
pH-Driven Polymorphic Behaviour of the Third PDZ Domain of PSD95: The Role of Electrostatic Interactions

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning, Expression and Purification of PSD95-PDZ3
2.2. Crystallisation and Data Collection
2.3. Structure Solution and Refinement
2.4. Structure Analysis
2.5. Dynamic Light Scattering
3. Results
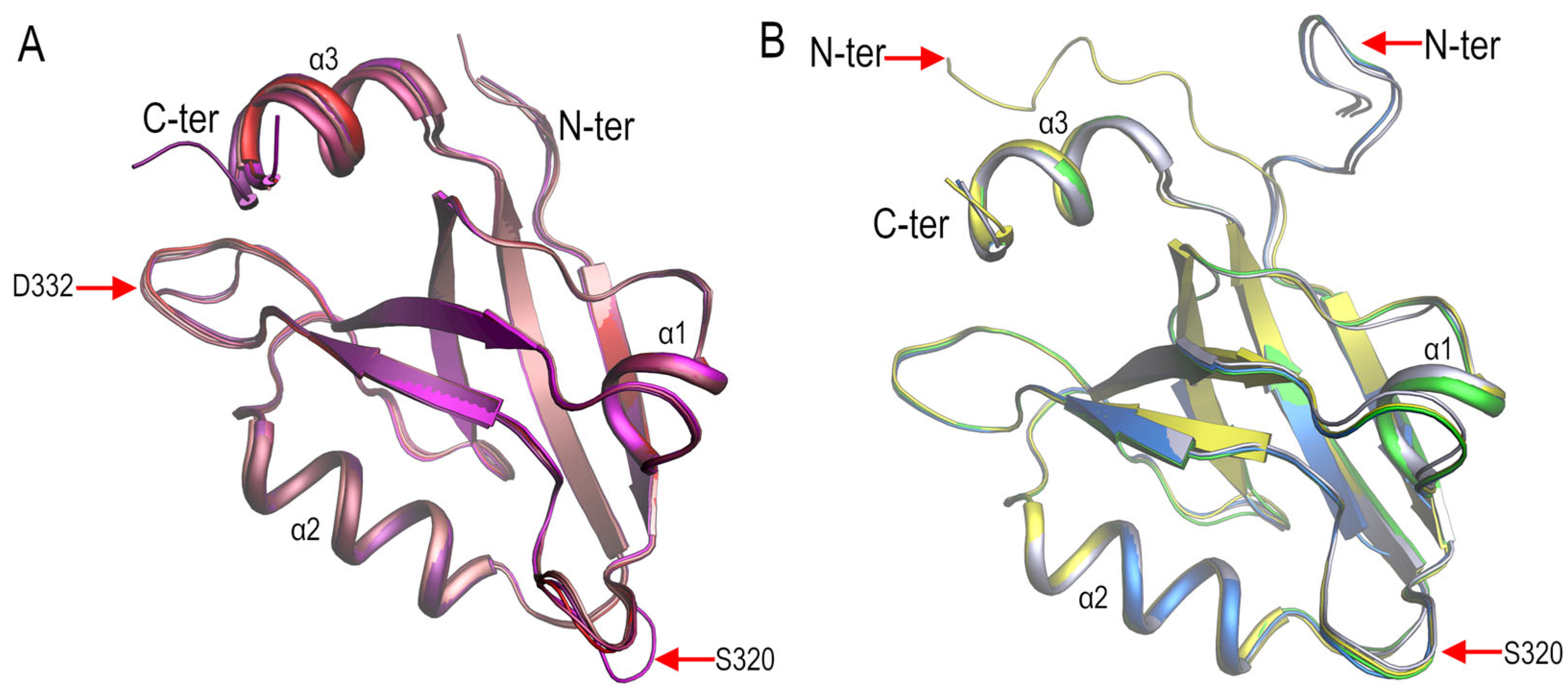
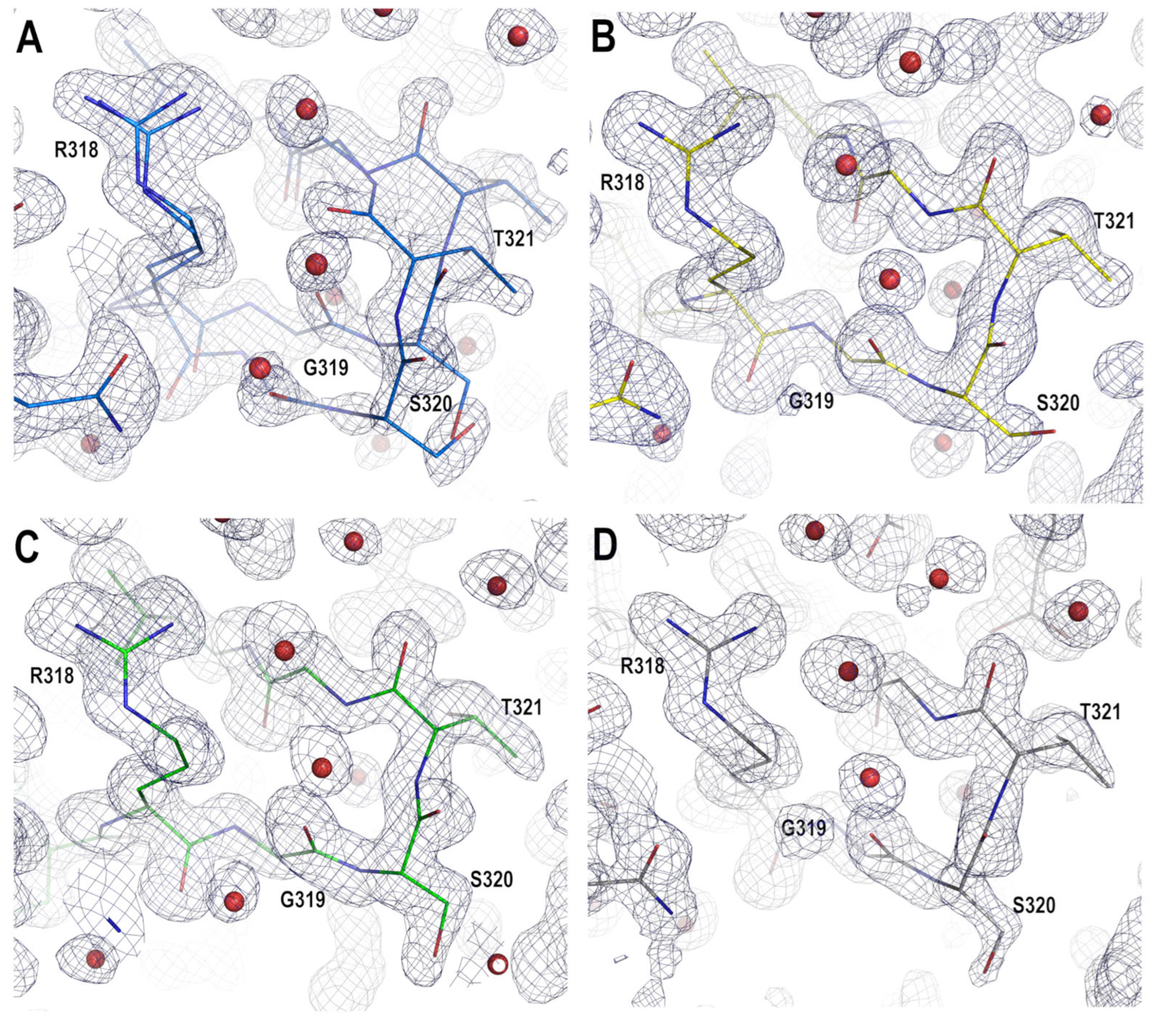
3.1. Crystal Polymorphism in the PSD95-PDZ3 at Mildly Acidic pH Conditions
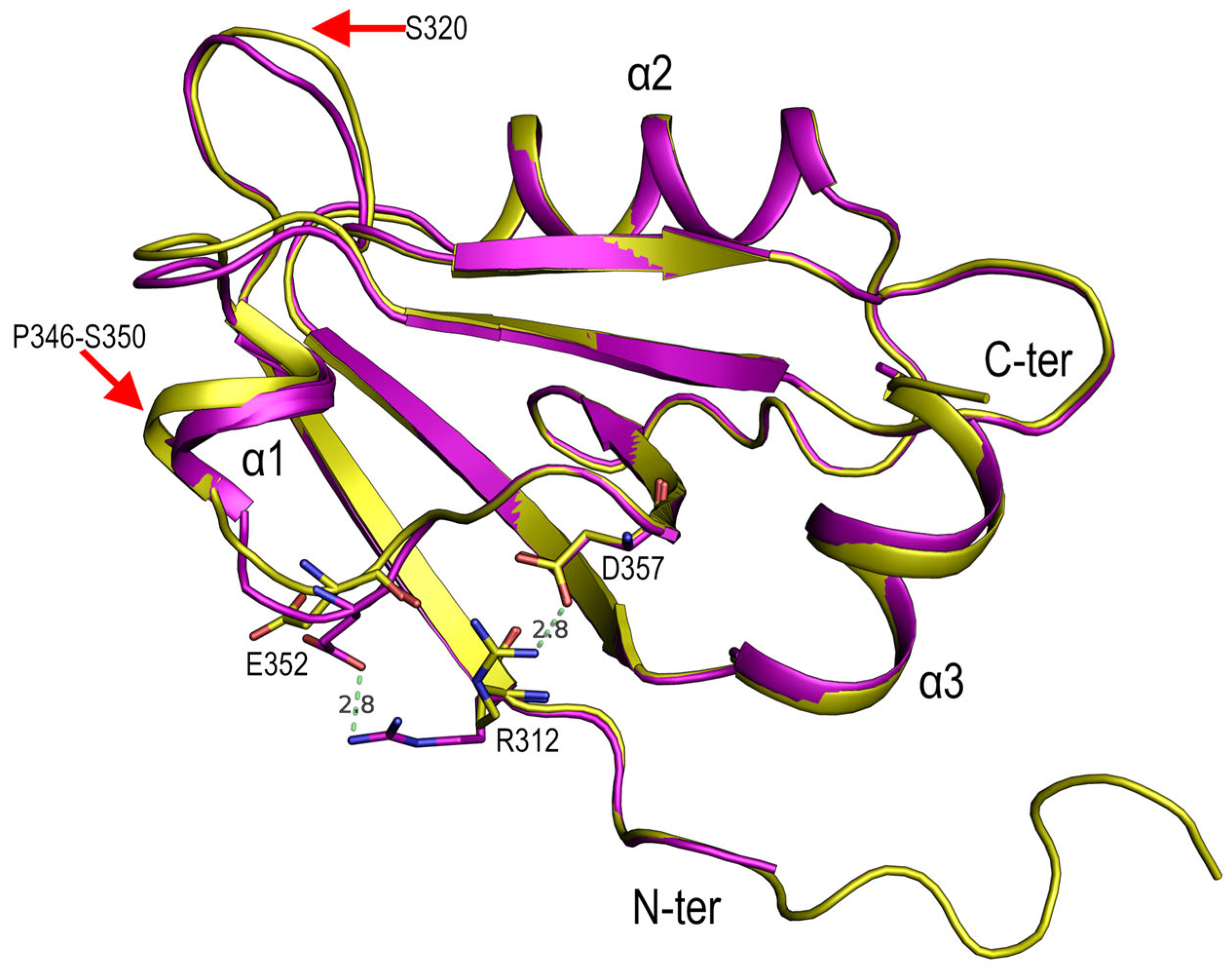
3.2. Structural Comparison of the PDZ3-PDS95 Polymorphs Obtained at Mildly Acidic pH Conditions
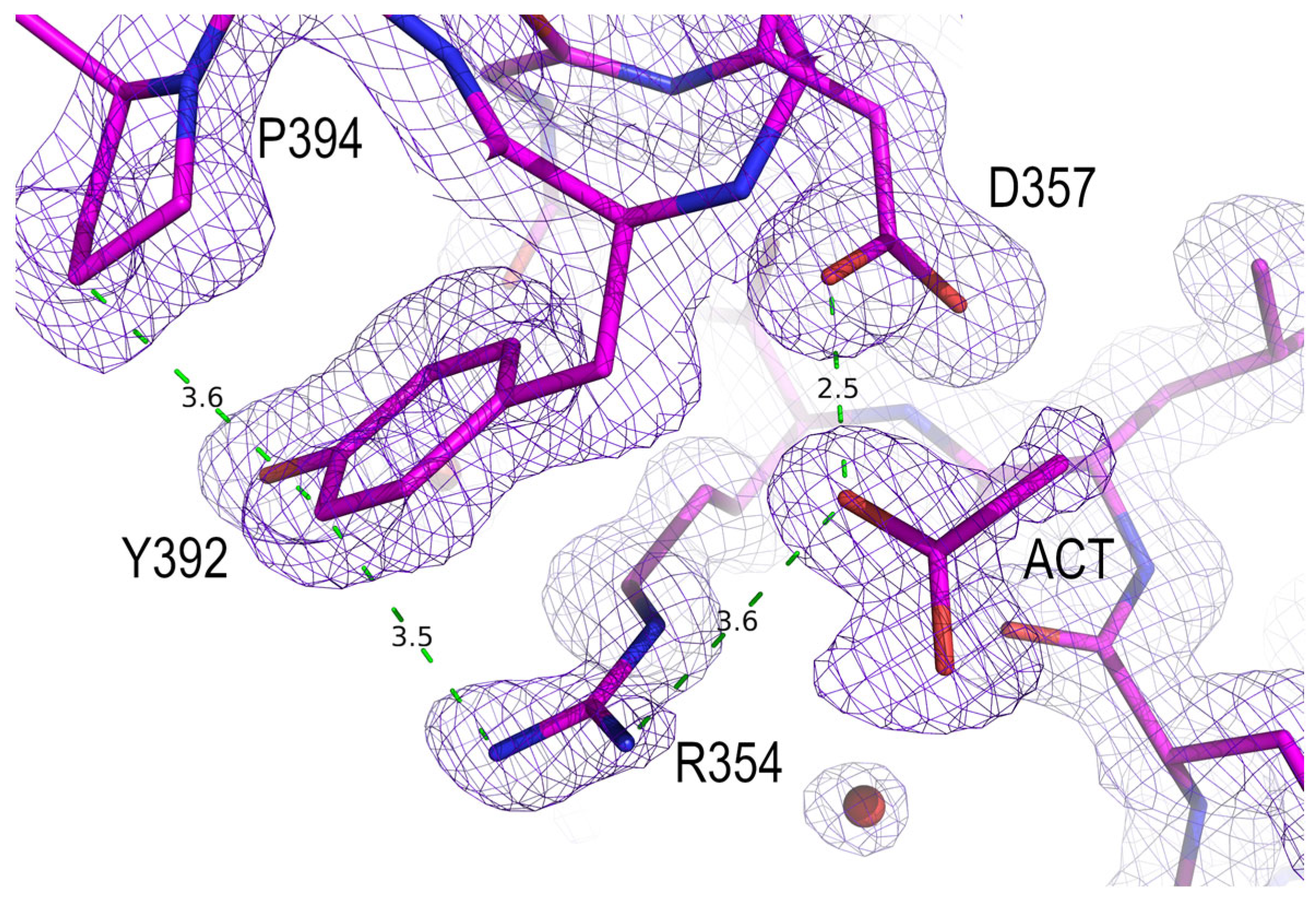
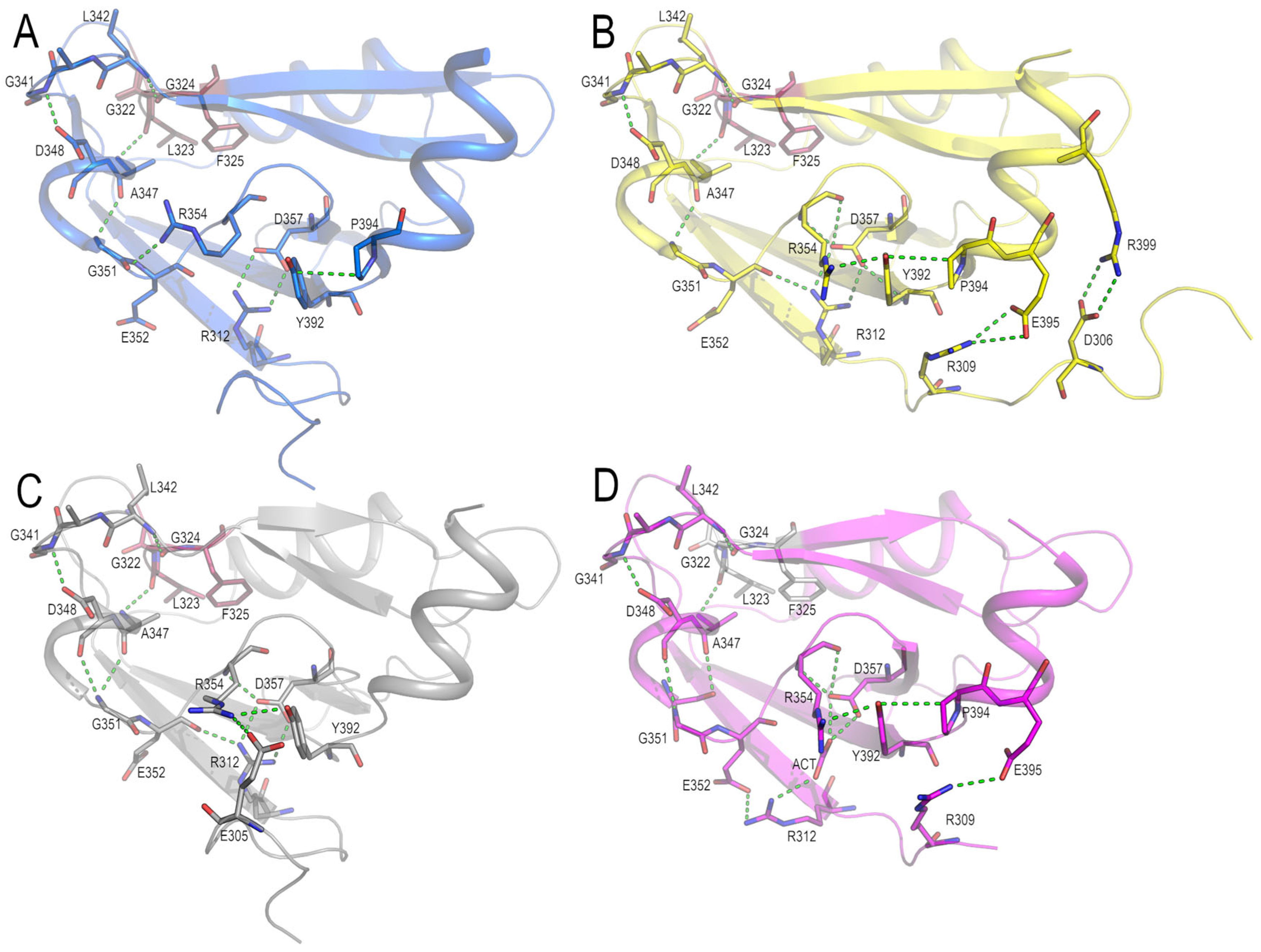
3.3. Analysis of the Electrostatic Interactions in the PSD95-PDZ3 Domain: Dependency on the pH
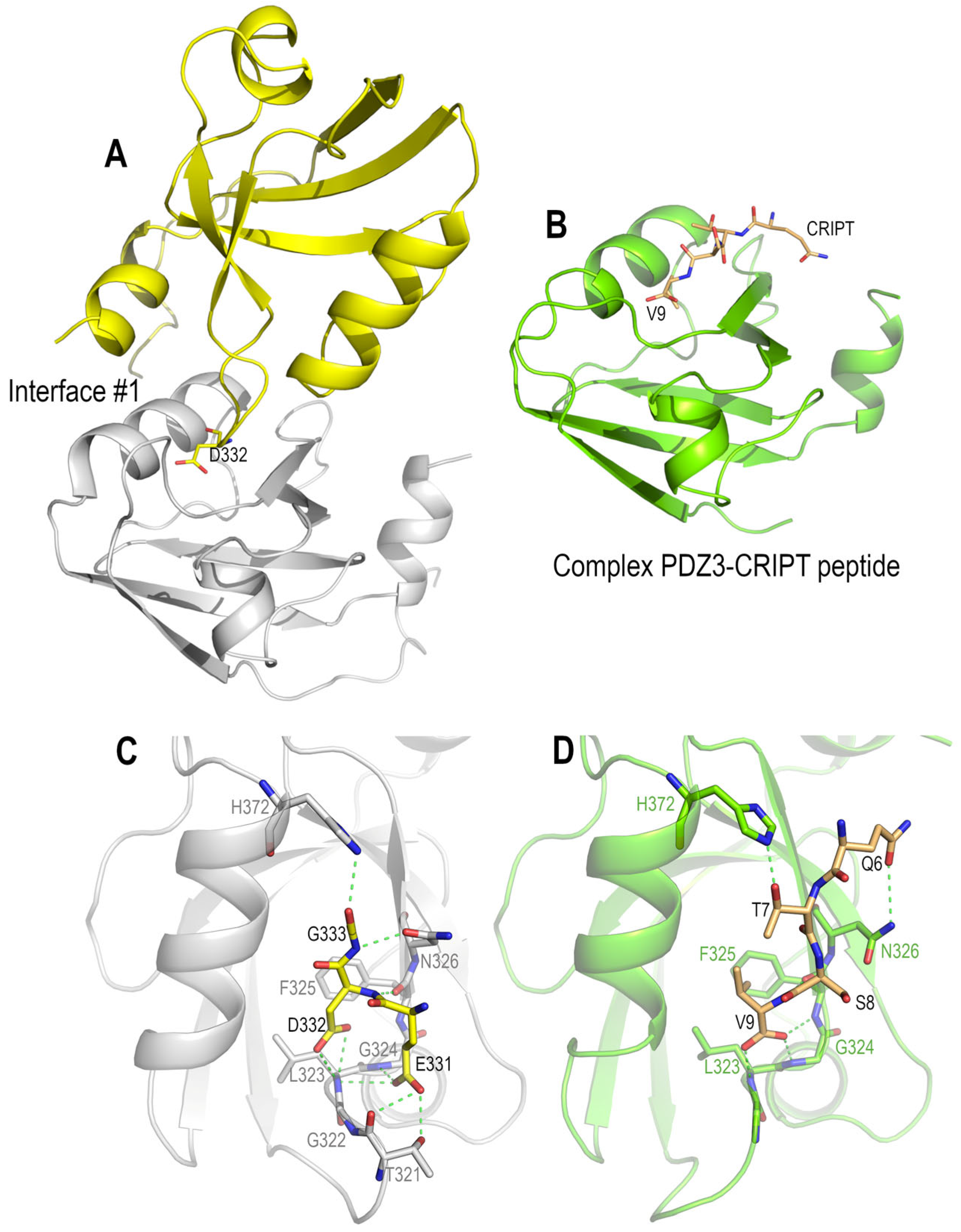
3.4. Polymorphism and Crystal Interfaces in the PSD95-PDZ3
4. Discussion
4.1. Polymorphism in the PSD95-PDZ3
4.2. Role of Asp332 in the Crystallisation of the PSD95-PDZ3
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kieffer, C.; Jourdan, J.P.; Jouanne, M.; Voisin-Chiret, A.S. Noncellular screening for the discovery of protein-protein interaction modulators. Drug Discov. Today 2020, 25, 1592–1603. [Google Scholar] [CrossRef]
- Camara-Artigas, A.; Andujar-Sanchez, M.; Ortiz-Salmeron, E.; Cuadri, C.; Casares, S. The effect of a proline residue on the rate of growth and the space group of alpha-spectrin SH3-domain crystals. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Camara-Artigas, A.; Gavira, J.A.; Casares, S.; Garcia-Ruiz, J.M.; Conejero-Lara, F.; Allen, J.P.; Martinez, J.C. Understanding the polymorphic behaviour of a mutant of the alpha-spectrin SH3 domain by means of two 1.1 A resolution structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Camara-Artigas, A.; Murciano-Calles, J.; Martinez, J.C. Conformational changes in the third PDZ domain of the neuronal postsynaptic density protein 95. Acta Crystallogr. D Struct. Biol. 2019, 75, 381–391. [Google Scholar] [CrossRef]
- Elkins, J.M.; Papagrigoriou, E.; Berridge, G.; Yang, X.; Phillips, C.; Gileadi, C.; Savitsky, P.; Doyle, D.A. Structure of PICK1 and other PDZ domains obtained with the help of self-binding C-terminal extensions. Protein Sci. 2007, 16, 683–694. [Google Scholar] [CrossRef]
- Borg, J.P. PDZ Mediated Interactions. In PDZ MEDIATED INTERACTIONS: Methods and Protocols; Borg, J.P., Ed.; Methods in Molecular Biology; Humana: New York, NY, USA, 2021; Volume 2256, pp. 1–292. [Google Scholar]
- Doyle, D.A.; Lee, A.; Lewis, J.; Kim, E.; Sheng, M.; MacKinnon, R. Crystal structures of a complexed and peptide-free membrane protein-binding domain: Molecular basis of peptide recognition by PDZ. Cell 1996, 85, 1067–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Zheng, J.J. PDZ domains and their binding partners: Structure, specificity, and modification. Cell Commun. Signal 2010, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murciano-Calles, J.; Coello, A.; Camara-Artigas, A.; Martinez, J.C. PDZ/PDZ interaction between PSD-95 and nNOS neuronal proteins: A thermodynamic analysis of the PSD95-PDZ2/nNOS-PDZ interaction. J. Mol. Recognit. 2020, 33, e2826. [Google Scholar] [CrossRef]
- Hillier, B.J.; Christopherson, K.S.; Prehoda, K.E.; Bredt, D.S.; Lim, W.A. Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science 1999, 284, 812–815. [Google Scholar] [CrossRef] [Green Version]
- Penkert, R.R.; DiVittorio, H.M.; Prehoda, K.E. Internal recognition through PDZ domain plasticity in the Par-6-Pals1 complex. Nat. Struct. Mol. Biol. 2004, 11, 1122–1127. [Google Scholar] [CrossRef]
- Zhang, J.; Lewis, S.M.; Kuhlman, B.; Lee, A.L. Supertertiary structure of the MAGUK core from PSD-95. Structure 2013, 21, 402–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murciano-Calles, J.; Marin-Argany, M.; Cobos, E.S.; Villegas, S.; Martinez, J.C. The impact of extra-domain structures and post-translational modifications in the folding/misfolding behaviour of the third PDZ domain of MAGUK neuronal protein PSD-95. PLoS ONE 2014, 9, e98124. [Google Scholar] [CrossRef] [Green Version]
- Murciano-Calles, J.; Corbi-Verge, C.; Candel, A.M.; Luque, I.; Martinez, J.C. Post-translational modifications modulate ligand recognition by the third PDZ domain of the MAGUK protein PSD-95. PLoS ONE 2014, 9, e90030. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.; Visconti, L.; Jemth, P.; Gianni, S. Addressing the role of the α-helical extension in the folding of the third PDZ domain from PSD-95. Sci. Rep. 2017, 7, 12593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murciano-Calles, J.; Cobos, E.S.; Mateo, P.L.; Camara-Artigas, A.; Martinez, J.C. A comparative analysis of the folding and misfolding pathways of the third PDZ domain of PSD95 investigated under different pH conditions. Biophys. Chem. 2011, 158, 104–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumawat, A.; Chakrabarty, S. Hidden electrostatic basis of dynamic allostery in a PDZ domain. Proc. Natl. Acad. Sci. USA 2017, 114, E5825–E5834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camara-Artigas, A.; Murciano-Calles, J.; Gavira, J.A.; Cobos, E.S.; Martinez, J.C. Novel conformational aspects of the third PDZ domain of the neuronal post-synaptic density-95 protein revealed from two 1.4A X-ray structures. J. Struct. Biol. 2010, 170, 565–569. [Google Scholar] [CrossRef]
- Petit, C.M.; Zhang, J.; Sapienza, P.J.; Fuentes, E.J.; Lee, A.L. Hidden dynamic allostery in a PDZ domain. Proc. Natl. Acad. Sci. USA 2009, 106, 18249–18254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Petit, C.M.; King, D.S.; Lee, A.L. Phosphorylation of a PDZ domain extension modulates binding affinity and interdomain interactions in postsynaptic density-95 (PSD-95) protein, a membrane-associated guanylate kinase (MAGUK). J. Biol. Chem. 2011, 286, 41776–41785. [Google Scholar] [CrossRef] [Green Version]
- Raman, A.S.; White, K.I.; Ranganathan, R. Origins of Allostery and Evolvability in Proteins: A Case Study. Cell 2016, 166, 468–480. [Google Scholar] [CrossRef] [Green Version]
- Murciano-Calles, J.; Cobos, E.S.; Mateo, P.L.; Camara-Artigas, A.; Martinez, J.C. An oligomeric equilibrium intermediate as the precursory nucleus of globular and fibrillar supramacromolecular assemblies in a PDZ domain. Biophys. J. 2010, 99, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.C.; von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Bowler, M.W.; Svensson, O.; Nurizzo, D. Fully automatic macromolecular crystallography: The impact of MASSIF-1 on the optimum acquisition and quality of data. Crystallogr. Rev. 2016, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Juanhuix, J.; Gil-Ortiz, F.; Cuni, G.; Colldelram, C.; Nicolas, J.; Lidon, J.; Boter, E.; Ruget, C.; Ferrer, S.; Benach, J. Developments in optics and performance at BL13-XALOC, the macromolecular crystallography beamline at the ALBA synchrotron. J. Synchrotron Radiat. 2014, 21, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Vonrhein, C.; Tickle, I.J.; Flensburg, C.; Keller, P.; Paciorek, W.; Sharff, A.; Bricogne, G. Advances in automated data analysis and processing within autoPROC, combined with improved characterisation, mitigation and visualisation of the anisotropy of diffraction limits using STARANISO. Acta Crystallogr. Sect. A 2018, 74, a360. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkoczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.-W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D 2019, 75, 861–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunkoczi, G.; Echols, N.; McCoy, A.J.; Oeffner, R.D.; Adams, P.D.; Read, R.J. Phaser.MRage: Automated molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 2276–2286. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joosten, R.P.; Long, F.; Murshudov, G.N.; Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 2014, 1, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A. PDBsum new things. Nucleic Acids Res. 2009, 37, D355–D359. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E. Macromolecular complexes in crystals and solutions. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. Sect. A 1976, 32, 922–923. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Olsson, M.H.; Sondergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.0; Schrodinger, LLC.: New York, NY, USA, 2015.
- Basdevant, N.; Weinstein, H.; Ceruso, M. Thermodynamic basis for promiscuity and selectivity in protein-protein interactions: PDZ domains, a case study. J. Am. Chem. Soc. 2006, 128, 12766–12777. [Google Scholar] [CrossRef] [Green Version]
- Mu, Y.; Cai, P.; Hu, S.; Ma, S.; Gao, Y. Characterization of diverse internal binding specificities of PDZ domains by yeast two-hybrid screening of a special peptide library. PLoS ONE 2014, 9, e88286. [Google Scholar] [CrossRef]
- McPherson, A.; Cudney, B. Optimization of crystallization conditions for biological macromolecules. Acta crystallogr. Sect. F, Struct. Biol. Commun. 2014, 70, 1445–1467. [Google Scholar] [CrossRef] [Green Version]
- McPherson, A.; Gavira, J.A. Introduction to protein crystallization. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 2–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantardjieff, K.A.; Rupp, B. Protein isoelectric point as a predictor for increased crystallization screening efficiency. Bioinformatics 2004, 20, 2162–2168. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [CrossRef]
- Hamiaux, C.; Perez, J.; Prange, T.; Veesler, S.; Ries-Kautt, M.; Vachette, P. The BPTI decamer observed in acidic pH crystal forms pre-exists as a stable species in solution. J. Mol. Biol. 2000, 297, 697–712. [Google Scholar] [CrossRef]
- Hamiaux, C.; Prange, T.; Ries-Kautt, M.; Ducruix, A.; Lafont, S.; Astier, J.P.; Veesler, S. The decameric structure of bovine pancreatic trypsin inhibitor (BPTI) crystallized from thiocyanate at 2.7 A resolution. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grouazel, S.; Bonneté, F.; Astier, J.-P.; Ferté, N.; Perez, J.; Veesler, S. Exploring Bovine Pancreatic Trypsin Inhibitor Phase Transitions. J. Phys. Chem. B 2006, 110, 19664–19670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubkowski, J.; Wlodawer, A. Decamers observed in the crystals of bovine pancreatic trypsin inhibitor. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 335–337. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, W.H.; Croker, K.M. Identification of a molecular switch that selects between two crystals forms of bovine pancreatic trypsin inhibitor. Protein Sci. 1994, 3, 1602–1604. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Appleton, B.A.; Ivarsson, Y.; Zhang, Y.; Gfeller, D.; Wiesmann, C.; Sidhu, S.S. A Structural Portrait of the PDZ Domain Family. J. Mol. Biol. 2014, 426, 3509–3519. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, K.A.; McLaughlin, R.N.; Ranganathan, R. Hot spots for allosteric regulation on protein surfaces. Cell 2011, 147, 1564–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Orthorhombic-A pH 4.6 | Orthorhombic-B pH 4.0 | Trigonal pH 4.0 | Monoclinic pH4.0 | |
|---|---|---|---|---|
| PDB entry | 8AH5 | 8AH7 | 8AH4 | 8AH6 |
| Wavelength (Å) | 0.9677 | 0.7749 | 0.9677 | 0.9677 |
| Resolution range (Å) | 19.35–1.25 (1.27–1.25) | 19.53–1.25 (1.27–1.25) | 48.41–1.48 (1.53–1.48) | 19.68–1.63 (1.66–1.63) |
| Space group | P212121 | P212121 | P3112 | P21 |
| Unit cell (Å, °) | 28.84 32.34 88.30 90 90 90 | 32.56 36.75 73.22 90 90 90 | 61.70 61.70 228.70 90 90 120 | 28.86 87.50 32.43 90 92.56 90 |
| Total reflections | 242,181 (3829) | 99,957 (4999) | 258,384 (12,939) | 83,547 (4147) |
| Unique reflections | 22,582 (754) | 24,458 (1205) | 53,125 (2631) | 19,832 (958) |
| Multiplicity | 10.7 (5.1) | 4.1 (4.1) | 4.9 (4.9) | 4.2 (4.3) |
| Completeness (%) | 95.6 (67.0) | 98.4 (99.6) | 98.6 (100) | 98.9 (100) |
| Mean I/sigma(I) | 9.9 (1.6) | 8.2 (1.0) | 12.1 (2.2) | 9.6 (2.1) |
| Wilson B-factor (Å2) | 10.43 | 16.37 | 16.73 | 19.06 |
| R-merge | 0.125 (0.700) | 0.040 (0.709) | 0.067 (0.675) | 0.074 (0.678) |
| CC1/2 | 0.996 (0.749) | 0.999 (0.762) | 0.999 (0.796) | 0.998 (0.854) |
| R-work | 0.150 (0.257) | 0.160 (0.310) | 0.199 (0.326) | 0.167 (0.246) |
| R-free | 0.185 (0.276) | 0.198 (0.358) | 0.238 (0.375) | 0.181 (0.265) |
| CC(work) | 0.97 (0.89) | 0.97 (0.91) | 0.96 (0.73) | 0.97 (0.91) |
| CC(free) | 0.96 (0.89) | 0.96 (0.86) | 0.95 (0.95) | 0.97 (0.89) |
| Protein residues | 102 | 102 | 566 | 203 |
| Solvent | 171 | 117 | 516 | 209 |
| RMS (bonds) | 0.011 | 0.014 | 0.012 | 0.004 |
| RMS (angles) | 1.20 | 1.26 | 1.09 | 0.71 |
| Ramachandran favored (%) | 100.00 | 100.00 | 98.38 | 100.00 |
| Ramachandran allowed (%) | 0.00 | 0.00 | 1.62 | 0.00 |
| Ramachandran outliers (%) | 0.00 | 0.00 | 0.00 | 0.00 |
| Average B-factor | 13.85 | 25.51 | 22.94 | 23.01 |
| Macromolecules | 11.86 | 24.23 | 21.69 | 22.01 |
| Ligands | 14.91 | 37.27 | 28.21 | 24.38 |
| Solvent | 23.40 | 33.37 | 29.88 | 30.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salinas-García, M.C.; Plaza-Garrido, M.; Gavira, J.A.; Murciano-Calles, J.; Andújar-Sánchez, M.; Ortiz-Salmerón, E.; Martinez, J.C.; Cámara-Artigas, A. pH-Driven Polymorphic Behaviour of the Third PDZ Domain of PSD95: The Role of Electrostatic Interactions. Crystals 2023, 13, 218. https://doi.org/10.3390/cryst13020218
Salinas-García MC, Plaza-Garrido M, Gavira JA, Murciano-Calles J, Andújar-Sánchez M, Ortiz-Salmerón E, Martinez JC, Cámara-Artigas A. pH-Driven Polymorphic Behaviour of the Third PDZ Domain of PSD95: The Role of Electrostatic Interactions. Crystals. 2023; 13(2):218. https://doi.org/10.3390/cryst13020218
Chicago/Turabian StyleSalinas-García, Mª Carmen, Marina Plaza-Garrido, Jose A. Gavira, Javier Murciano-Calles, Montserrat Andújar-Sánchez, Emilia Ortiz-Salmerón, Jose C. Martinez, and Ana Cámara-Artigas. 2023. "pH-Driven Polymorphic Behaviour of the Third PDZ Domain of PSD95: The Role of Electrostatic Interactions" Crystals 13, no. 2: 218. https://doi.org/10.3390/cryst13020218