
Crystal Structure and Spectroscopic Analysis of 3-Diethoxyphosphoryl-28-[1-(1-deoxy-β-D-glucopyranosyl)-1H-1,2,3-triazol-4-yl]carbonylbetulin
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of 3-Diethoxyphosphoryl-28-[1-(1-deoxy-β-D-glucopyranosyl)-1H-1,2,3-triazol-4-yl]carbonylbetulin
2.2. X-ray Diffraction Analysis
2.3. Hirshfeld Surface Analysis
2.4. FT-IR Spectra
2.5. Computational Details
3. Results and Discussion
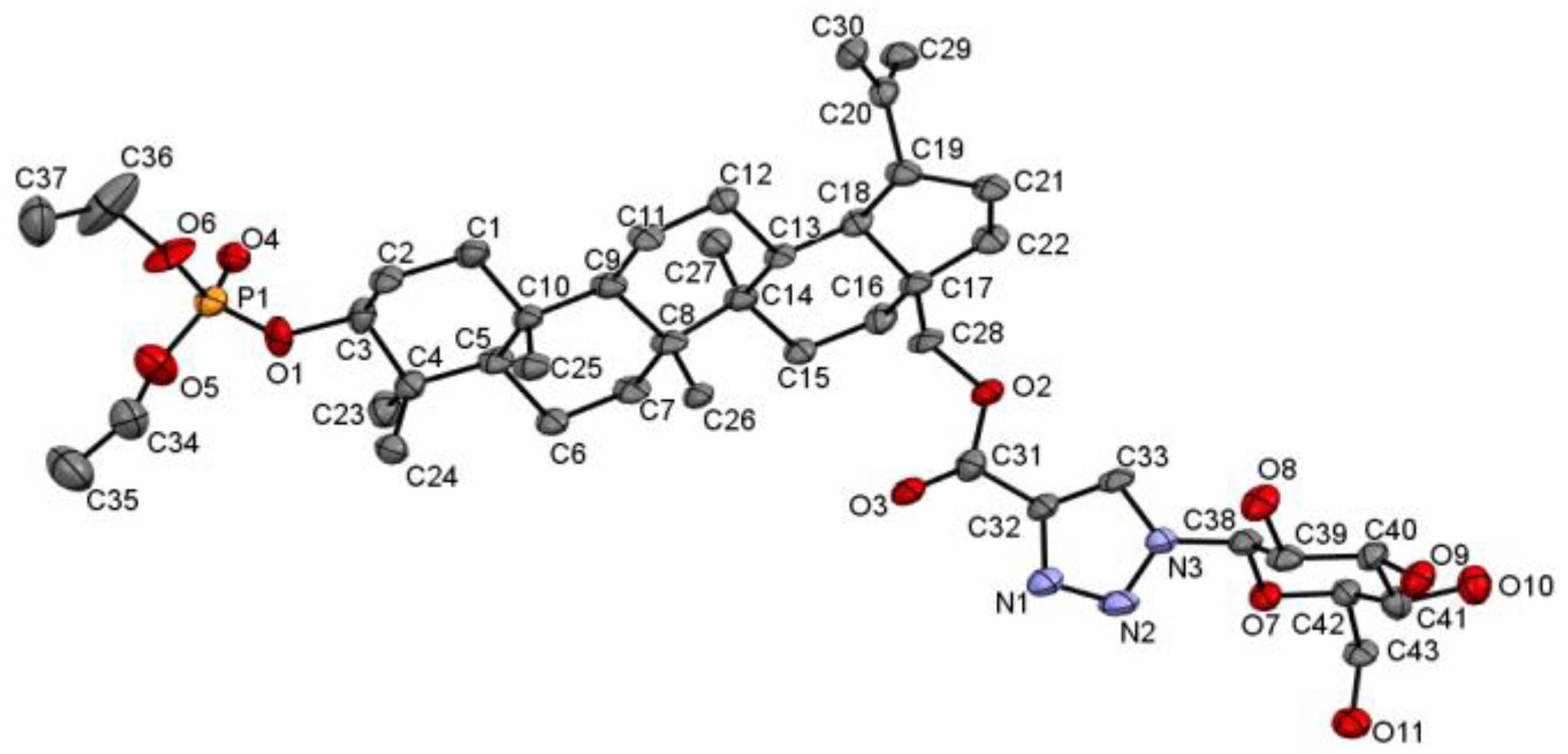
3.1. Crystal Structure
3.2. Hirshfeld Surface
3.3. Vibrational Assignments
3.4. Chemical Reactivity Descriptors
3.5. MEP Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhao, J.; Li, R.; Pawlak, A.; Henklewska, M.; Sysak, A.; Wen, L.; Yi, J.E.; Obmińska-Mrukowicz, B. Antitumor activity of betulinic acid and betulin in canine cancer cell lines. In Vivo 2018, 32, 1081–1088. [Google Scholar] [CrossRef]
- Schwiebs, A.; Radeke, H.H. Immunopharmacological activity of betulin in inflammation-associated carcinogenesis. Anticancer Agents Med. Chem. 2018, 18, 645–651. [Google Scholar] [CrossRef]
- Buko, V.; Zavodnik, I.; Palecz, B.; Stepniak, A.; Kirko, S.; Shlyahtun, A.; Misiuk, W.; Belonovskaya, E.; Lukivskaya, O.; Naruta, E.; et al. Betulin/2-hydroxypropyl-β-cyclodextrin inclusion complex: Physicochemical characterization and hepatoprotective activity. J. Mol. Liq. 2020, 309, 113118. [Google Scholar] [CrossRef]
- Chaniad, P.; Sudsai, T.; Septama, A.W.; Chukaew, A.; Tewtrakul, S. Evaluation of anti-HIV-1 integrase and anti-Inflammatory activities of compounds from betula alnoides buch-ham. Adv. Pharmacol. Sci. 2019, 2019, 2573965. [Google Scholar] [CrossRef] [PubMed]
- Javed, S.; Mahmood, Z.; Khan, K.M.; Sarker, S.D.; Javaid, A.; Khan, I.H.; Shoaib, A. Lupeol acetate as a potent antifungal compound against opportunistic human and phytopathogenic mold Macrophomina phaseolina. Sci. Rep. 2021, 11, 8417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ma, Z.; Wang, R.; Zhu, M. Synthesis and characterization of methacrylate-functionalized betulin derivatives as antibacterial comonomer for dental restorative resins. ACS Biomater. Sci. Eng. 2021, 7, 3132–3140. [Google Scholar] [CrossRef]
- Li, J.; Jiang, B.; Chen, C.; Fan, B.; Huang, H.; Chen, G. Biotransformation of betulin by Mucor subtilissimus to discover anti-inflammatory derivatives. Phytochemistry 2019, 166, 112076. [Google Scholar] [CrossRef]
- Wiemer, A.J. Metabolic efficacy of phosphate prodrugs and the remdesivir paradigm. ACS Pharmacol. Transl. Sci. 2020, 3, 613–626. [Google Scholar] [CrossRef]
- Chrobak, E.; Kadela-Tomanek, M.; Bębenek, E.; Marciniec, K.; Wietrzyk, J.; Trynda, J.; Pawełczak, B.; Kusz, J.; Kasperczyk, J.; Chodurek, E.; et al. New phosphate derivatives of betulin as anticancer agents: Synthesis, crystal structure, and molecular docking study. Bioorg. Chem. 2019, 87, 613–628. [Google Scholar] [CrossRef]
- Chrobak, E.; Jastrzębska, M.; Bębenek, E.; Kadela-Tomanek, M.; Marciniec, K.; Latocha, M.; Wrzalik, R.; Kusz, J.; Boryczka, S. Molecular structure, in vitro anticancer study and molecular docking of new phosphate derivatives of betulin. Molecules 2021, 26, 737. [Google Scholar] [CrossRef] [PubMed]
- Talele, T.T. Acetylene group, friend or foe in medicinal chemistry. J. Med. Chem. 2020, 63, 5625–5663. [Google Scholar] [CrossRef]
- Boryczka, S.; Bębenek, E.; Wietrzyk, J.; Kempińska, K.; Jastrzębska, M.; Kusz, J.; Nowak, M. Synthesis, structure and cytotoxic activity of new acetylenic derivatives of betulin. Molecules 2013, 18, 4526–4543. [Google Scholar] [CrossRef]
- Bębenek, E.; Kadela-Tomanek, M.; Chrobak, E.; Wietrzyk, J.; Sadowska, J.; Boryczka, S. New acetylenic derivatives of betulin and betulone, synthesis and cytotoxic activity. Med. Chem. Res. 2017, 26, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khadem, F.; Behrouj, H.; Aghanoori, M.R.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and its derivatives as novel compounds with different pharmacological effects. Biotechnol. Adv. 2020, 38, 107409. [Google Scholar] [CrossRef]
- Jiang, X.; Hao, X.; Jing, L.; Wu, G.; Kang, D.; Liu, X.; Zhan, P. Recent applications of click chemistry in drug discovery. Expert. Opin. Drug Discov. 2019, 14, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M. 1,2,3-Triazole hybrids as anticancer agents: A review. Arch. Pharm. 2022, 355, e2100158. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhao, S.J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef]
- Bębenek, E.; Jastrzębska, M.; Kadela-Tomanek, M.; Chrobak, E.; Orzechowska, B.; Zwolińska, K.; Latocha, M.; Mertas, A.; Czuba, Z.; Boryczka, S. Novel triazole hybrids of betulin: Synthesis and biological activity profile. Molecules 2017, 22, 1876–1898. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Jastrzębska, M.; Marciniec, K.; Chrobak, E.; Bębenek, E.; Latocha, M.; Kuśmierz, D.; Boryczka, S. Design, synthesis and biological activity of 1,4-quinone moiety attached to betulin derivatives as potent DT-diaphorase substrate. Bioorg. Chem. 2021, 106, 104478. [Google Scholar] [CrossRef]
- Grishko, V.V.; Tolmacheva, I.A.; Nebogatikov, V.O.; Galaiko, N.V.; Nazarov, A.V.; Dmitriev, M.V.; Ivshina, I.B. Preparation of novel ring-A fused azole derivatives of betulin and evaluation of their cytotoxicity. Eur. J. Med. Chem. 2017, 125, 629–639. [Google Scholar] [CrossRef]
- Csuk, R.; Deigner, H.P. The potential of click reactions for the synthesis of bioactive triterpenes. Bioorg. Med. Chem. Lett. 2019, 29, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Kadela-Tomanek, M.; Jastrzębska, M.; Marciniec, K.; Chrobak, E.; Bębenek, E.; Boryczka, S. Lipophilicity, pharmacokinetic properties, and molecular docking study on SARS-CoV-2 target for betulin triazole derivatives with attached 1,4-quinone. Pharmaceutics 2021, 13, 781. [Google Scholar] [CrossRef]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. Engl. 2002, 4, 49–76. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Sugiyama, J.; Chanzy, H.; Langan, P. Crystal structure and hydrogen bonding system in cellulose I(alpha) from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 2003, 125, 14300–14306. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Wolff, S.; Grimwood, D.J.; McKinnon, J.; Jayatilaka, D.; Spackman, M. CrystalExplorer 3.0.; University of Western Australia: Perth, Australia, 2001. [Google Scholar]
- Spackman, M.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision A. 03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Foresman, J.; Frisch, E. Exploring Chemistry with Electronic Structure Methods: A Guide to Using Gaussian; Gaussian, Inc.: Pittsburg, PA, USA, 1996. [Google Scholar]
- Politzer, P.; Laurence, P.; Jayasuriya, K. Molecular electrostatic potentials: An effective tool for the elucidation of biochemical phenomena. Environ. Health Perspect. 1985, 61, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.; Millam, J. GaussView Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Guerroudj, A.R.; Boukabcha, N.; Benmohammed, A.; Dege, N.; Belkafouf, N.E.H.; Khelloul, N.; Djafri, A.; Chouaih, A. Synthesis, crystal structure, vibrational spectral investigation, intermolecular interactions, chemical reactivity, NLO properties and molecular docking analysis on (E)-N-(4-nitrobenzylidene)-3-chlorobenzenamine: A combined experimental and theoretical study. J. Mol. Struct. 2021, 1240, 130589. [Google Scholar]
- Anthony, L.A.; Rajaraman, D.; Sundararajan, G.; Suresh, M.; Nethaji, P.; Jaganathan, R.; Poomani, K. Synthesis, crystal structure, Hirshfeld surface analysis, DFT, molecular docking and molecular dynamic simulation studies of (E)-2,6-bis(4-chlorophenyl)-3-methyl-4-(2-(2,4,6-trichlorophenyl)hydrazono)piperidine derivatives. J. Mol. Struct. 2022, 1266, 133483. [Google Scholar] [CrossRef]
- Silverstein, R.; Webster, F.; Kiemle, D.; Bryce, D. Spectrometric Identification of Organic Compounds, 8th ed.; Wiley: New York, NY, USA, 2014. [Google Scholar]
- Krishnakumar, V.; Dheivamalar, S.; Xavier, R.J.; Balachandran, V. Analysis of vibrational spectra of 4-amino-2,6-dichloropyridine and 2-chloro-3,5-dinitropyridine based on density functional theory calculations. Spectrochim. Acta A Biomol. Spectosc. 2006, 65, 147–154. [Google Scholar] [CrossRef]
- Kalsi, P.S. Spectroscopy of Organic Compounds; New Age International: New York, NY, USA, 2007. [Google Scholar]
- Boukabcha, N.; Djafri, A.; Megrouss, Y.; Tamer, O.; Avcı, D.; Tuna, M.; Dege, N.; Chouaih, A.; Atalay, Y.; Djafri, A.F. Hamzaoui, Synthesis, crystal structure, spectroscopic characterization and nonlinear optical properties of (Z)-N’-(2,4-dinitrobenzylidene)-2-(quinolin-8-yloxy) acetohydrazide. J. Mol. Struct. 2019, 1194, 112–123. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies; Wiley-Blackwell: Hoboken, NJ, USA, 2004. [Google Scholar]
- Muruganantham, N.; Sivakumar, R.; Anbalagan, N.; Gunasekaran, V.; Leonard, J.T. Synthesis, anticonvulsant and antihypertensive activities of 8-substituted quinoline derivatives. Biol. Pharm. Bull. 2004, 27, 1683–1687. [Google Scholar] [CrossRef]
- Saral, A.; Sudha, P.; Muthu, S.; Sevvanthi, S.; Sangeetha, P.; Selvakumari, S. Vibrational spectroscopy, quantum computational and molecular docking studies on 2-chloroquinoline-3-carboxaldehyde. Heliyon 2021, 7, e07529. [Google Scholar] [CrossRef] [PubMed]
- Manoj, K.P.; Elangovan, N.; Chandrasekar, S. Synthesis, XRD, hirshfeld surface analysis, ESP, HOMO-LUMO, quantum chemical modeling and anticancer activity of di(p-methyl benzyl)(dibromo)(1,10-phenanthroline) tin(IV) complex. Inorg. Chem. Commun. 2022, 139, 109324. [Google Scholar] [CrossRef]
- Parvathy, G.; Kaliammal, R.; Velsankar, K.; Kumar, M.K.; Sankaranarayanan, K.; Sudhahar, S. Studies on structural, optical, homo-lumo and mechanical properties of piperazinium p-hydroxybenzoate monohydrate single crystal for nonlinear optical applications. Chem. Phys. Lett. 2020, 758, 137934. [Google Scholar] [CrossRef]
- Aihara, J. Reduced HOMO−LUMO gap as an Index of kinetic stability for polycyclic aromatic hydrocarbons. J. Phys. Chem. A 1999, 103, 7487–7495. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A.; Chermette, H. Is hyper-hardness more chemically relevant than expected? J. Mol. Model. 2013, 19, 2893–2900. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Buvaneswari, M.; Santhakumari, R.; Usha, C.; Jayasree, R.; Sagadevan, S. Synthesis, growth, structural, spectroscopic, optical, thermal, DFT, HOMO–LUMO, MEP, NBO analysis and thermodynamic properties of vanillin isonicotinic hydrazide single crystal. J. Mol. Struct. 2021, 1243, 130856. [Google Scholar] [CrossRef]
- Kumar, C.S.C.; Balachandran, V.; Fun, H.K.; Chandraju, S.; Quah, C.K. Synthesis, crystal growth, single crystal X-ray analysis and vibrational spectral studies of (2E)-3-(2-chloro-4-fluorophenyl)-1-(3,4-dimethoxyphenyl)prop-2-en-1-one: A combined DFT study. J. Mol. Struct. 2015, 1100, 299–310. [Google Scholar] [CrossRef]
- Pęcak, P.; Chrobak, E.; Kadela-Tomanek, M. The applicat ion of the RP-TLC method and in silico studies to the evaluation of pharmacokinet ic parameters of acylated betulin derivatives. In Badania i rozwój Młodych Naukowców w Polsce. Nauki Medyczne i Nauki o Zdrowiu. Część 3; Młodzi Naukowcy: Poznan, Poland, 2021; pp. 72–79. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D–H···A | D–H | H···A | D···A | <(DHA) |
|---|---|---|---|---|
| O8-H8···O4 i | 0.84 | 1.950 | 2.780 | 169.47 |
| O9-H9···O4 i | 0.84 | 1.962 | 2.796 | 172.56 |
| Experimental (cm−1) | Calculated (cm−1) | Assignment |
|---|---|---|
| 3387 | 3509 3413 | νO-H νC-H |
| 2968 | 2970 | νC-Hisopropenyl |
| 2940 | 2945 | νC-Hbetulin |
| 2872 | 2849 | νC-Hbetulin |
| 1721 | 1669 | νC=O |
| 1450 | 1496 | δC-Hbetulin |
| 1391 | 1427 | νC-Ntriazole |
| 1367 | 1409 | δC-Hethyl |
| 1230 | 1209 | δC-Hsugar |
| 1167 | 1166 | δC-Htriazol |
| 1132 | 1126 | νN=N νC-O |
| 1065 | 1087 | δC-Hbetulin |
| 1016 | 1036 | νP=O |
| 983 | 993 | δC-Hsugar |
| 880 | 897 | δC-O |
| Parameters [eV] | 6-311+G(d.p) |
|---|---|
| EHOMO | −6.526 |
| ELUMO | −1.580 |
| ΔELUMO−EHOMO | 4.946 |
| Ionization potential (I) | 6.526 |
| Electron affinity (A) | 1.580 |
| Hardness (η) | 2.473 |
| Softness (S) | 0.202 |
| Chemical potential (μ) | −4.053 |
| Electronegativity (ϰ) | 4.053 |
| Electrophilicity index (ω) | 3.321 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadela-Tomanek, M.; Bębenek, E.; Sokal, A.; Książek, M.; Chrobak, E. Crystal Structure and Spectroscopic Analysis of 3-Diethoxyphosphoryl-28-[1-(1-deoxy-β-D-glucopyranosyl)-1H-1,2,3-triazol-4-yl]carbonylbetulin. Crystals 2023, 13, 1488. https://doi.org/10.3390/cryst13101488
Kadela-Tomanek M, Bębenek E, Sokal A, Książek M, Chrobak E. Crystal Structure and Spectroscopic Analysis of 3-Diethoxyphosphoryl-28-[1-(1-deoxy-β-D-glucopyranosyl)-1H-1,2,3-triazol-4-yl]carbonylbetulin. Crystals. 2023; 13(10):1488. https://doi.org/10.3390/cryst13101488
Chicago/Turabian StyleKadela-Tomanek, Monika, Ewa Bębenek, Arkadiusz Sokal, Maria Książek, and Elwira Chrobak. 2023. "Crystal Structure and Spectroscopic Analysis of 3-Diethoxyphosphoryl-28-[1-(1-deoxy-β-D-glucopyranosyl)-1H-1,2,3-triazol-4-yl]carbonylbetulin" Crystals 13, no. 10: 1488. https://doi.org/10.3390/cryst13101488