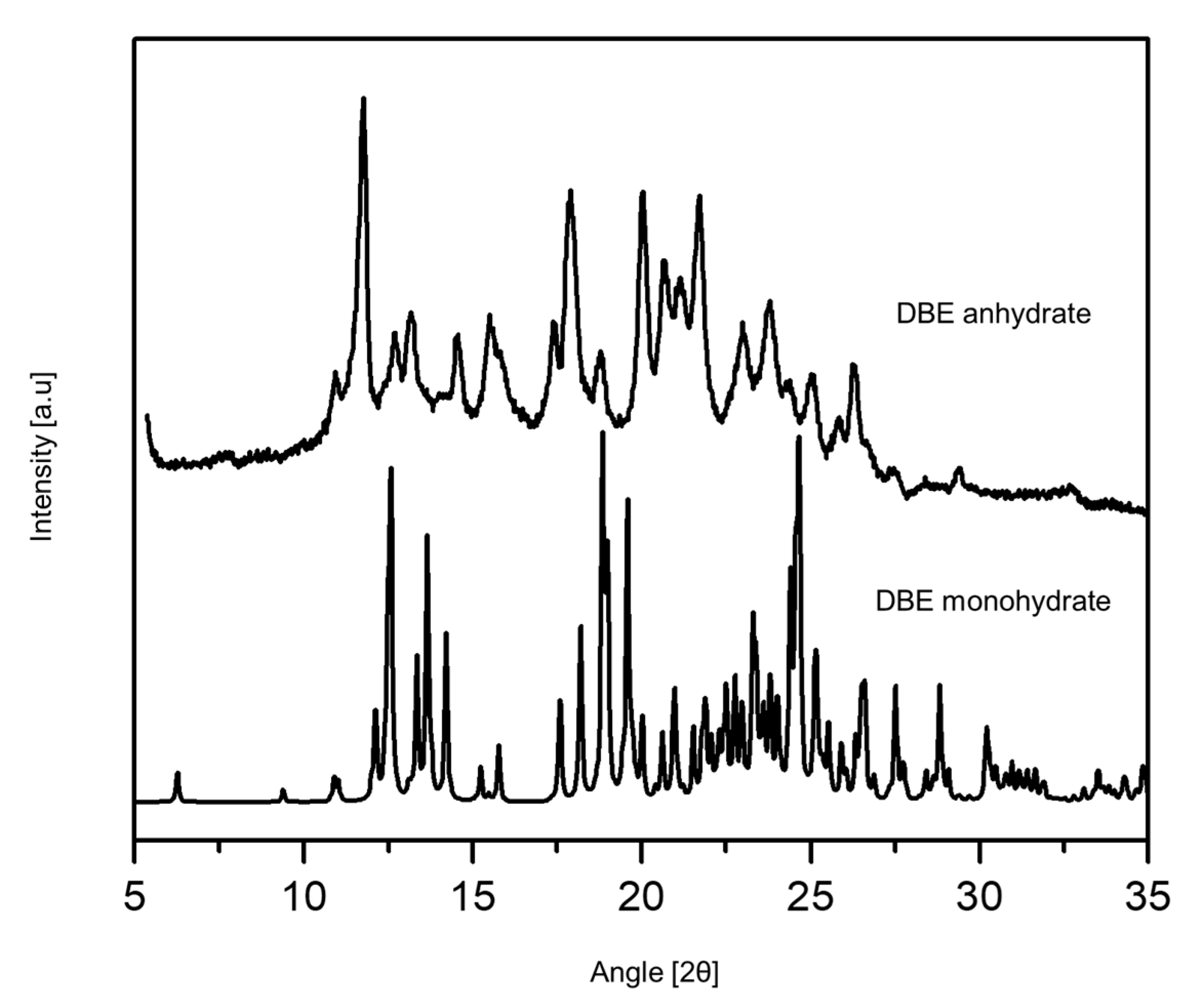
3.2. Characterization of DBE Crystal Forms
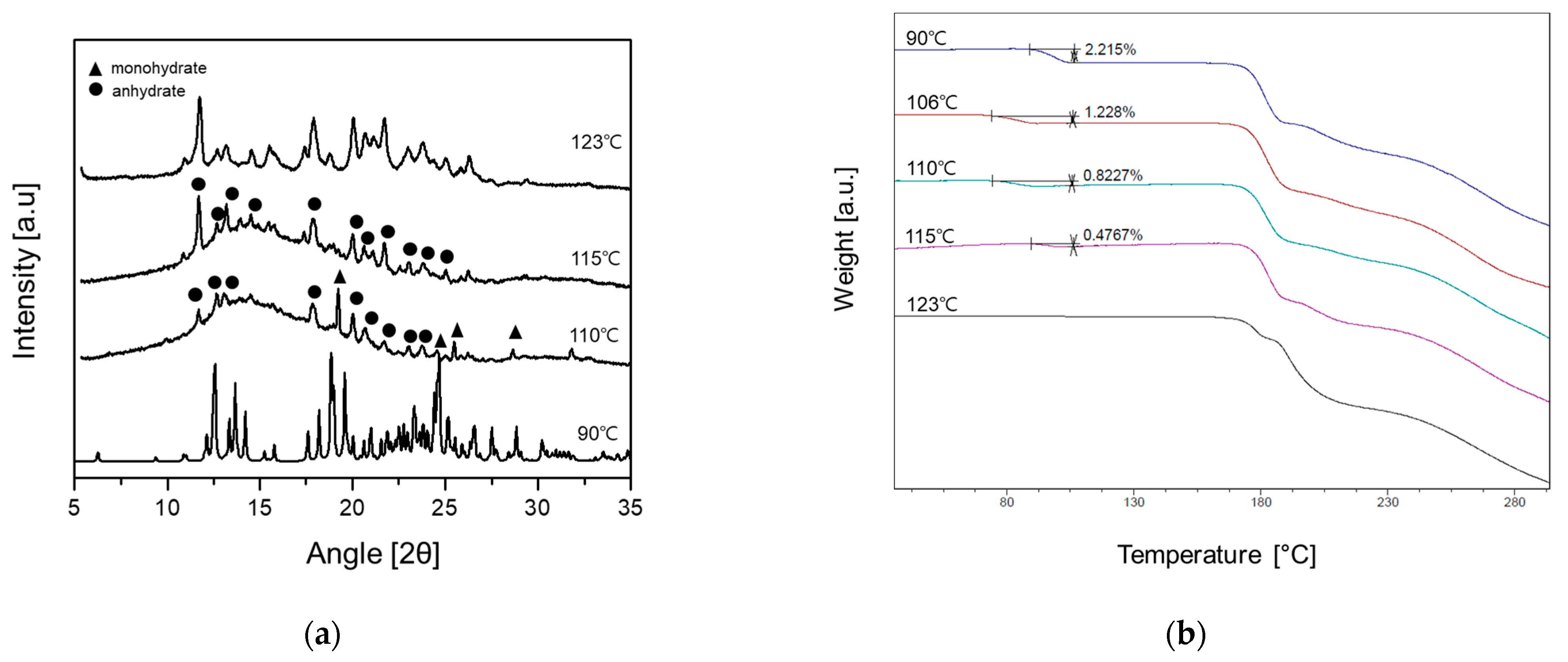
3.3. Solid State Phase Transformation of DBE Crystal Forms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hilfiker, R. Polymorphism in the Pharmaceutical Industry; Wiley-VCH: Weinheim, Germany, 2006; Chapter 1; pp. 1–19. [Google Scholar]
- Hu, S.; Mishra, M.K.; Sun, C.C. Twistable Pharmaceutical Crystal Exhibiting Exceptional Plasticity and Tabletability. Chem. Mater. 2019, 31, 3818–3822. [Google Scholar] [CrossRef]
- Bolla, G.; Sarma, B.; Namgia, A.K. Crystal Engineering of Pharmaceutical Cocrystals in the Discovery and Development of Improved Drugs. Chem. Rev. 2022, 13, 11514–11603. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Unruh, D.K.; Hutchins, K.M. Cocrystallization of Trimethoprim and Solubility Enhancement via Salt Formation. Cryst. Growth Des. 2021, 21, 1507–1517. [Google Scholar] [CrossRef]
- Bharate, S.S. Recrnt developments in pharmaceutical aslts: FDA approvals from 2015 to 2019. Drug Discov. Today. 2020, 26, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Gunnam, A.; Nangia, A.K. High-Solubility Salts of the Multiple Sclerosis Drug Teriflunomide. Cryst. Growth Des. 2019, 19, 5407–5417. [Google Scholar] [CrossRef]
- Chemburkar, S.R.; Bauer, J.; Deming, K.; Spiwek, H.; Patel, K.; Morris, J.; Henry, R.; Spanton, S.; Dziki, W.; Porter, W.; et al. Dealing with the Impact of Ritonavir Polymorphs on the Late Stages of Bulk Drug Process Development. Org. Process Res. Dev. 2000, 4, 413–417. [Google Scholar] [CrossRef]
- Rietveld, I.B.; Céolin, R. Rotigotine: Unexpected Polymorphism with Predictable Overall Monotropic Behavior. J. Pharm. Sci. 2015, 104, 4117–4122. [Google Scholar] [CrossRef]
- Santos, O.M.M.; Reis, M.E.D.; Jacon, J.T.; Lino, M.E.S.; Simões, J.S.; Doriguetto, A.C. Polymorphism: An evaluation of the potential risk to the quality of drug products from the Farmácia Popular Rede Própria. Braz. J. Pharm. Sci. 2014, 50, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Waters, C. The Development of the Rotigotine Transdermal Patch A Historical Perspective. Neurol. Clin. 2013, 31, S37–S50. [Google Scholar] [CrossRef] [Green Version]
- An, J.-H.; Jin, F.; Kim, H.S.; Ryu, H.C.; Kim, J.S.; Kim, H.M.; Kim, K.H.; Kiyonga, A.N.; Jung, K. Investigation of the Polymorphic Transformation of the Active Pharmaceutical Ingredient Clopidogrel Bisulfate Using the Ionic Liquid AEImBF4. Cryst. Growth Des. 2016, 16, 1829–1836. [Google Scholar] [CrossRef]
- An, J.-H.; Jin, F.; Kim, H.S.; Ryu, H.C.; Kim, J.S.; Kim, H.M.; Kiyonga, A.N.; Min, D.S.; Youn, W.; Kim, K.H.; et al. Application of ionic liquid to polymorphic transformation of antiviral/HIV drug adefovir dipivoxil. Arch. Pharm. Res. 2016, 39, 646–659. [Google Scholar] [CrossRef]
- An, J.-H.; Choi, G.J.; Kim, W.S. Polymorphic and kinetic investigation of adefovir dipivoxil during phase transformation. Int. J. Pharm. 2012, 422, 185–193. [Google Scholar] [CrossRef]
- Davey, R.; Garside, J. From Molecules to Crystallizers an Introduction to Crystallization; Oxford University Press: Oxford, NY, USA, 2000; Chapter 6; pp. 48–52. [Google Scholar]
- Yada, S.; Ohya, M.; Ohuchi, Y.; Hamaura, T.; Wakiyama, N.; Usui, F.; Kusai, A.; Yamamoto, K. Solid phase transition of CS-891 enantiotropes during grinding. Int. J. Pharm. 2003, 255, 69–79. [Google Scholar] [CrossRef]
- Mura, P.; Cirri, M.; Faucci, M.; Ginès-Dorado, J.; Bettinetti, G. Investigation of the effects of grinding and co-grinding on physicochemical properties of glisentide. J. Pharm. Biomed. Anal. 2002, 30, 227–237. [Google Scholar] [CrossRef]
- Miyamae, A.; Kema, H.; Kawabata, T.; Yasuda, T.; Otsuka, M.; Matsuda, Y. X-ray powder diffraction study on the grinding effect of the polymorphs of a novel and orally effective uricosuric agent: FR76505. Drug Dev. Ind. Pharm. 1994, 20, 2881–2897. [Google Scholar] [CrossRef]
- Otsuka, M.; Otsuka, K.; Kaneniwa, N. Relation Between Polymorphic Transformation Pathway During Grinding and the Physicochemical Properties of Bulk Powders for Pharmaceutical Preparations. Drug Dev. Ind. Pharm. 1994, 20, 1649–1660. [Google Scholar] [CrossRef]
- Bartolomei, M.; Bertocchi, P.; Ramusino, M.C.; Santucci, N.; Valvo, L. Physico-chemical characterisation of the modifications I and II of (R,S) propranolol hydrochloride: Solubility and dissolution studies. J. Pharm. Biomed. Anal. 1999, 21, 299–309. [Google Scholar] [CrossRef]
- Chieng, N.; Rades, T.; Aaltonen, J. An overview of recent studies on the analysis of pharmaceutical polymorphs. J. Pharm. Biomed. Anal. 2011, 55, 618–644. [Google Scholar] [CrossRef]
- Puigjaner, C.; Barbas, R.; Portell, A.; Font-Bardia, M.; Alcobé, X.; Prohens, R. Revisiting the Solid State of Norfloxacin. Cryst. Growth Des. 2010, 10, 2948–2953. [Google Scholar] [CrossRef]
- Maher, A.; Seaton, C.C.; Hudson, S.; Croker, D.M.; Rasmuson, Å.C.; Hodnett, B.K. Investigation of the Solid-State Polymorphic Transformations of Piracetam. Cryst. Growth Des. 2012, 12, 6223–6233. [Google Scholar] [CrossRef] [Green Version]
- Chernyshev, V.V.; Yatsenko, A.V.; Pirogov, S.V.; Nikulenkova, T.F.; Tumanova, E.V.; Lonin, I.S.; Paseshnichenko, K.A.; Mironov, A.V.; Velikodny, Y.A. Two Anhydrous and a Trihydrate Form of Tilorone Dihydrochloride: Hydrogen-Bonding Patterns and Reversible Hydration/Dehydration Solid-State Transformation. Cryst. Growth Des. 2012, 12, 6118–6125. [Google Scholar] [CrossRef]
- Teychene, S.; Autret, J.; Biscans, B.J. Determination of solubility profiles of eflucimibe polymorphs: Experimental and modeling. Pharm. Sci. 2006, 95, 871–882. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, M.A.; Maher, A.; Croker, D.M.; Rasmuson, Å.C.; Hodnett, B.K. Examining Solution and Solid State Composition for the Solution-Mediated Polymorphic Transformation of Carbamazepine and Piracetam. Cryst. Growth Des. 2012, 12, 1925–1932. [Google Scholar] [CrossRef] [Green Version]
- Bannigan, P.; Zeglinski, J.; Lusi, M.; O’Brien, J.; Hudson, S.P. Investigation into the Solid and Solution Properties of Known and Novel Polymorphs of the Antimicrobial Molecule Clofazimine. Cryst. Growth Des. 2016, 16, 7240–7250. [Google Scholar] [CrossRef]
- Nyström, M.; Roine, J.; Murtomaa, M.; Sankaran, R.M.; Santos, H.A.; Salonen, J. Solid state transformations in consequence of electrospraying—A novel polymorphic form of piroxicam. Eur. J. Pharm. Biopharm. 2015, 89, 182–189. [Google Scholar] [CrossRef]
- Pallipurath, A.R.; Skelton, J.M.; Warren, M.R.; Kamali, N.; McArdle, P.; Erxleben, A. Sulfamerazine: Understanding the Influence of Slip Planes in the Polymorphic Phase Transformation through X-Ray Crystallographic Studies and ab Initio Lattice Dynamics. Mol. Pharm. 2015, 12, 3735–3748. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Wang, J.-R.; Mei, X. Insight into the Phase Transformation among Various Solid Forms of Baicalein. Cryst. Growth Des. 2015, 15, 4959–4968. [Google Scholar] [CrossRef]
- Hauel, N.; Priepke, H.; Ries, U.; Stassen, J.M.; Wienen, W. Disubstituted Bicyclic Heterocycles, the Preparations and the Use Thereof as Pharmaceutical Compositions. U.S. Patent 6087380, 11 July 2000. [Google Scholar]
- Schmid, R.; Sieger, P.; Sobotta, R. 3-[(2-{[4-(hexyloxycarbonylaminoiminomethyl) phenylamino] methyl}-1-methyl-1h-benzimidazol-5-carbonyl) pyridin-2-ylamino] Propionic Acid Ethylester Methansulfonate and Its Use as a Medicament. U.S. Patent 7932273 B2, 26 April 2011. [Google Scholar]
- An, J.-H.; Ko, M.J. The Polymorph of Novel Dabigatran Etexilate Hemi-Salt and the Manufcturung Method Thereof. K.R. Patent 102147600 B1, 25 August 2020. [Google Scholar]
- Liu, H.-Q.; Zhang, W.-G.; Cai, Z.-Q.; Xu, W.-R.; Shen, X.-P. Dabigatran etexilate tetrahydrate. Acta Crystallogr. Sect. E. Struct. Rep. Online 2012, E68, o3385. [Google Scholar] [CrossRef]
- Nagadeep, J.; Kamaraj, P.; Arthanareeswari, M. Gradient RP-HPLC method for the determination of potential impurities in dabigatran etexilate in bulk drug and capsule formulations. Arab. J. Chem. 2019, 12, 3431–3443. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.K. Applications of solid-state NMR to pharmaceutical polymorphism and related matters. J. Pharm. Pharmacol. 2007, 59, 225–239. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | DBE Hydrate Crystal |
|---|---|
| Chemical formula sum | C35 H46 N7 O9 S |
| Formula weight, amu | 740.85 |
| Crystal system | Triclinic |
| Space group | P |
| a, Å | 7.7793 (4) |
| b, Å | 8.7050 (5) |
| c, Å | 28.3967 (15) |
| α (°) | 84.172 (2) |
| β (°) | 88.831 (1) |
| γ (°) | 69.599 (2) |
| Cell volume (Å3) | 1792.83 (17) |
| Z | 2 |
| Temperature, K | 150 (1) |
| Radiation | Graphite monochromated MoKα (λ = 0.71073 Å) |
| Linear absorption coefficient, mm−1 | 0.155 |
| Crystal size, mm3 | 0.50 × 0.38 × 0.08 |
| Scan type | Ω |
| θ limits, deg. | 3.0° < θ < 26.00° |
| No. unique data | 7025 |
| No. unique data with I > 2σ(I) | 5228 |
| wR2 (all data) | 0.1328 |
| R (on for I > 2σ(I)) | 0.0492 |
| Goodness-of-fit on F2 | 1.079 |
| Min. and Max. residual electron density (e/Å3) | −0.746 and 0.333 |
| Type | Donor–H…Acceptor | D–H (Å) | H…A (Å) | D…A (Å) | D–H…A (°) |
|---|---|---|---|---|---|
| Inter | DN(1)–H(1B) … EO(6)i | 0.86 | 2.02 | 2.8299(2) | 156 |
| DN(2)–H(2) … WO(9)iii | 0.86 | 1.95 | 2.7670(2) | 158 | |
| DN(3)–H(3A) … EO(7)v | 0.86 | 2.18 | 2.9450(2) | 148 | |
| WO(9)–H(9C) … DN(5)ii | 0.87(3) | 2.00(3) | 2.8552(2) | 168(3) | |
| WO(9)–H(9D) … EO(6) | 0.94(6) | 2.04(5) | 2.9339(2) | 159(4) | |
| Intra | DN(1)–H(1A) … DO(1) | 0.86 | 2.03 | 2.6643(2) | 130 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, J.; Lim, C.; Nam, S.; Yoon, W.; Yun, H.; Kim, W.-S.; An, J.-H. Investigation of Solid-State Hydrate-Anhydrous Phase Transformations of Dabigatran Etexilate Hemi-Edisylate. Crystals 2023, 13, 424. https://doi.org/10.3390/cryst13030424
Feng J, Lim C, Nam S, Yoon W, Yun H, Kim W-S, An J-H. Investigation of Solid-State Hydrate-Anhydrous Phase Transformations of Dabigatran Etexilate Hemi-Edisylate. Crystals. 2023; 13(3):424. https://doi.org/10.3390/cryst13030424
Chicago/Turabian StyleFeng, Jin, Changjin Lim, Sihyun Nam, Woojin Yoon, Hoseop Yun, Woo-Sik Kim, and Ji-Hun An. 2023. "Investigation of Solid-State Hydrate-Anhydrous Phase Transformations of Dabigatran Etexilate Hemi-Edisylate" Crystals 13, no. 3: 424. https://doi.org/10.3390/cryst13030424