
Centrosymmetric Nickel(II) Complexes Derived from Bis-(Dithiocarbamato)piperazine with 1,1′-Bis-(Diphenylphosphino)ferrocene and 1,2-Bis-(Diphenylphosphino)ethane as Ancillary Ligands: Syntheses, Crystal Structure and Computational Studies
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
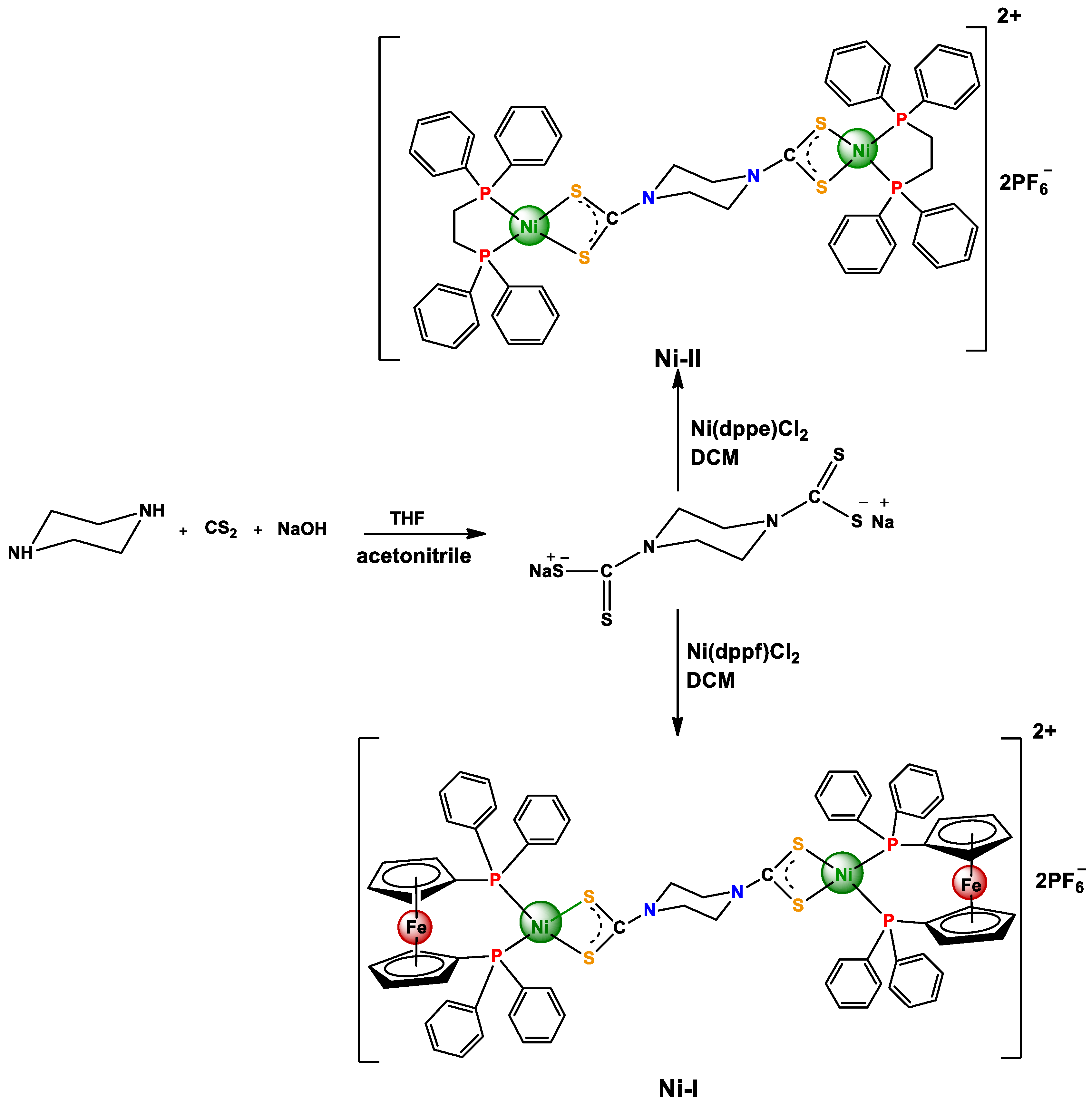
2.1. Synthesis
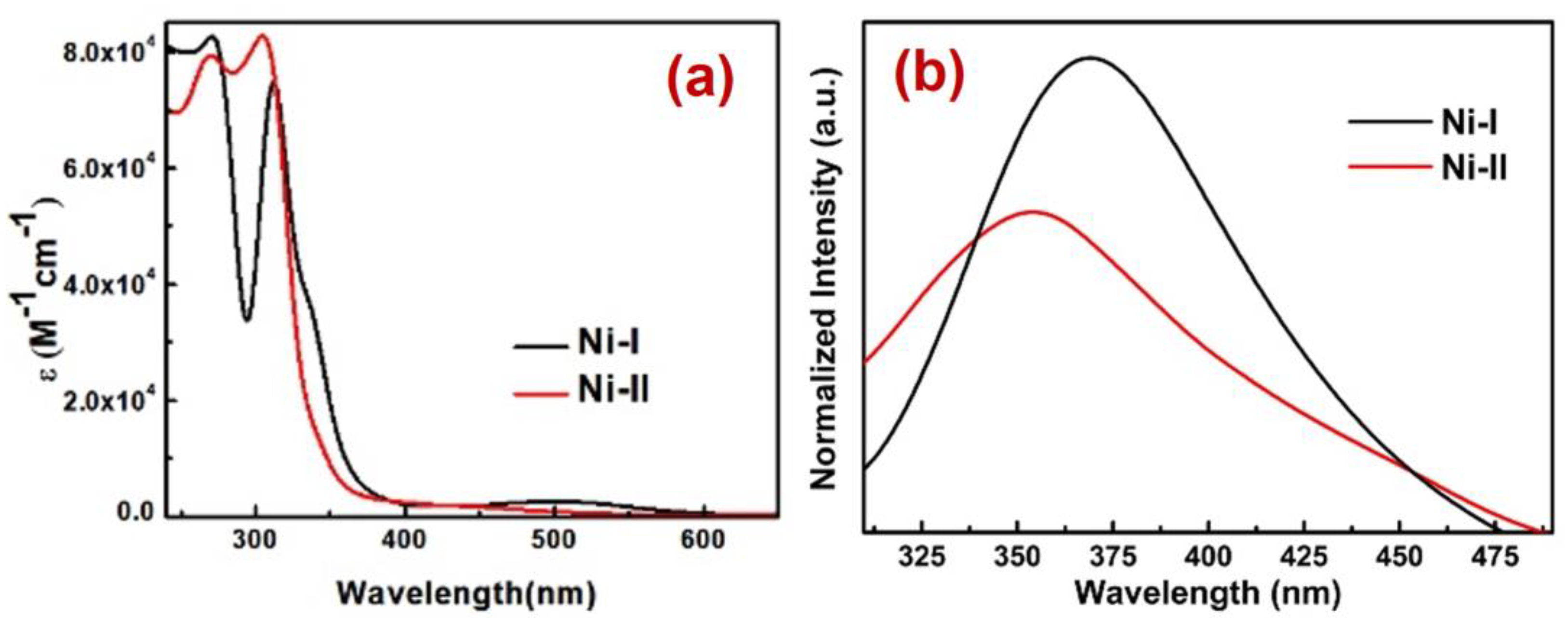
2.2. Spectroscopy
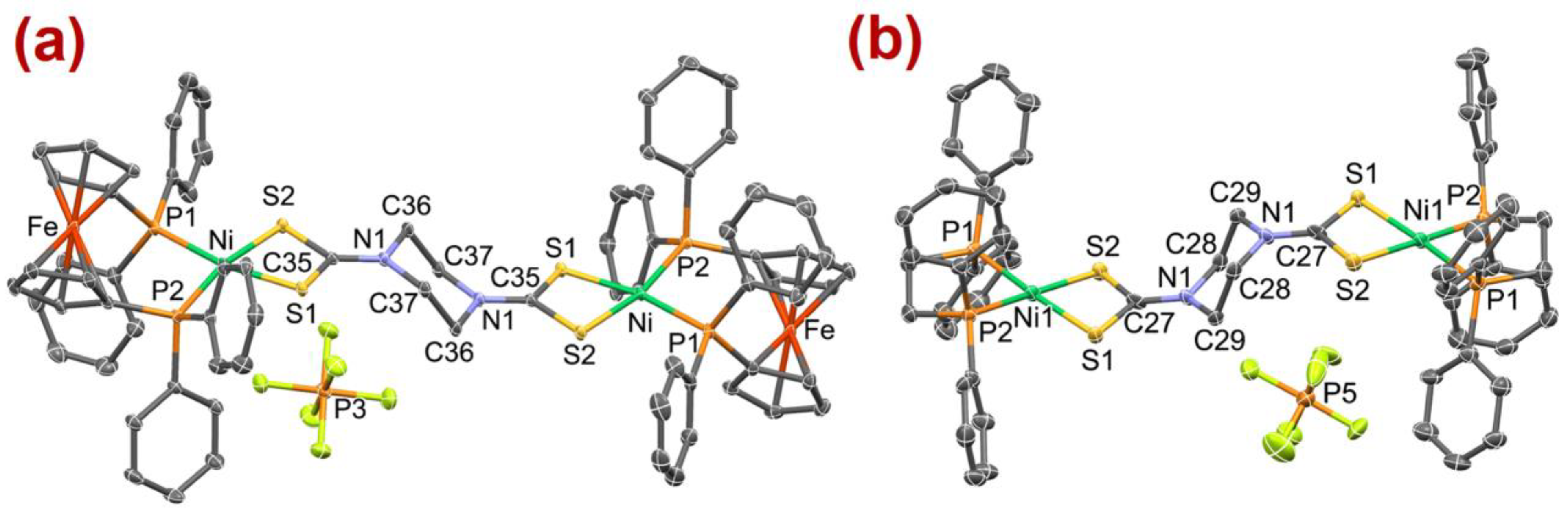
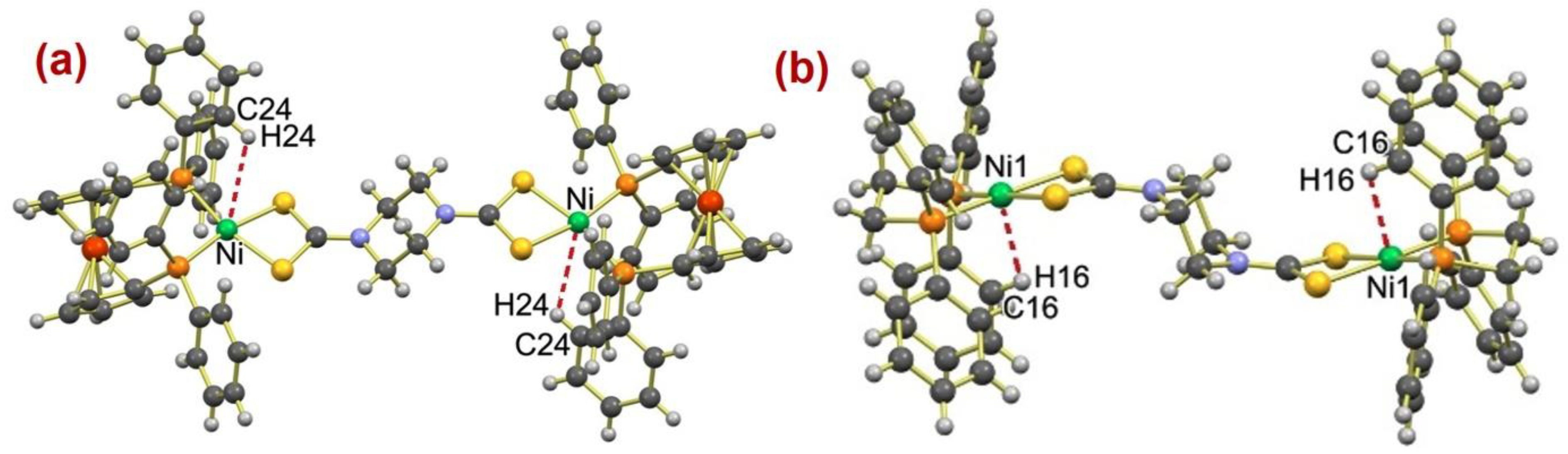
2.3. Molecular Structure Description
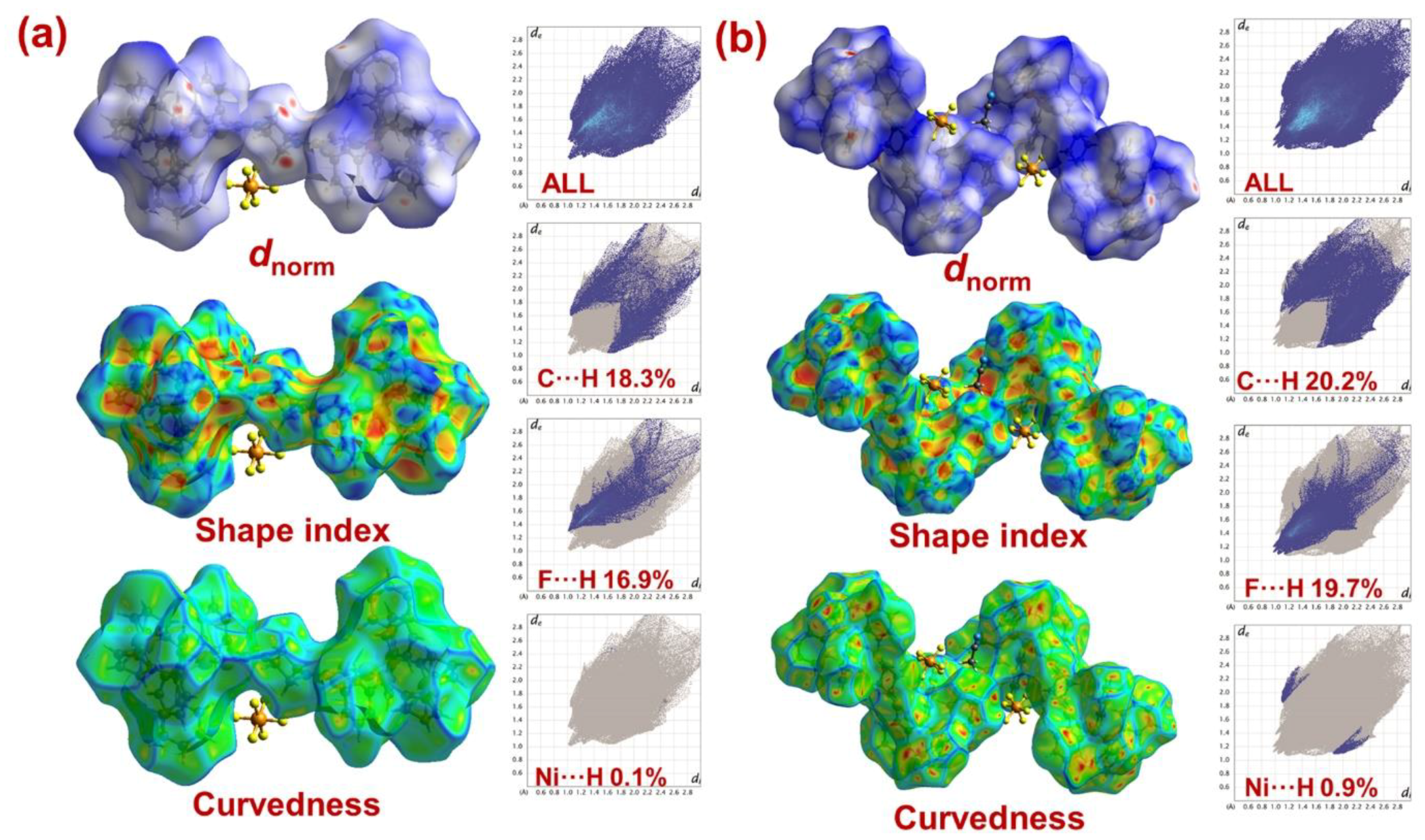
2.4. Hirshfeld Surface Analyses
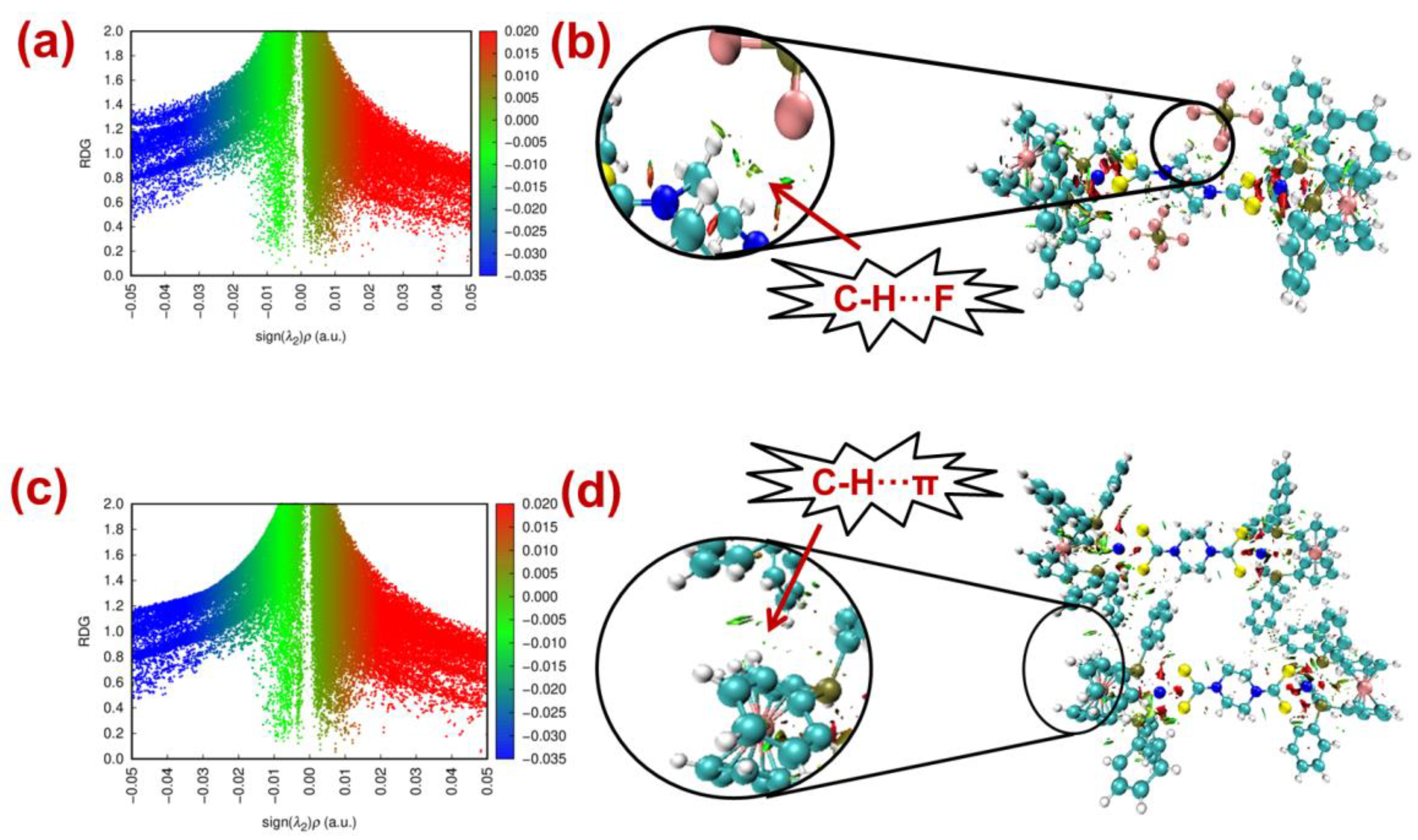
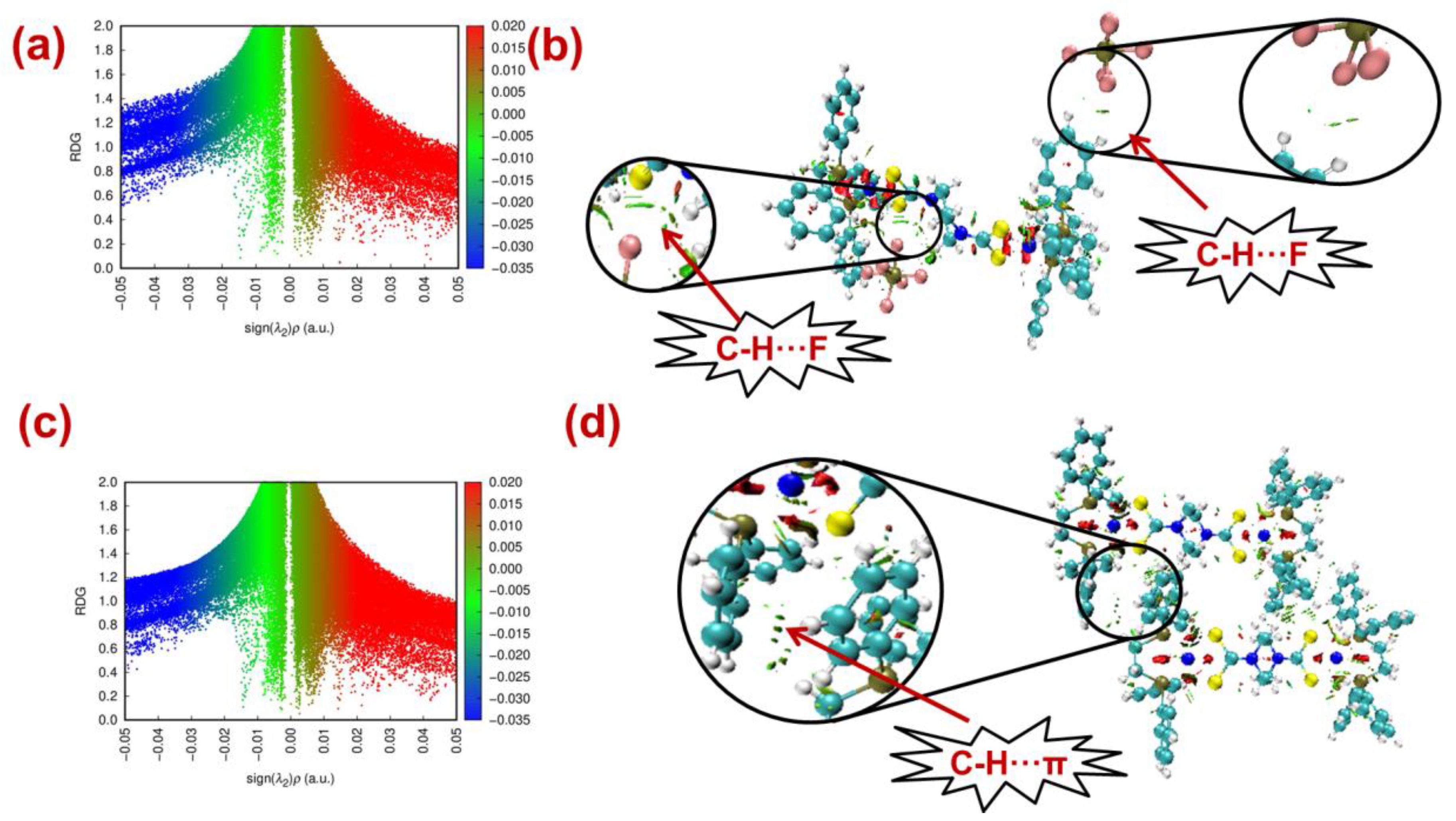
2.5. NCI-RDG
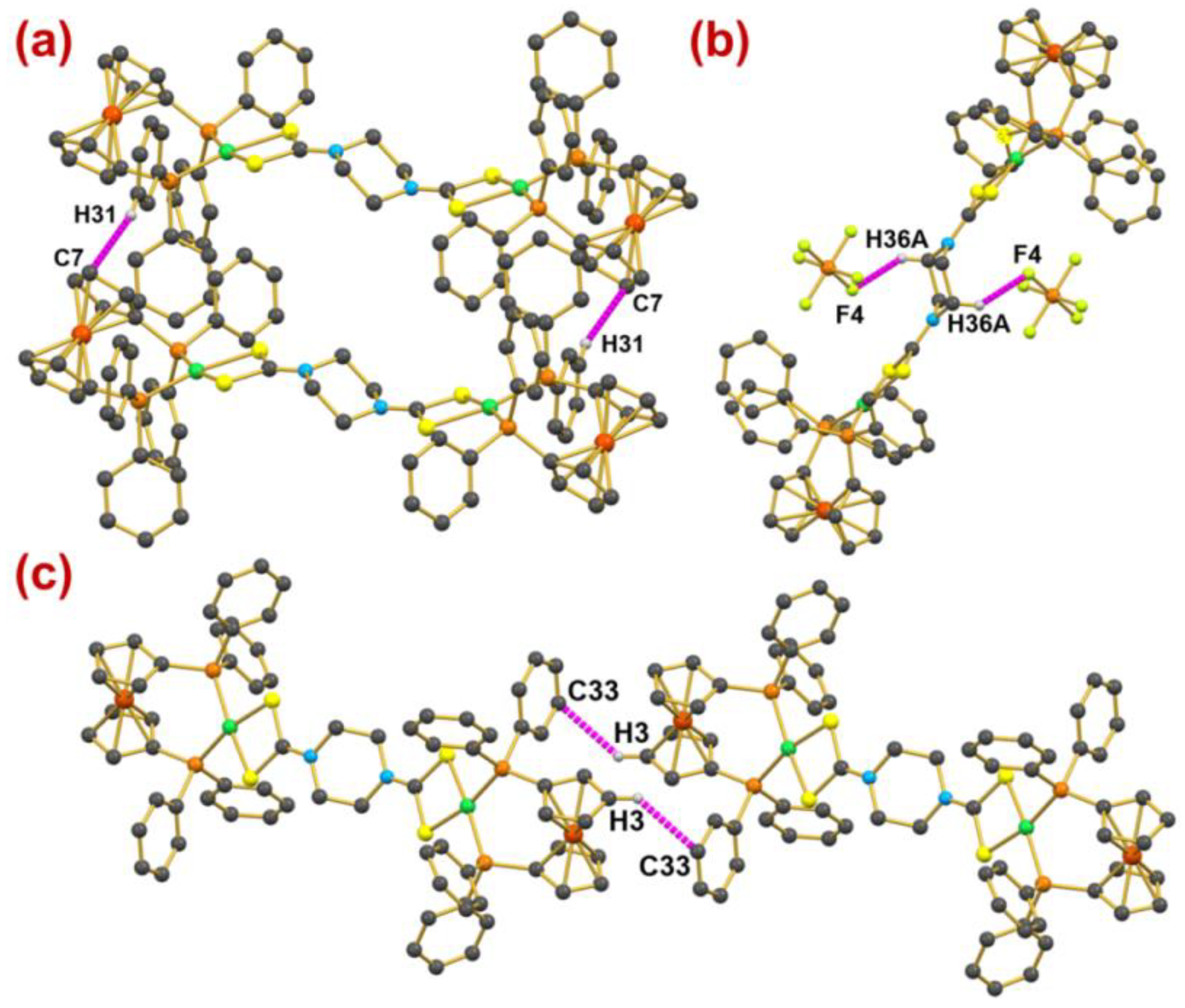
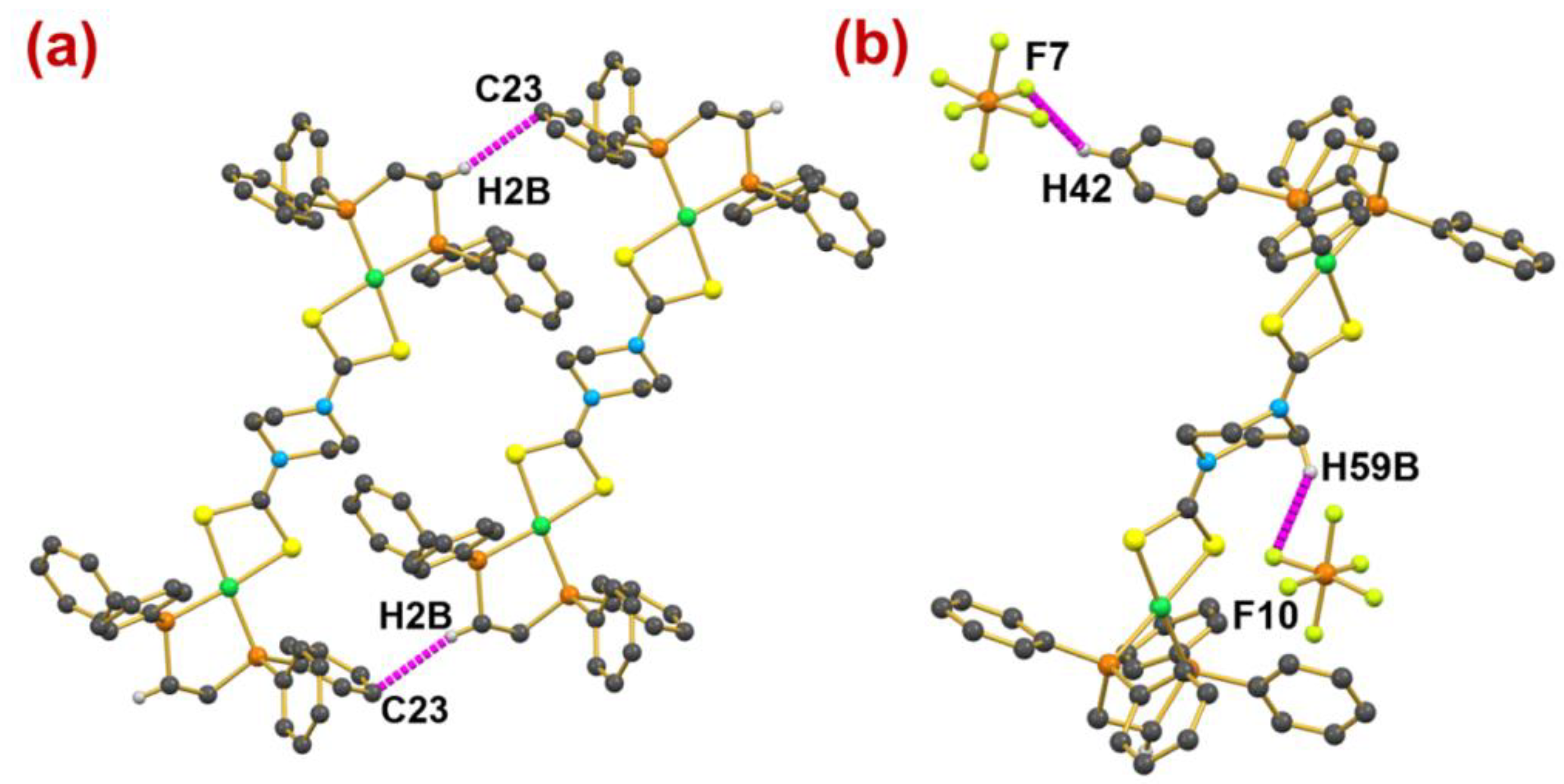
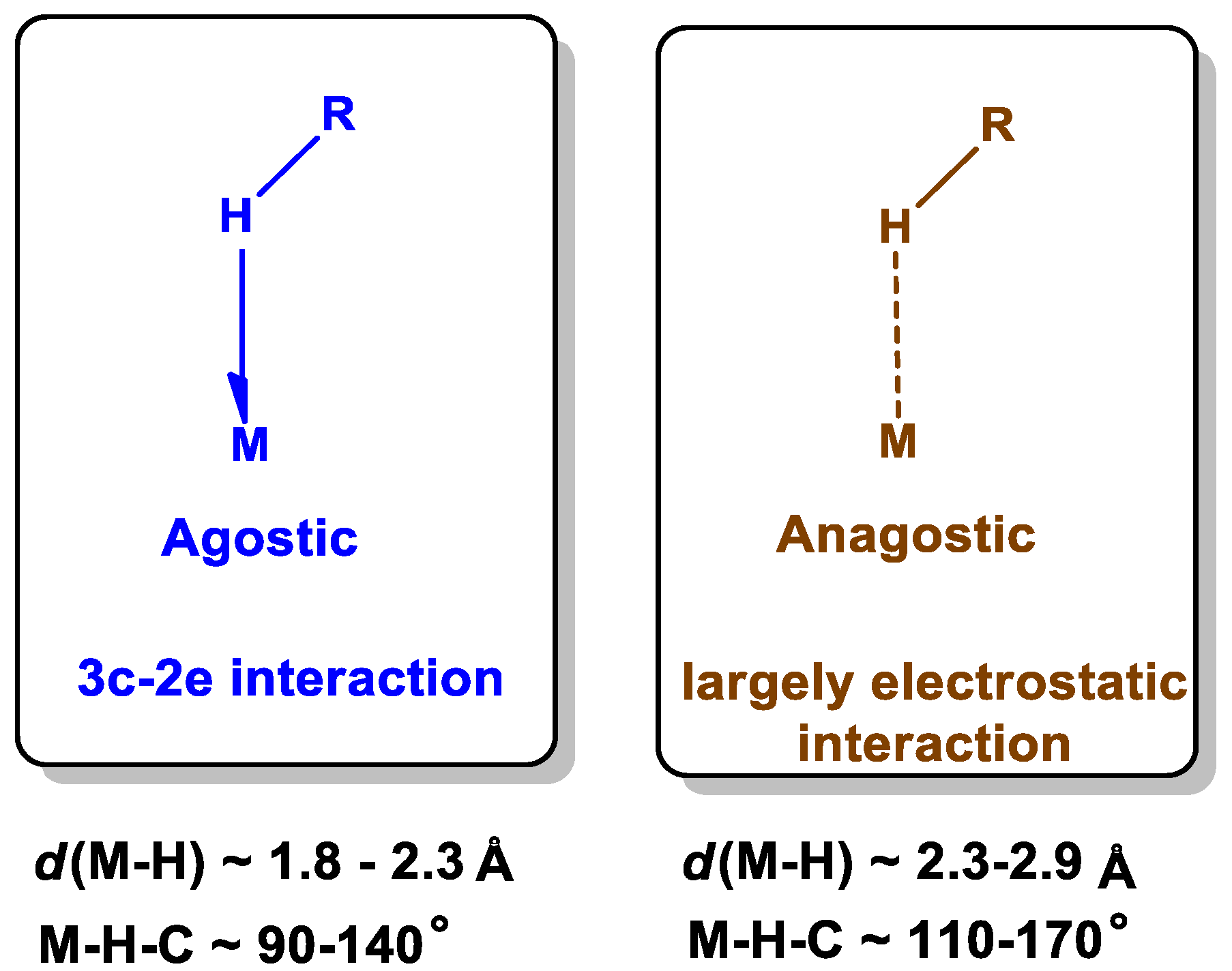
2.6. Computational Studies for Non-Covalent Interaction
2.7. Wiberg Bond Indices, Mayer Bond Order and Delocalization Index Calculations
3. Experimental Procedures
3.1. Materials and Methods
3.2. Synthesis
3.2.1. Synthesis of ({Ni(dppf)}2(piperdtc)) (PF6) (Ni-I)
3.2.2. Synthesis of ({Ni(dppe)}2(piperdtc))(PF6)(Ni-II)
3.3. X-ray Crystallography
3.3.1. Crystallographic Data for Ni-I
3.3.2. Crystallographic Data for Ni-II
3.4. Computational Details
3.5. Hirshfeld Surface Analyses
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, J.-Q.; Luo, Z.-D.; Pan, Y.; Singh, A.K.; Trivedi, M.; Kumar, A. Recent developments in luminescent coordination polymers: Designing strategies, sensing application and theoretical evidences. Coord. Chem. Rev. 2020, 406, 213145. [Google Scholar] [CrossRef]
- Dutta, A.; Singh, A.; Wang, X.; Kumar, A.; Liu, J. Luminescent sensing of nitroaromatics by crystalline porous materials. CrystEngComm 2020, 22, 7736–7781. [Google Scholar] [CrossRef]
- Silva, P.; Vilela, S.M.F.; Tomé, J.P.C.; Paz, F.A.A. Multifunctional metal–organic frameworks: From academia to industrial applications. Chem. Soc. Rev. 2015, 44, 6774–6803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes, R.F.; Figueira, F.; Leite, J.P.; Gales, L.; Paz, F.A.A. Metal–organic frameworks: A future toolbox for biomedicine? Chem. Soc. Rev. 2020, 49, 9121–9153. [Google Scholar] [CrossRef]
- Feng, X.; Liu, J.; Li, J.; Ma, L.-F.; Wang, L.-Y.; Ng, S.-W.; Qin, G.-Z. Series of coordination polymers based on 4-(5-sulfo-quinolin-8-yloxy) phthalate and bipyridinyl coligands: Structure diversity and properties. J. Sol. State Chem. 2015, 230, 80–89. [Google Scholar] [CrossRef]
- Tan, Y.S.; Yeo, C.I.; Tiekink, E.R.T.; Heard, P.J. Dithiocarbamate complexes of platinum group metals: Structural aspects and applications. Inorganics 2021, 9, 60. [Google Scholar] [CrossRef]
- Lee, S.M.; Tiekink, E.R.T. A structural survey of poly-functional dithiocarbamate ligands and the aggregation patterns they sustain. Inorganics 2021, 9, 7. [Google Scholar] [CrossRef]
- Lippard, S.J. Chemistry and molecular biology of platinum anticancer drugs. Pure Appl. Chem. 1987, 59, 731–742. [Google Scholar] [CrossRef] [Green Version]
- Cardell, D.; Hogarth, G.; Faulkner, S. A dithiocarbamate-stabilized copper(I) cube. Inorg. Chim. Acta. 2006, 359, 1321–1324. [Google Scholar] [CrossRef]
- Oyaizu, K.; Yamamoto, K.; Ishii, Y.; Tsuchida, E. Synthesis and characterization of nickel dithiocarbamate complexes bearing ferrocenyl subunits. Chem. Eur. J. 1999, 5, 3193–3207. [Google Scholar] [CrossRef]
- Gimeno, M.C.; Jones, P.G.; Laguna, A.; Sarroca, S.; Calherda, M.J.; Veiros, J. Gold(i) and Gold(iii) Complexes with the 1,1′-Bis (diethyldithiocarbamate) ferrocene Ligand. Chem. Eur. J. 1998, 4, 2308–2314. [Google Scholar] [CrossRef]
- Chant, R.; Hendrickson, A.R.; Martin, R.L.; Rohde, N.M. Metal ion and ligand dependency of the redox behaviour of some first row transition metal dithiocarbamates. Aust. J. Chem. 1973, 26, 2533–2536. [Google Scholar] [CrossRef]
- Beer, P.D.; Berry, N.G.; Cowley, A.R.; Hayes, E.J.; Oates, E.C.; Wong, W.W.H. Metal-directed self-assembly of bimetallic dithiocarbamate transition metal cryptands and their binding capabilities. Chem. Commun. 2003, 19, 2408–2409. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.W.H.; Curiel, D.; Cowley, A.R.; Beer, P.D. Dinuclear zinc(II) dithiocarbamate macrocycles: Ditopic receptors for a variety of guest molecules. Dalton Trans. 2005, 2, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Beer, P.D.; Cheetham, A.G.; Drew, M.G.B.; Fox, O.D.; Hayes, E.J.; Rolls, T.D. Pyrrole-based metallo-macrocycles and cryptands. Dalton Trans. 2003, 4, 603–611. [Google Scholar] [CrossRef]
- Beer, P.D.; Berry, N.; Drew, M.G.B.; Fox, O.D.; Padilla-Tosta, M.E.; Patell, S. Self-assembled dithiocarbamate–copper(II) macrocycles for electrochemical anion recognition. Chem. Commun. 2001, 2, 199–200. [Google Scholar] [CrossRef]
- Padilla-Tosta, M.E.; Fox, O.D.; Drew, M.G.B.; Beer, P.D. Self-assembly of a mixed-valence copper(II)/copper(III) dithiocarbamate catenane. Angew. Chem. Int. Ed. 2001, 40, 4235–4239. [Google Scholar] [CrossRef]
- Fox, O.D.; Drew, M.G.B.; Beer, P.D. Resorcarene-based nanoarchitectures: Metal-directed assembly of a molecular loop and tetrahedron. Angew. Chem. Int. Ed. 2000, 39, 135–140. [Google Scholar] [CrossRef]
- Wilton-Ely, J.D.E.T.; Solanki, D.; Hogarth, G. Multifunctional dithiocarbamates as ligands towards the rational synthesis of polymetallic arrays: An example based on a piperizine-derived dithiocarbamate ligand. Eur. J. Inorg. Chem. 2005, 2005, 4027–4030. [Google Scholar] [CrossRef]
- Knight, E.R.; Leung, N.H.; Lin, Y.H.; Cowley, A.R.; Watkin, D.J.; Thompson, A.L.; Hogarth, G.; Wilton-Ely, J.D.E.T. Multimetallic arrays: Symmetrical bi-, tri-and tetrametallic complexes based on the group 10 metals and the functionalisation of gold nanoparticles with nickel-phosphine surface units. Dalton Trans. 2009, 19, 3688–3697. [Google Scholar] [CrossRef]
- Singh, N.; Kumar, A.; Molloy, K.C.; Mahon, M.F. Syntheses, crystal and molecular structures, and properties of some new phenylmercury(ii) dithiolate complexes. Dalton Trans. 2008, 37, 4999–5007. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Banerjee, S.; Radha, A.; Firdoos, T.; Sahoo, S.C.; Pandey, S.K. Role of non-covalent interactions in the supramolecular architectures of mercury(ii) diphenyldithiophosphates: An experimental and theoretical investigation. New J. Chem. 2021, 45, 2249–2263. [Google Scholar] [CrossRef]
- Arnam, B.V.; Dougherty, D.A. Functional Probes of Drug–Receptor Interactions Implicated by Structural Studies: Cys-Loop Receptors Provide a Fertile Testing Ground: Miniperspective. J. Med. Chem. 2014, 57, 6289–6300. [Google Scholar] [CrossRef]
- Uhlenheuer, D.A.; Petkau, K.; Brunsveld, L. Combining supramolecular chemistry with biology. Chem. Soc. Rev. 2010, 39, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Pieters, B.J.G.E.; van Eldijk, M.B.; Nolte, R.J.M.; Mecinović, J. Natural supramolecular protein assemblies. Chem. Soc. Rev. 2016, 45, 24–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, R.; Trivedi, M.; Singh, J.; Molloy, K.C.; Kociok-Köhn, G.; Mulik, U.P.; Amalnerkar, D.P.; Kumar, A. 1,2-Bis (diphenylphosphino) ethane nickel(II) dithiocarbamate as potential precursor for nickel sulfide: Effect of counter anion on phase and morphology. Inorg. Chim. Acta 2014, 415, 69–74. [Google Scholar] [CrossRef]
- Singh, A.; Singh, A.; Kociok-Köhn, G.; Molloy, K.C.; Singh, A.K.; Kumar, A.; Muddassir, M. Ni(ii) dithiolate anion composites with two-dimensional materials for electrochemical oxygen evolution reactions (OERs). New J. Chem. 2021, 45, 16264–16270. [Google Scholar] [CrossRef]
- Yadav, R.; Singh, A.; Waghadkar, Y.; Kociok-Köhn, G.; Kumar, A.; Chauhan, R.; Rane, S.; Gosavi, S. 1,2-Bis (diphenylphosphino) ethane nickel(II) O, O′-dialkyldithiophosphates as potential precursors for nickel sulfides. New J. Chem. 2017, 41, 1327–1333. [Google Scholar] [CrossRef]
- Singh, A.; Yadav, R.; Kociok-Köhn, G.; Trivedi, M.; Azad, U.P.; Singh, A.K.; Kumar, A. Syntheses of nickel sulfides from 1,2-bis(diphenylphosphino) ethane nickel(II) dithiolates and their application in the oxygen evolution reaction. Int. J. Hydrogen Energy 2018, 43, 5985–5995. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ 4. Dalton Trans. 2007, 955–964. [Google Scholar] [CrossRef]
- Singh, A.; Dutta, A.; Singh, A.K.; Trivedi, M.; Kociok-Köhn, G.; Muddasir, M.; Kumar, A. Tertiary phosphine-appended transition metal ferrocenyl dithiocarbamates: Syntheses, Hirshfeld surface, and electrochemical analyses. Appl. Organomet. Chem. 2020, 34, e5879. [Google Scholar] [CrossRef]
- Singh, A.; Singh, P.; Kociok-Köhn, G.; Trivedi, M.; Kumar, A.; Chauhan, R.; Rane, S.B.; Terashima, C.; Gosavi, S.W.; Fujishma, A. 1,1′-Bis(diphenylphosphino) ferrocene-appended nickel(II) dithiolates as sensitizers in dye-sensitized solar cells. New J. Chem. 2018, 42, 9306–9316. [Google Scholar] [CrossRef]
- Singh, A.; Dutta, A.; Srivastava, D.; Kociok-Köhn, G.; Chauhan, R.; Gosavi, S.; Kumar, A.; Muddassir, M. Effect of different aromatic groups on photovoltaic performance of 1,1′-bis(diphenylphosphino) ferrocene functionalized Ni(II) dithiolates as sensitizers in dye sensitized solar cells. Appl. Organomet. Chem. 2021, 35, e64022021. [Google Scholar] [CrossRef]
- Yadav, R.; Trivedi, M.; Kociok-Köhn, G.; Prasad, R.; Kumar, A. New Ni(II) 1,2-bis(diphenylphosphino) ethane dithiolates: Crystallographic, computational and Hirshfeld surface analyses. CrystEngComm 2015, 17, 9175–9184. [Google Scholar] [CrossRef]
- Singh, A.; Singh, A.; Kociok-Köhn, G.; Chauhan, R.; Gosavi, S.W.; Singh, A.; Singh, A.K.; Kumar, A.; Muddassir, M. Phase-controlled solvothermal syntheses and oxygen evolution reaction (OER) activity of nickel sulfide nanoparticles obtained from 1,2-bis(diphenylphosphino) ethane nickel(ii) acetylacetonatedithiolate. New J. Chem. 2022, 46, 10246–10255. [Google Scholar] [CrossRef]
- Singh, V.; Chauhan, R.; Kumar, A.; Bahadur, L.; Singh, N. Efficient phenylmercury(II) methylferrocenyldithiocarbamate functionalized dye-sensitized solar cells. Dalton Trans. 2010, 39, 9779–9788. [Google Scholar] [CrossRef]
- Barnsley, J.E.; Findlay, J.A.; Shillito, G.E.; Pellet, W.S.; Scottwell, S.; Mcintyre, S.M.; Tay, E.T.; Gordon, K.C.; Crowley, J.D. Long-lived MLCT states for Ru(II) complexes of ferrocene-appended 2,2′-bipyridines. Dalton Trans. 2019, 48, 15713–15722. [Google Scholar] [CrossRef]
- Lever, A.B.P. Inorganic Electronic Spectroscopy; Elsevier: Amsterdam, The Netherlands, 1984; pp. 479–507. [Google Scholar]
- Singh, A.; Singh, A.; Srivastava, D.; Kociok-Köhn, G.; Köhn, R.D.; Kumar, A.; Muddassir, M. New di-n-butyltin(iv)-bis-(1-alkoxy-isoquinoline-4-nitrile thiolate): Crystallographic and computational studies. CrystEngComm 2022, 24, 4274–4282. [Google Scholar] [CrossRef]
- Singh, P.; Singh, A.; Singh, A.; Singh, A.K.; Kociok-Köhn, G.; Alowais, A.; Abduh, N.A.Y.; Muddassir, M.; Kumar, A. New 1D diorganotin(IV) dithiolate coordination polymers: Crystallographic, computational, Hirshfeld surface and thermal analyses. CrystEngComm 2020, 22, 2049–2059. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A.; Yadav, R.; Singh, S.; Kociok-Köhn, G.; Trivedi, M. Supramolecular architecture of organotin(IV) N-methyl ferrocenyl N-ethanol dithiocarbamates: Crystallographic and computational studies. Inorg. Chim. Acta 2018, 471, 234–243. [Google Scholar] [CrossRef]
- Yadav, R.; Trivedi, M.; Chauhan, R.; Prasad, R.; Kociok- Köhn, G.; Kumar, A. Supramolecular architecture of organotin(IV) 4-hydroxypiperidine dithiocarbamates: Crystallographic, computational and Hirshfeld surface analyses. Inorg. Chim. Acta 2016, 450, 57–68. [Google Scholar] [CrossRef]
- Jelsh, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ashfaq, M.; Din, Z.U.; Ibrahim, M.; Khalid, M.; Assiri, M.A.; Riaz, A.; Tahir, M.N.; Rodrigues-Filho, E.; Imran, M.; et al. Synthesis, Structural, and Intriguing Electronic Properties of Symmetrical Bis-Aryl-α, β-Unsaturated Ketone Derivatives. ACS Omega 2022, 7, 39294–39309. [Google Scholar] [CrossRef]
- Askerov, R.K.; Ashfaq, M.; Chipinsky, E.V.; Osmanov, V.K.; Tahir, M.N.; Baranov, E.V.; Fukin, G.K.; Khrustalev, V.N.; Nazarov, R.H.; Borisova, G.N.; et al. Synthesis, crystal structure, exploration of the supramolecular assembly through Hirshfeld surface analysis and bactericidal activity of the cadmium organometallic complexes obtained from the heterocyclic ligand. Results Chem. 2022, 4, 100600. [Google Scholar] [CrossRef]
- Chiter, C.; Bouchama, A.; Nardjes Mouas, T.; Allal, H.; Yahiaoui, M.; Warad Il Zarrouk, A.; Djedouani, A. Synthesis, crystal structure, spectroscopic and hirshfeld surface analysis, NCI-RDG, DFT computations and antibacterial activity of new asymmetrical azines. J. Mol. Struc. 2020, 1217, 128376. [Google Scholar] [CrossRef]
- Foroutan-Nejad, C.; Shahbazian, S.; Marek, R. Toward a consistent interpretation of the QTAIM: Tortuous link between chemical bonds, interactions, and bond/line paths. Chem. Eur. J. 2014, 20, 10140. [Google Scholar] [CrossRef]
- Sowlati-Hashjin, S.; Šadek, V.; Sadjadi, S.; Karttunen, M.; Martín-Pendás, A.; Foroutan-Nejad, C. Collective interactions among organometallics are exotic bonds hidden on lab shelves. Nat. Commun. 2022, 13, 2069. [Google Scholar] [CrossRef]
- Matito, E.; Poater, J.; Solà, M.; Duran, M.; Salvador, P. Comparison of the AIM delocalization index and the mayer and fuzzy atom bond orders. J. Phys. Chem. A 2005, 43, 9904–9910. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Matta, C.F.; Boyd, R.J. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; Wiley VCH, Verlag Gmbh & Co.: KGaA, Germany, 2007. [Google Scholar]
- Matta, C.F.; Hernandez-Tryjillo, J.; Tang, T.H.; Bader, R.F.W. Hydrogen–hydrogen bonding: A stabilizing interaction in molecules and crystals. Chem. Eur. J. 2003, 9, 1940. [Google Scholar] [CrossRef]
- Kumar, A.; Mayer-Figge, H.; Sheldrick, W.S.; Singh, N. Synthesis, structure, conductivity, and calculated nonlinear optical properties of two novel bis(triphenylphosphane)copper(I) dithiocarbamates. Eur. J. Inorg. Chem. 2009, 2009, 2720–2725. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction, version 1.171.40.67a; CrysAlisPro; Rigaku Oxford Diffraction: Tokyo, Japan, 2019.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T., Jr.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 09 Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Keith, T.A.; Gristmill, T.K. Software; TK Gristmill Software: Overland Park, KS, USA; Available online: Aim.tkgristmill.com (accessed on 31 January 2023).
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K.J. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Williams, T.; Kelley, C. 1986–1993, 1998, 2004, 2007–2022. Available online: www.gnuplot.info (accessed on 31 January 2023).
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378. [Google Scholar] [CrossRef]
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 309. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Mitchell, A.S.; Spackman, M.A. Hirshfeld surfaces: A new tool for visualising and exploring molecular crystals. Chem-Eur. J. 1998, 4, 2136. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. Sec. B 2004, 60, 627. [Google Scholar] [CrossRef]
- Rohl, A.L.; Moret, M.; Kaminsky, W.; Claborn, K.; McKinnon, J.J.; Kahr, B. Hirshfeld surfaces identify inadequacies in computations of intermolecular interactions in crystals: Pentamorphic 1,8-dihydroxyanthraquinone. Cryst. Growth Des. 2008, 8, 4517. [Google Scholar] [CrossRef]
- Parkin, A.; Barr, G.; Dong, W.; Gilmore, C.J.; Jayatilaka, D.; McKinnon, J.J.; Spackman, M.A.; Wilson, C.C. Comparing entire crystal structures: Structural genetic fingerprinting. CrystEngComm 2007, 9, 648. [Google Scholar] [CrossRef]
- Wolff, S.K.; Greenwood, D.J.; McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer 3.1; University of Western Australia: Perth, Australia, 2012. [Google Scholar]
- Koenderink, J.J.; Van Doorn, A.J. Surface shape and curvature scales. Image Vision Comput. 1992, 10, 557. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contact % | Atom | H | C | F | N | S | Ni |
|---|---|---|---|---|---|---|---|
| H | 57.7 | 18.3 | 16.9 | 0.3 | 4.6 | 0.1 | |
| C | 18.3 | 1.1 | 0.5 | - | - | - | |
| F | 16.9 | 0.5 | - | 0.1 | 0.4 | - | |
| N | 0.3 | - | 0.1 | - | - | - | |
| S | 4.6 | - | 0.4 | - | - | - | |
| Ni | 0.1 | - | - | - | - | - | |
| Surface% | 77.8 | 10.5 | 8.95 | 2.5 | 0.2 | 0.45 | |
| Random contacts % | Atom | H | C | F | N | S | Ni |
| H | 60.52 | ||||||
| C | 16.33 | 1.10 | |||||
| F | 13.92 | 1.87 | 0.80 | ||||
| N | 0.46 | 0.04 | 0.05 | ||||
| S | 3.89 | 0.52 | 0.44 | 0.01 | 0.06 | ||
| Ni | 0.70 | 0.09 | 0.08 | 0.001 | 0.02 | 0.002 | |
| Enrichment ratio | Atom | H | C | F | N | S | Ni |
| H | 0.95 | ||||||
| C | 1.12 | 1.00 | |||||
| F | 1.21 | 0.26 | |||||
| N | |||||||
| S | 1.18 | ||||||
| Ni |
| Contact % | Atom | H | C | F | N | S | Ni |
|---|---|---|---|---|---|---|---|
| H | 44.9 | 20.2 | 19.7 | 4.2 | 5.4 | 0.9 | |
| C | 20.2 | 2.0 | 0.5 | 0.4 | 1.7 | - | |
| F | 19.7 | 0.5 | - | 0.4 | - | - | |
| N | 4.2 | 0.4 | 0.2 | - | - | - | |
| S | 5.4 | 1.7 | - | - | - | - | |
| Ni | 0.9 | - | - | - | - | - | |
| Surface % | 69.75 | 13.4 | 10.2 | 2.45 | 3.55 | 0.45 | |
| Random contacts % | Atom | H | C | F | N | S | Ni |
| H | 48.65 | - | - | - | - | - | |
| C | 18.69 | 1.79 | - | - | - | - | |
| F | 14.22 | 2.73 | 1.04 | - | - | - | |
| N | 3.35 | 0.64 | 0.49 | 0.06 | - | - | |
| S | 4.95 | 0.95 | 0.72 | 0.17 | 0.12 | 0.03 | |
| Ni | 0.62 | 0.1 | 0.09 | 0.02 | 0.03 | - | |
| Enrichment ratio | Atom | H | C | F | N | S | Ni |
| H | 0.92 | ||||||
| C | 1.08 | 1.11 | |||||
| F | 1.38 | 0.18 | |||||
| N | 1.20 | ||||||
| S | 1.09 | ||||||
| Ni |
| Interaction Type | ρbcp | ∇2 ρbcp | (ε) | H | E (kJ/mol) |
|---|---|---|---|---|---|
| Ni-I | |||||
| C31-H31⋯C7 | +0.0063 | +0.0212 | +1.525 | 0.0011 | 4.04 |
| C3-H3⋯C33 | +0.0043 | +0.0319 | +1.325 | 0.0012 | 4.12 |
| C36-H36A⋯F4 | +0.0090 | +0.0361 | +0.028 | 0.0023 | 7.38 |
| Ni-II | |||||
| C2-H2B⋯C23 | +0.0060 | +0.0190 | +0.604 | 0.0010 | 3.52 |
| C42-H42⋯F7 | +0.0066 | +0.0307 | +0.148 | 0.0007 | 7.18 |
| C59-H59B⋯F10 | +0.0052 | +0.0265 | +0.296 | 0.0016 | 4.65 |
| Bond Type | Wiberg Bond Index | Mayer Atomic Bond Order | Delocalization Index (Atom Basin) | Natural Charges | |
|---|---|---|---|---|---|
| Ni-I | |||||
| C-H24⋯Ni | 0.0041 | 0.0073 | 0.0209 | H24 | 0.2216 |
| H28 (not displaying C-H⋯Ni) | 0.2134 | ||||
| Ni-II | |||||
| C-H14⋯Ni | 0.0059 | 0.0112 | 0.0198 | H14 | 0.2287 |
| H10 (not displaying C-H⋯Ni) | 0.2102 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, D.; Prakash, O.; Kociok-Köhn, G.; Kumar, A.; Alarifi, A.; Abduh, N.A.Y.; Afzal, M.; Muddassir, M. Centrosymmetric Nickel(II) Complexes Derived from Bis-(Dithiocarbamato)piperazine with 1,1′-Bis-(Diphenylphosphino)ferrocene and 1,2-Bis-(Diphenylphosphino)ethane as Ancillary Ligands: Syntheses, Crystal Structure and Computational Studies. Crystals 2023, 13, 343. https://doi.org/10.3390/cryst13020343
Srivastava D, Prakash O, Kociok-Köhn G, Kumar A, Alarifi A, Abduh NAY, Afzal M, Muddassir M. Centrosymmetric Nickel(II) Complexes Derived from Bis-(Dithiocarbamato)piperazine with 1,1′-Bis-(Diphenylphosphino)ferrocene and 1,2-Bis-(Diphenylphosphino)ethane as Ancillary Ligands: Syntheses, Crystal Structure and Computational Studies. Crystals. 2023; 13(2):343. https://doi.org/10.3390/cryst13020343
Chicago/Turabian StyleSrivastava, Devyani, Om Prakash, Gabriele Kociok-Köhn, Abhinav Kumar, Abdullah Alarifi, Naaser A. Y. Abduh, Mohd Afzal, and Mohd Muddassir. 2023. "Centrosymmetric Nickel(II) Complexes Derived from Bis-(Dithiocarbamato)piperazine with 1,1′-Bis-(Diphenylphosphino)ferrocene and 1,2-Bis-(Diphenylphosphino)ethane as Ancillary Ligands: Syntheses, Crystal Structure and Computational Studies" Crystals 13, no. 2: 343. https://doi.org/10.3390/cryst13020343