
Interatomic Potential to Predict the Favored Glass-Formation Compositions and Local Atomic Arrangements of Ternary Al-Ni-Ti Metallic Glasses


Abstract
:1. Introduction
2. Model and Methods
2.1. Potential Formalism
2.2. Atomic Simulation Models
3. Results and Discussion
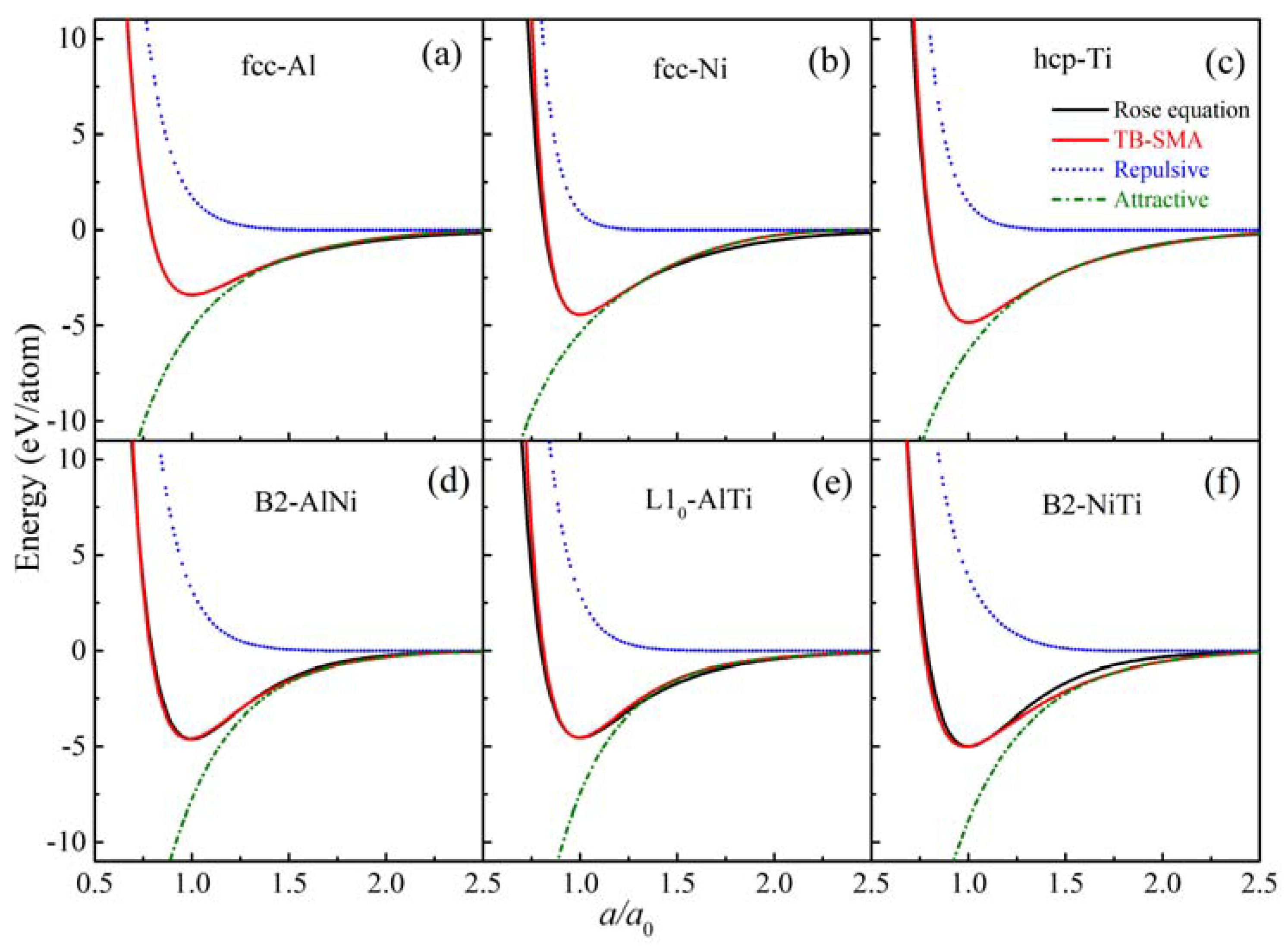
3.1. Construction of Al-Ni-Ti Interatomic Potential
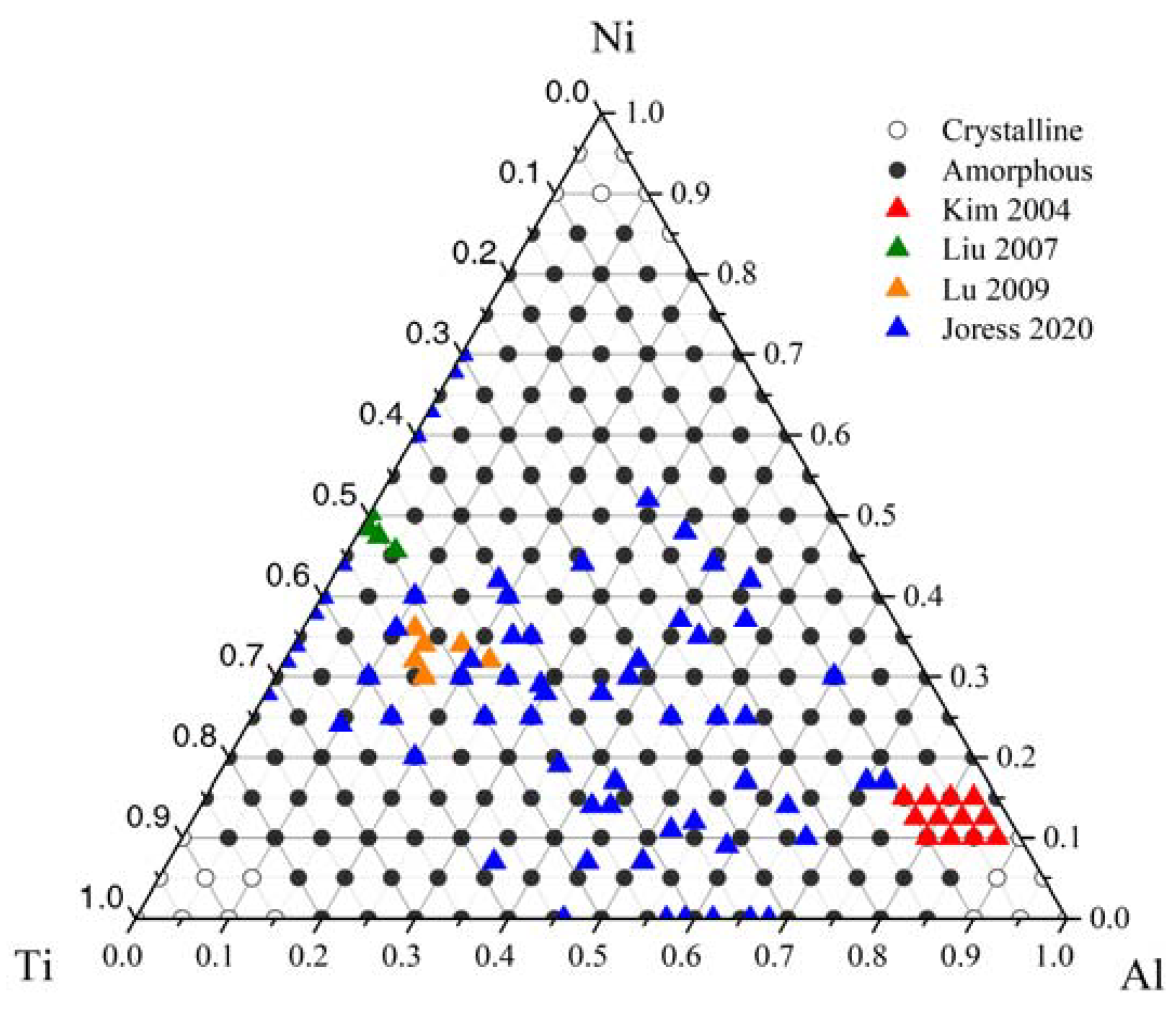
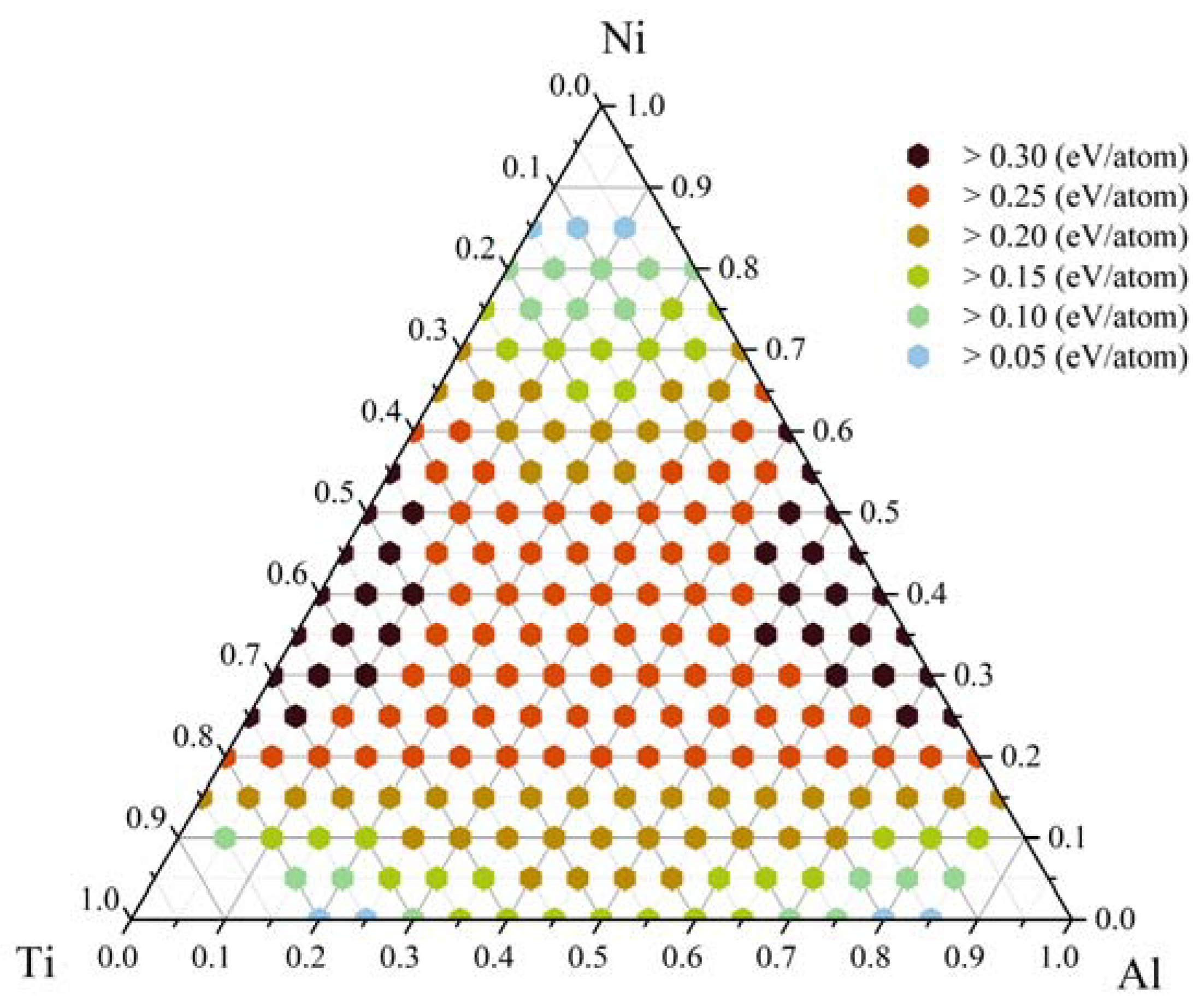
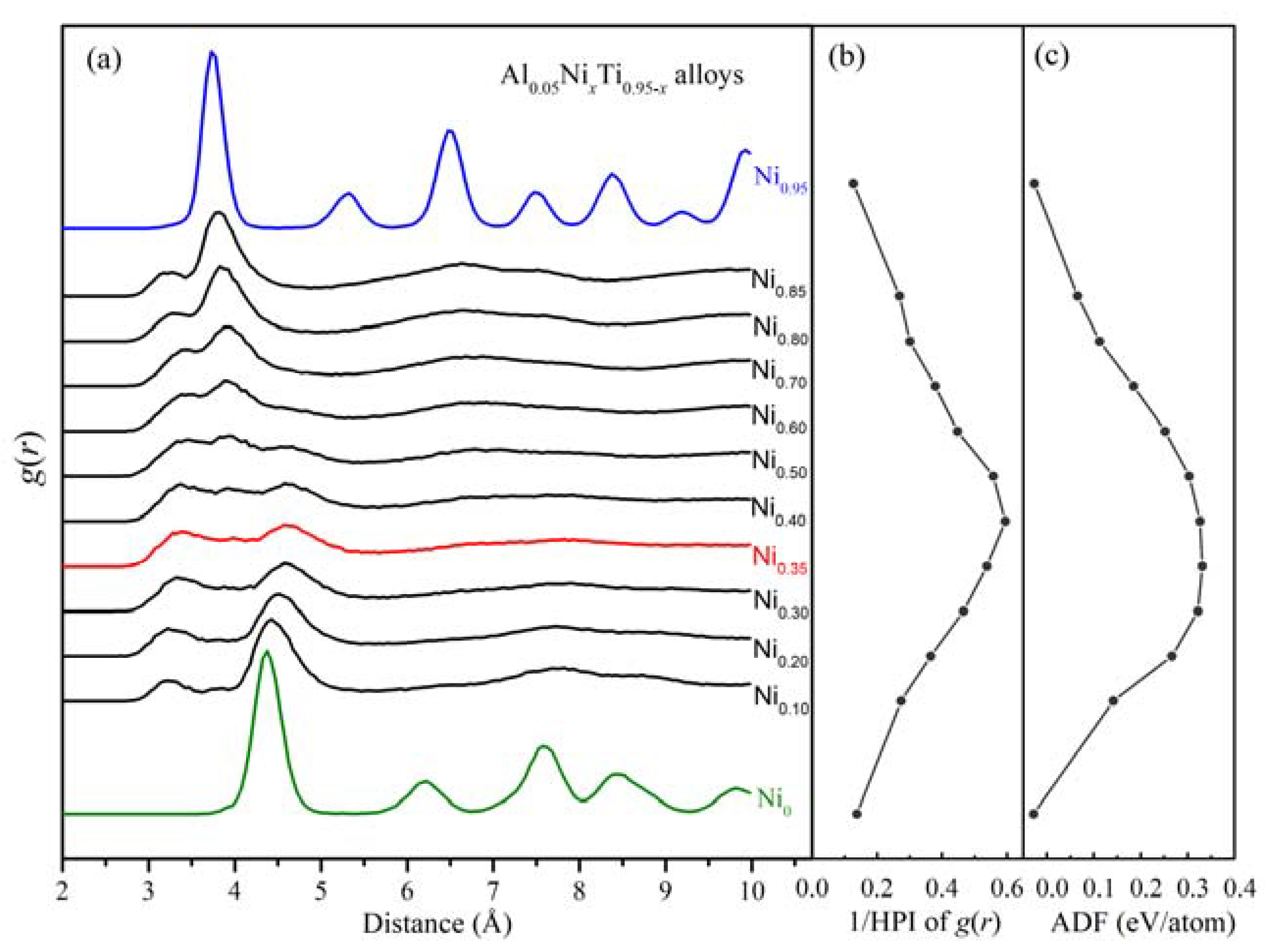
3.2. Glass-Formation Range and Ability of the Al-Ni-Ti System
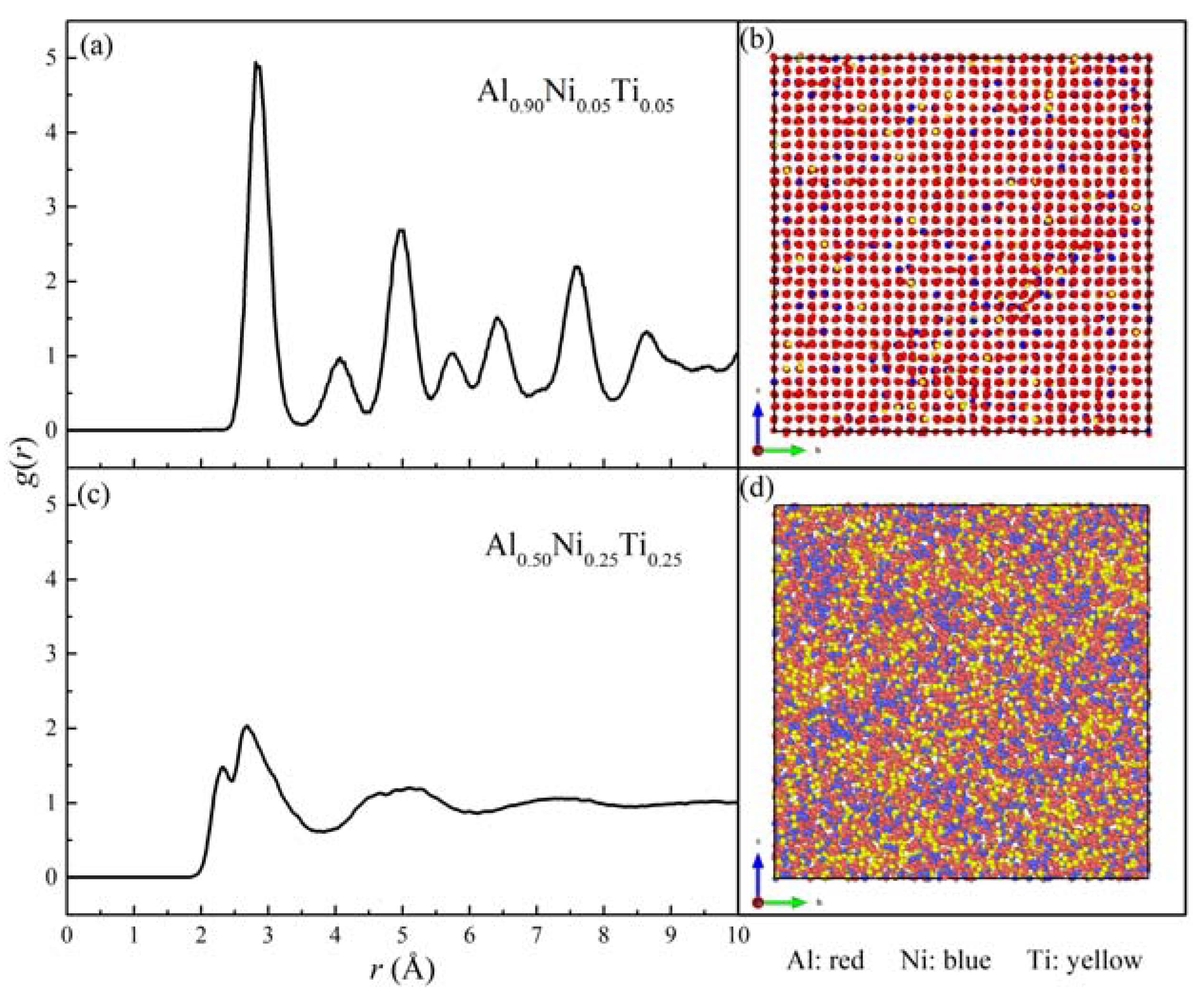
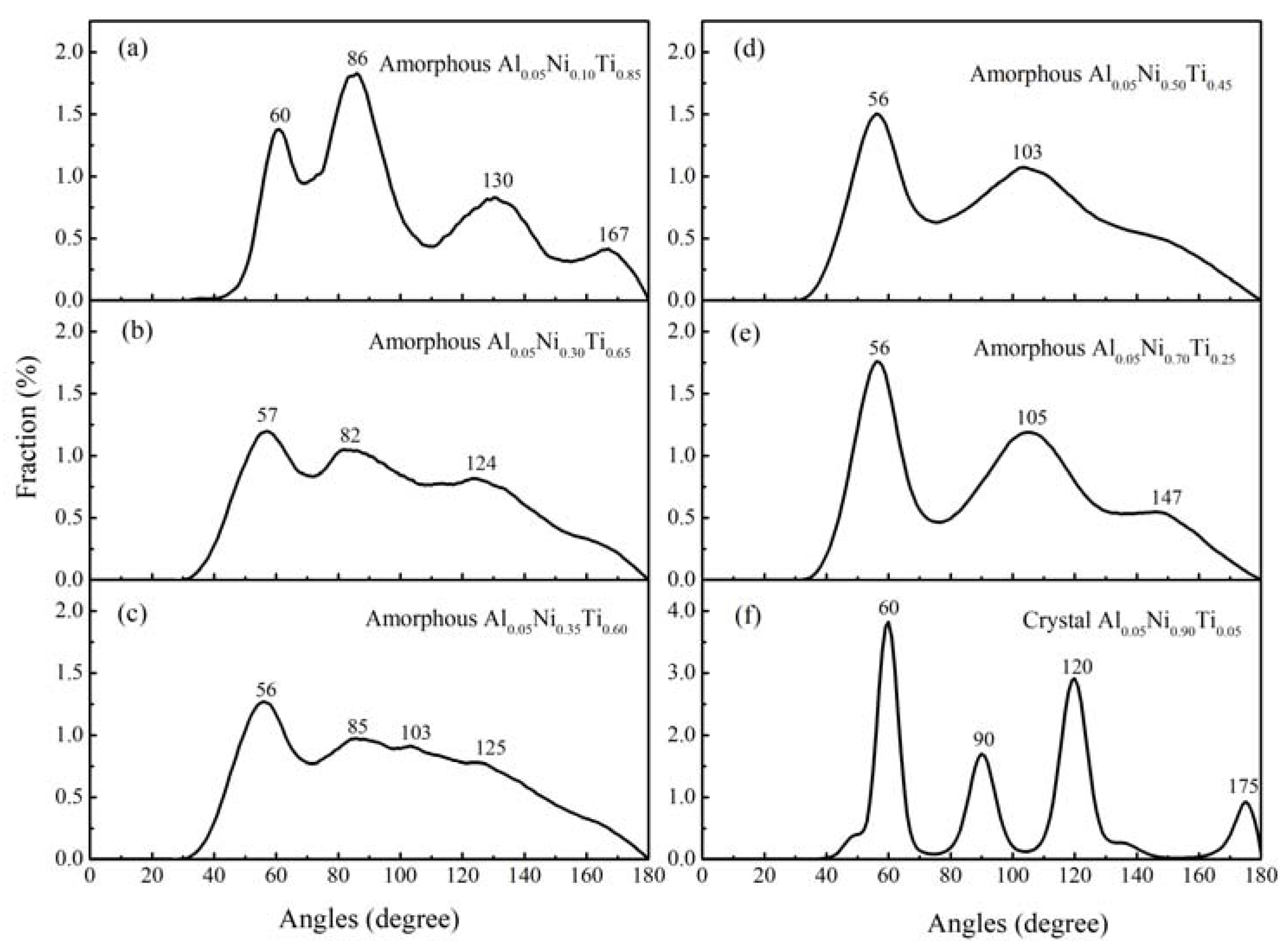
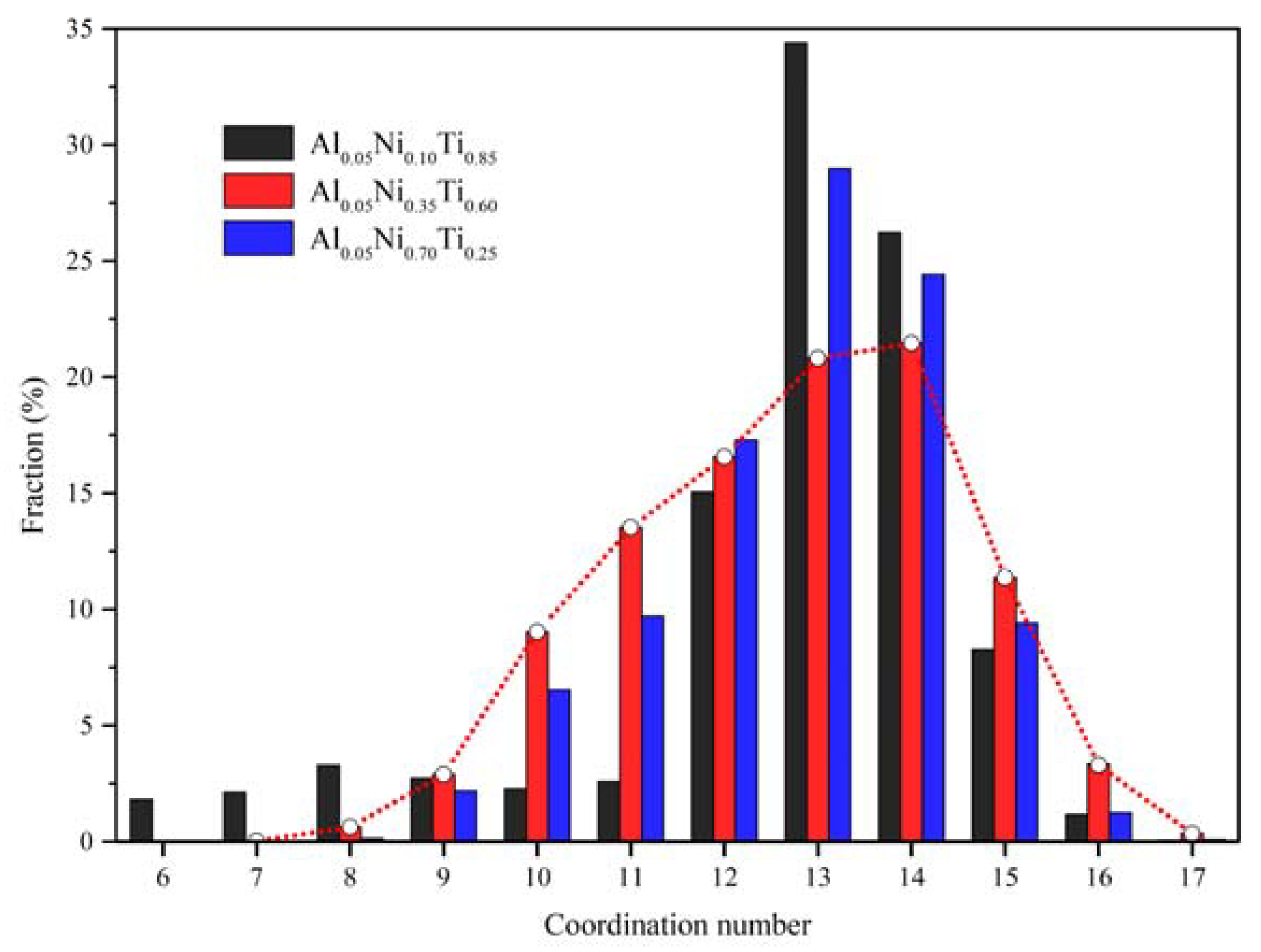
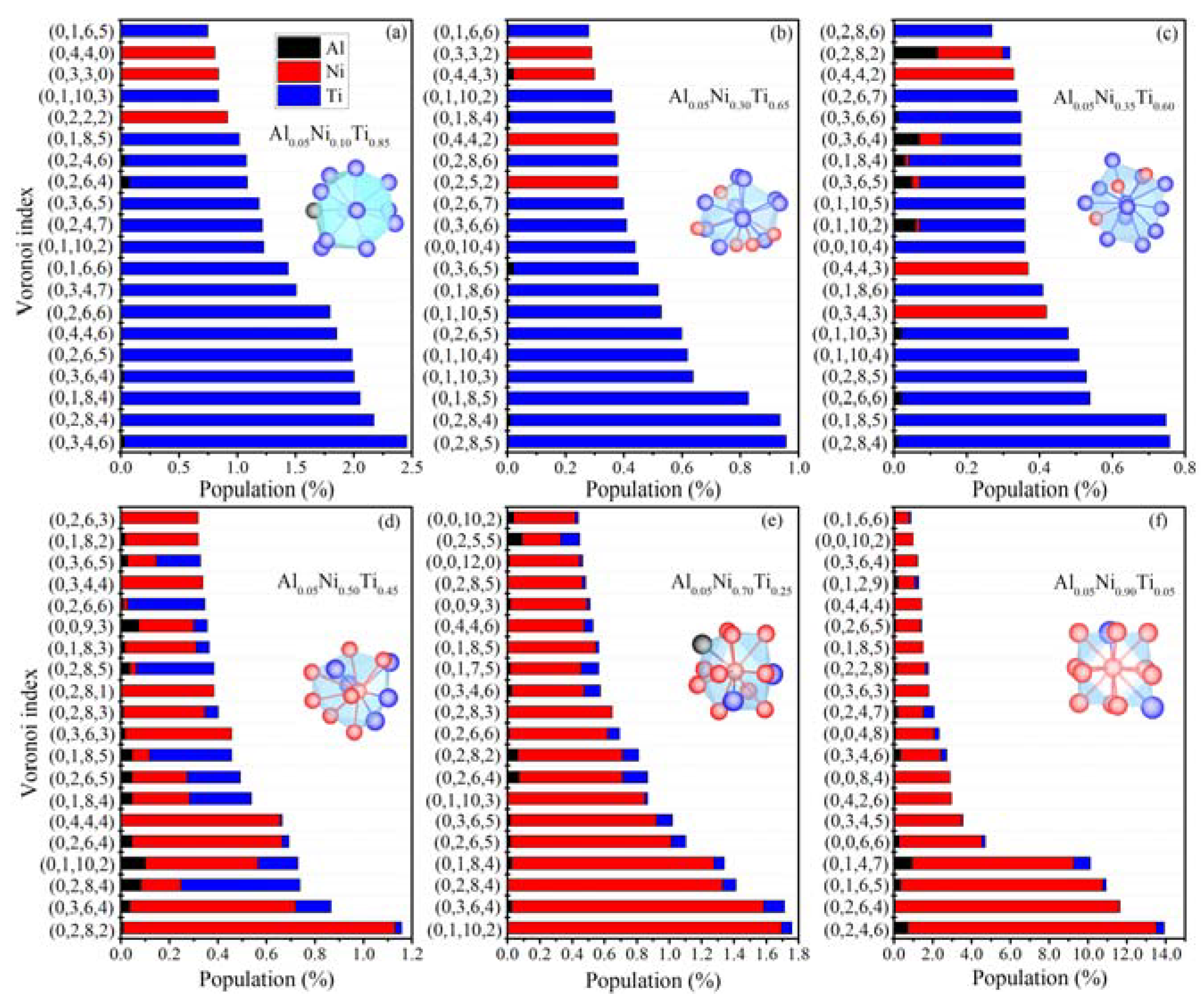
3.3. Local Atomic Arrangements of Al-Ni-Ti Metallic Glasses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Pan, J.; Ivanov, Y.P.; Zhou, W.H.; Li, Y.; Greer, A.L. Strain-hardening and suppression of shear-banding in rejuvenated bulk metallic glass. Nature 2020, 578, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Halim, Q.; Mohamed, N.A.N.; Rejab, M.R.M.; Naim, W.N.W.A.; Ma, Q. Metallic glass properties, processing method and development perspective: A review. Int. J. Adv. Manuf. Technol. 2021, 112, 1231–1258. [Google Scholar] [CrossRef]
- Huaa, N.; Wang, W.; Wang, Q.; Ye, Y.; Lin, S.; Zhang, L.; Guo, Q.; Brechtl, J.; Liaw, P.K. Mechanical, corrosion, and wear properties of biomedical Ti–Zr–Nb–Ta–Mo high entropy alloys. J. Alloys Compd. 2021, 861, 157997. [Google Scholar] [CrossRef]
- Klement, W.; Willens, R.H.; Duwez, P. Non-crystalline structure in solidified gold–silicon alloys. Nature 1960, 187, 869–870. [Google Scholar] [CrossRef]
- Schroers, J. Bulk Metallic Glasses. Phys. Today 2013, 66, 32–37. [Google Scholar] [CrossRef]
- Turnbull, D. Under what conditions can a glass be formed? Contemp. Phys. 1969, 10, 473–488. [Google Scholar] [CrossRef]
- Liu, B.; Johnson, W.L.; Nicolet, M.; Lau, S.S. Structural difference rule for amorphous alloy formation by ion mixing. Appl. Phys. Lett. 1983, 42, 45–47. [Google Scholar] [CrossRef]
- Egami, T.; Waseda, Y. Atomic size effect on the formability of metallic glasses. J. Non-Cryst. Solids 1984, 64, 113–134. [Google Scholar] [CrossRef]
- Inoue, A. Recent progress of Zr-based bulk amorphous alloys. Sci. Rep.-Res. Inst. Tohoku Univ. Ser. A 1996, 42, 1–12. [Google Scholar]
- Inoue, A. Stabilization of supercooled liquid and opening-up of bulk glassy alloys. Proc. Jpn. Acad. Ser. B 1997, 73, 19–24. [Google Scholar] [CrossRef]
- Inoue, A. Bulk Amorphous Alloys: Preparation and Functional Characteristics; Trans Tech Publications Ltd.: Zurich, Switzerland, 1998; pp. 1–15. [Google Scholar]
- Li, J.H.; Zhao, S.Z.; Dai, Y.; Cui, Y.Y.; Liu, B.X. Formation and structure of Al-Zr metallic glasses studied by Monte Carlo simulations. J. Appl. Phys. 2011, 109, 113538. [Google Scholar] [CrossRef]
- Luo, S.; Li, J.; Liu, J.; Liu, B. Atomic modeling to design favored compositions for the ternary Ni–Nb–Zr metallic glass formation. Acta Mater. 2014, 76, 482–492. [Google Scholar] [CrossRef]
- Wada, T.; Jiang, J.; Yubuta, K.; Kato, H.; Takeuchi, A. Septenary Zr–Hf–Ti–Al–Co–Ni–Cu high-entropy bulk metallic glasses with centimeter-scale glass-forming ability. Materialia 2019, 7, 100372. [Google Scholar] [CrossRef]
- Joress, H.; DeCost, B.L.; Sarker, S.; Braun, T.M.; Jilani, S.; Smith, R.; Ward, L.; Laws, K.J.; Mehta, A.; Hattrick-Simpers, J.R. A high-throughput structural and electrochemical study of metallic glass formation in Ni–Ti–Al. ACS Comb. Sci. 2020, 22, 330–338. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, Z.; Chen, Y.; Liang, X.; Shen, B. Composition optimization of Al-Ni-Ti alloys based on glass-forming ability and preparation of amorphous coating with good wear resistance by plasma spray. Surf. Coat. Technol. 2021, 408, 126800. [Google Scholar] [CrossRef]
- Yang, B.; Yao, J.; Zhang, J.; Yang, H.; Wang, J.; Ma, E. Al-rich bulk metallic glasses with plasticity and ultrahigh specific strength. Scr. Mater. 2009, 61, 423–426. [Google Scholar] [CrossRef]
- Fan, C.; Yue, X.; Inoue, A.; Liu, C.-T.; Shen, X.; Liaw, P.K. Recent topics on the structure and crystallization of Al-based glassy alloys. Mater. Res. 2019, 22. [Google Scholar] [CrossRef]
- Pang, S.; Zhang, T.; Asami, K.; Inoue, A. Bulk glassy Ni (Co–) Nb–Ti–Zr alloys with high corrosion resistance and high strength. Mater. Sci. Eng. A 2004, 375, 368–371. [Google Scholar] [CrossRef]
- Ishida, M.; Takeda, H.; Nishiyama, N.; Kita, K.; Shimizu, Y.; Saotome, Y.; Inoue, A. Wear resistivity of super-precision microgear made of Ni-based metallic glass. Mater. Sci. Eng. A 2007, 449–451, 149–154. [Google Scholar] [CrossRef]
- Calin, M.; Gebert, A.; Ghinea, A.C.; Gostin, P.F.; Abdi, S.; Mickel, C.; Eckert, J. Designing biocompatible Ti-based metallic glasses for implant applications. Mater. Sci. Eng. C 2013, 33, 875–883. [Google Scholar] [CrossRef]
- Li, J.; Bai, J.; Wang, J.; Kou, H.; Hu, R.; Fu, H. Deformation behavior of a Ti-based bulk metallic glass composite with excellent cryogenic mechanical properties. Mater. Des. 2014, 53, 737–740. [Google Scholar] [CrossRef]
- Inoue, A.; Takeuchi, A. Recent development and application products of bulk glassy alloys. Acta Mater. 2011, 59, 2243–2267. [Google Scholar] [CrossRef]
- Gong, P.; Yao, K.F.; Shao, Y. Lightweight Ti–Zr–Be–Al bulk metallic glasses with improved glass-forming ability and compressive plasticity. J. Non-Cryst. Solids 2012, 358, 2620–2625. [Google Scholar] [CrossRef]
- Guo, F.; Wang, H.-J.; Poon, S.J.; Shiflet, G.J. Ductile titanium-based glassy alloy ingots. Appl. Phys. Lett. 2005, 86, 091907. [Google Scholar] [CrossRef]
- Rosato, V.; Guillope, M.; Legrand, B. Thermodynamical and structural properties of fcc transition metals using a simple tight-binding model. Philos. Mag. A 1989, 59, 321–336. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B Condens. Matter 1993, 48, 13115–13118. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B Condens. Matter 1996, 54, 11169. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Li, J.; Dai, Y.; Cui, Y.; Liu, B. Atomistic theory for predicting the binary metallic glass formation. Mater. Sci. Eng. R Rep. 2011, 72, 1–28. [Google Scholar] [CrossRef]
- Cai, B.; Li, J.; Lai, W.; Liu, J.; Liu, B. Construction of Al-Mg-Zn Interatomic Potential and the Prediction of Favored Glass Formation Compositions and Associated Driving Forces. Materials 2022, 15, 2062. [Google Scholar] [CrossRef] [PubMed]
- Hellenbrandt, M. The inorganic crystal structure database (ICSD)—Present and future. Crystallogr. Rev. 2004, 10, 17–22. [Google Scholar] [CrossRef]
- News, S.T.B. CRC Handbook of Chemistry and Physics, A Ready-Reference Book of Chemical and Physical Data, 88th ed.; Scitech Book News: Portland, OR, USA, 2007. [Google Scholar]
- Kittel, C. Introduction to solid state physics. Am. J. Phys. 1967, 35, 547–548. [Google Scholar] [CrossRef]
- Bradley, A.J.; Taylor, A. An X-ray Analysis of the Nickel-Aluminium System. Proc. R. Soc. A 1937, 159, 56–72. [Google Scholar]
- Ageev, N.V.; Guseva, L.N. Experimental study of the electron density in crystals. Bull. Acad. Sci. USSR Div. Chem. Sci. 1952, 1, 31–39. [Google Scholar] [CrossRef]
- Rao, P.V.M.; Suryanarayana, S.V.; Murthy, K.S.; Naidu, S.V.N. The high-temperature thermal expansion of Ni3Al measured by X-ray diffraction and dilation methods. J. Phys. Condens. Matter 1989, 1, 5357. [Google Scholar] [CrossRef]
- Norby, P.; Nørlund Christensen, A. Preparation and Structure of Al3Ti. Acta Chem. Scand. 1986, 40, 157–159. [Google Scholar] [CrossRef]
- Duwez, P.; Taylor, J.L. Crystal structure of TiAl. JOM-J. Miner. Met. Mater. Soc. 1952, 4, 70–71. [Google Scholar] [CrossRef]
- Penaloza, V.A.; Houska, C.R. Refinements on the X-ray intensities from Ti3-2Al. An. Congr. Nac. Metal. 3rd 1983, 54, A55–A59. [Google Scholar]
- Laves, F.; Wallbaum, H.J. Die Kristallstruktur von Ni3Ti und Si2Ti. Z. Krist.—Cryst. Mater. 1939, 101, 78–93. [Google Scholar] [CrossRef]
- Stüwe, H.P.; Shimomura, Y. Gitterkonstanten der kubisch raumzentrierten Phasen FeTi, CoTi, NiTi. Z. Met. 1960, 51, 180–181. [Google Scholar]
- Yurko, G.A.; Barton, J.W.; Parr, J.G. The crystal structure of Ti2Ni. Acta Crystallogr. 1959, 12, 909–911. [Google Scholar] [CrossRef]
- Kubaschewski, O.; Dench, W.A. The heats of formation in the systems titanium-aluminium and titanium-iron. Acta Metall. 1955, 3, 339–346. [Google Scholar] [CrossRef]
- Mercier, O.; Melton, K.N.; Gremaud, G.; Hägi, J. Single-crystal elastic constants of the equiatomic NiTi alloy near the martensitic transformation. J. Appl. Phys. 1980, 51, 1833–1834. [Google Scholar] [CrossRef]
- Kubaschewski, O.; Villa, H.; Dench, W.A. The reaction of titanium tetrachloride with hydrogen in contact with various refractories. Trans. Faraday Soc. 1956, 52, 214–222. [Google Scholar] [CrossRef]
- Ghosh, G. First-principles calculations of structural energetics of Cu–TM (TM = Ti, Zr, Hf) intermetallics. Acta Mater. 2007, 55, 3347–3374. [Google Scholar] [CrossRef]
- Prikhodko, S.V.; Ma, Y.; Ardell, A.J.; Isaak, D.G.; Carnes, J.D.; Moser, S. Elastic constants of face-centered cubic and L12 Ni-Si alloys: Composition and temperature dependence. Metall. Mater. Trans. A 2003, 34, 1863–1868. [Google Scholar] [CrossRef]
- Ko, W.-S.; Grabowski, B.; Neugebauer, J. Development and application of a Ni-Ti interatomic potential with high predictive accuracy of the martensitic phase transition. Phys. Rev. B 2015, 92, 134107. [Google Scholar] [CrossRef]
- Yan, X.; Grytsiv, A.; Rogl, P.; Pomjakushin, V.; Palm, M. The Heusler Phase Ti25(Fe50−x Ni x) Al25 (0 ≤ x≤ 50); Structure and Constitution. J. Phase Equilibria Diffus. 2008, 29, 500–508. [Google Scholar] [CrossRef]
- Lazurenko, D.V.; Lozanov, V.V.; Stark, A.; Pyczak, F.; Ruktuev, A.A.; Emurlaev, K.I.; Song, L.; Bataev, I.A.; Ivanov, I.V.; Ogneva, T.S.; et al. In situ synchrotron X-ray diffraction study of reaction routes in Ti-Al3Ti-based composites: The effect of transition metals on L12 structure stabilization. J. Alloys Compd. 2021, 875, 160004. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, W.T.; Kim, D.H. Formation and crystallization of Al–Ni–Ti amorphous alloys. Mater. Sci. Eng. A 2004, 385, 44–53. [Google Scholar] [CrossRef]
- Liu, K.; Duh, J. Effect of aluminum on the corrosion behavior of NiTiAl thin films. Appl. Surf. Sci. 2007, 253, 5268–5273. [Google Scholar] [CrossRef]
- Lu, B.-C.; Li, Y.; Xu, J. Optimal glass-forming composition and its correlation with eutectic reaction in the Ti–Ni–Al ternary system. J. Alloys Compd. 2009, 467, 261–267. [Google Scholar] [CrossRef]
- Chen, Q.J.; Fan, H.B.; Ye, L.; Ringer, S.; Sun, J.F.; Shen, J.; McCartney, D.G. Enhanced glass forming ability of Fe–Co–Zr–Mo–W–B alloys with Ni addition. Mater. Sci. Eng. A 2005, 402, 188–192. [Google Scholar] [CrossRef]
- Li, Y.H.; Zhang, W.; Dong, C.; Qiang, J.B.; Fukuhara, M.; Makino, A.; Inoue, A. Effects of Ni addition on the glass-forming ability, mechanical properties and corrosion resistance of Zr–Cu–Al bulk metallic glasses. Mater. Sci. Eng. A 2011, 528, 8551–8556. [Google Scholar] [CrossRef]
- Wang, A.; Zhang, M.; Zhang, J.; Men, H.; Shen, B.; Pang, S.; Zhang, T. Effect of Ni addition on the glass-forming ability and soft-magnetic properties of FeNiBPNb metallic glasses. Chin. Sci. Bull. 2011, 56, 3932–3936. [Google Scholar] [CrossRef]
- Rapaport, D.C.; Blumberg, R.L.; McKay, S.R.; Christian, W. The Art of Molecular Dynamics Simulation; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Sheng, H.W.; Luo, W.K.; Alamgir, F.; Bai, J.M.; Ma, E. Atomic packing and short-to-medium-range order in metallic glasses. Nature 2006, 439, 419–425. [Google Scholar] [CrossRef]
- Lazar, E.A.; Han, J.; Srolovitz, D.J. Topological framework for local structure analysis in condensed matter. Proc. Natl. Acad. Sci. USA 2015, 112, E5769–E5776. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.M.; Schmidt, S.; Schiøtz, J. Robust structural identification via polyhedral template matching. Model. Simul. Mater. Sci. Eng. 2016, 24, 055007. [Google Scholar] [CrossRef]
- Hirata, A.; Wada, T.; Obayashi, I.; Hiraoka, Y. Structural changes during glass formation extracted by computational homology with machine learning. Commun. Mater. 2020, 1, 98. [Google Scholar] [CrossRef]
- Ramos, P.; Herranz, M.; Foteinopoulou, K.; Karayiannis, N.; Laso, M. Identification of Local Structure in 2-D and 3-D Atomic Systems through Crystallographic Analysis. Crystals 2020, 10, 1008. [Google Scholar] [CrossRef]
- Pascuet, M.I.; Fernández, J.R. Atomic interaction of the MEAM type for the study of intermetallics in the Al–U alloy. J. Nucl. Mater. 2015, 467, 229–239. [Google Scholar] [CrossRef]
- Winey, J.M.; Kubota, A.; Gupta, Y.M. Thermodynamic approach to determine accurate potentials for molecular dynamics simulations: thermoelastic response of aluminum. Model. Simul. Mater. Sci. Eng. 2010, 18, 055004. [Google Scholar] [CrossRef]
- Zhakhovskii, V.V.; Inogamov, N.A.; Petrov, Y.V.; Ashitkov, S.I.; Nishihara, K. Molecular dynamics simulation of femtosecond ablation and spallation with different interatomic potentials. Appl. Surf. Sci. 2009, 255, 9592–9596. [Google Scholar]
- Mendelev, M.I.; Kramer, M.J.; Becker, C.A.; Asta, M. Analysis of semi-empirical interatomic potentials appropriate for simulation of crystalline and liquid Al and Cu. Philos. Mag. 2008, 88, 1723–1750. [Google Scholar]
- Liu, X.-Y.; Ercolessi, F.; Adams, J.B. Aluminium interatomic potential from density functional theory calculations with improved stacking fault energy. Model. Simul. Mater. Sci. Eng. 2004, 12, 665–670. [Google Scholar] [CrossRef]
- Zhou, X.W.; Johnson, R.A.; Wadley, H.N.G. Misfit-energy-increasing dislocations in vapor-deposited CoFe/NiFe multilayers. Phys. Rev. B 2004, 69, 144113. [Google Scholar]
- Lee, B.-J.; Shim, J.-H.; Baskes, M.I. Semiempirical atomic potentials for the fcc metals Cu, Ag, Au, Ni, Pd, Pt, Al, and Pb based on first and second nearest-neighbor modified embedded atom method. Phys. Rev. B 2003, 68, 144112. [Google Scholar] [CrossRef]
- Zope, R.R.; Mishin, Y. Interatomic potentials for atomistic simulations of the Ti-Al system. Phys. Rev. B 2003, 68, 024102. [Google Scholar]
- Sturgeon, J.B.; Laird, B.B. Adjusting the melting point of a model system via Gibbs-Duhem integration: Application to a model of aluminum. Phys. Rev. B 2000, 62, 14720. [Google Scholar] [CrossRef]
- Martienssen, W.; Warlimont, H. Springer Handbook of Condensed Matter and Materials Data; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Etesami, S.A. Asadi, E. Molecular dynamics for near melting temperatures simulations of metals using modified embedded-atom method. J. Phys. Chem. Solids 2018, 112, 61–72. [Google Scholar] [CrossRef]
- Stoller, R.E.; Tamm, A.; Béland, L.K.; Samolyuk, G.D.; Stocks, G.M.; Caro, A.; Slipchenko, L.V.; Osetsky, Y.N.; Aabloo, A.; Klintenberg; et al. Impact of Short-Range Forces on Defect Production from High-Energy Collisions. J. Chem Theory Comput. 2016, 12, 2871–2879. [Google Scholar] [CrossRef]
- Asadi, E.; Zaeem, M.A.; Nouranian, S.; Baskes, M.I. Two-phase solid–liquid coexistence of Ni, Cu, and Al by molecular dynamics simulations using the modified embedded-atom method. Acta Mater. 2015, 86, 169–181. [Google Scholar] [CrossRef]
- Mendelev, M.I.; Kramer, M.J.; Hao, S.G.; Ho, K.M.; Wang, C.Z. Development of interatomic potentials appropriate for simulation of liquid and glass properties of NiZr2 alloy. Philos. Mag. 2012, 92, 4454–4469. [Google Scholar] [CrossRef]
- Mendelev, M.I.; Underwood, T.L.; Ackland, G.J. Development of an interatomic potential for the simulation of defects, plasticity, and phase transformations in titanium. J Chem. Phys. 2016, 145, 154102. [Google Scholar] [CrossRef] [PubMed]
- Hennig, R.G.; Lenosky, T.J.; Trinkle, D.R.; Rudin, S.P.; Wilkins, J.W. Classical potential describes martensitic phase transformations between the α, β, and ω titanium phases. Phys. Rev. B 2008, 78, 054121. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Lee, B.-J.; Baskes, M.I. Modified embedded-atom method interatomic potentials for Ti and Zr. Phys. Rev. B 2006, 74, 014101. [Google Scholar] [CrossRef]
- Mahata, A.; Mukhopadhyay, T.; Zaeem, M.A. Modified embedded-atom method interatomic potentials for Al-Cu, Al-Fe and Al-Ni binary alloys: From room temperature to melting point. Comput. Mater. Sci. 2022, 201, 110902. [Google Scholar] [CrossRef]
- Purja Pun, G.P.; Mishin, Y. Development of an interatomic potential for the Ni-Al system. Philos. Mag. 2009, 89, 3245–3267. [Google Scholar] [CrossRef]
- e Silva, A.C.; Ågren, J.; Clavaguera-Mora, M.T.; Djurovic, D.; Gomez-Acebo, T.; Lee, B.J.; Liu, Z.K.; Miodownik, P.; Seifert, H.J. Applications of computational thermodynamics—the extension from phase equilibrium to phase transformations and other properties. Calphad 2007, 31, 53–74. [Google Scholar] [CrossRef]
- Mishin, Y. Atomistic modeling of the γ and γ′-phases of the Ni–Al system. Acta Mater. 2004, 52, 1451–1467. [Google Scholar] [CrossRef]
- Mishin, Y.; Mehl, M.J.; Papaconstantopoulos, D.A. Embedded-atom potential forB2−NiAl. Phys. Rev. B 2002, 65, 224114. [Google Scholar] [CrossRef]
- Ageev, N.V.; Guseva, L.N. Experimental study of the electron density in crystals. Bull. Acad. Sci. Ussr Div. Chem. Sci. 1952, 1, 31–39. [Google Scholar] [CrossRef]
- Kim, Y.K.; Kim, H.K.; Jung, W.S.; Lee, B.J. Atomistic modeling of the Ti–Al binary system. Comput. Mater. Sci. 2016, 119, 1–8. [Google Scholar] [CrossRef]
- Kavousi, S.; Novak, B.R.; Baskes, M.I.; Zaeem, M.A.; Moldovan, D. Modified embedded-atom method potential for high-temperature crystal-melt properties of Ti–Ni alloys and its application to phase field simulation of solidification. Model. Simul. Mater. Sci. Eng. 2019, 28, 015006. [Google Scholar] [CrossRef]
- Kim, Y.K.; Kim, H.K.; Jung, W.S.; Lee, B.J. Development and application of Ni-Ti and Ni-Al-Ti 2NN-MEAM interatomic potentials for Ni-base superalloys. Comput. Mater. Sci. 2017, 139, 225–233. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Al-Al | Ni-Ni | Ti-Ti | Al-Ni | Al-Ti | Ni-Ti | |
|---|---|---|---|---|---|---|
| A1 (eV) | 0.4569 | 0.5586 | 0.3172 | 0.3564 | 1.6994 | 2.4855 |
| p1 | 7.5838 | 11.5942 | 9.9892 | 8.0628 | 7.4878 | 5.1036 |
| A2 (eV) | 0.4854 | 1.3281 | 2.0865 | 0.7006 | 0.0694 | 0.9432 |
| p2 | 2.7347 | 1.5300 | 0.1108 | 2.0128 | 2.2728 | 0.7224 |
| n1 | 4 | 4 | 4 | 4 | 4 | 4 |
| rm1 (Å) | 3.2824 | 2.9785 | 3.4976 | 3.3593 | 4.7839 | 3.0663 |
| rc1 (Å) | 5.6445 | 3.9029 | 4.7025 | 5.2709 | 6.8037 | 5.2128 |
| B1 (eV2) | 4.9693 | 6.7236 | 6.3238 | 5.6417 | 22.9707 | 35.0220 |
| q1 | 4.8330 | 3.2568 | 3.9940 | 7.3234 | 6.5876 | 4.9950 |
| B2 (eV2) | 0.1363 | 0.5683 | 0.1181 | 0.4487 | 1.6521 | 0.2281 |
| q2 | 0.6337 | 0.1000 | 0.1993 | 0.6539 | 2.9662 | 1.3153 |
| n2 | 5 | 5 | 5 | 5 | 5 | 5 |
| rm2 (Å) | 4.7207 | 2.4520 | 5.0156 | 4.6133 | 3.3745 | 4.5877 |
| rc2 (Å) | 8.1302 | 6.1358 | 8.9364 | 6.7808 | 7.0103 | 7.7823 |
| r0 (Å) | 2.8635 | 2.3258 | 2.9757 | 2.8913 | 2.6333 | 2.3510 |
| Al | Ni | Ti | |||||||
|---|---|---|---|---|---|---|---|---|---|
| fcc | bcc | hcp | fcc | bcc | hcp | hcp | bcc | fcc | |
| a0 (Å) | 4.05 | 3.22 | 2.85 | 3.52 | 2.81 | 2.48 | 2.95 | 3.28 | 4.13 |
| 4.05 [33] | 3.23 ab | 2.85 ab | 3.52 [33] | 2.79 ab | 2.47 ab | 2.95 [33] | 3.25 ab | 4.10 ab | |
| c0 (Å) | - | - | 4.72 | - | - | 4.09 | 4.68 | - | - |
| - | - | 4.73 ab | - | - | 4.09 ab | 4.68 [33] | - | - | |
| Ec (eV) | 3.39 | 3.37 | 3.39 | 4.44 | 4.39 | 4.44 | 4.85 | 4.82 | 4.85 |
| 3.39 [34] | 3.29 ab | 3.36 ab | 4.44 [34] | 4.39 ab | 4.41 ab | 4.85 [34] | 4.74 ab | 4.79 ab | |
| B0 (GPa) | 72.2 | 70.7 | 72.0 | 189.6 | 177.4 | 189.6 | 106.4 | 102.1 | 106.2 |
| 72.2 [34] | 68.4 ab | 74.4 ab | 186.0 [34] | 200.9 ab | 203.2 ab | 105.1 [34] | 109.6 ab | 111.5 ab | |
| C11 (GPa) | 83.1 | 70.6 | 97.6 | 242.5 | 162.7 | 303.1 | 131.2 | 81.8 | 169.8 |
| 106.8 [33] | 41.3 ab | 106.1 ab | 248.1 [33] | 178.5 ab | 300.5 ab | 162.4 [33] | 98.6 ab | 140.0 ab | |
| C12 (GPa) | 66.7 | 70.8 | 66.2 | 163.1 | 184.8 | 158.4 | 94.0 | 112.2 | 74.5 |
| 60.4 [33] | 81.9 ab | 67.8 ab | 154.9 [33] | 212.0 ab | 180.1 ab | 92.0 [33] | 115.0 ab | 97.2 ab | |
| C44 (GPa) | 29.1 | 33.2 | 15.0 | 123.4 | 121.0 | 67.6 | 61.9 | 97.8 | 33.8 |
| 28.3 [33] | 41.3 ab | 6.5 ab | 124.2 [33] | 147.6 ab | 48.9 ab | 46.7 [33] | 45.4 ab | 59.2 ab | |
| Al3Ni | AlNi | AlNi3 | Al3Ti | AlTi | AlTi3 | Ni3Ti | NiTi | NiTi2 | |
|---|---|---|---|---|---|---|---|---|---|
| D011 | B2 | L12 | D022 | L10 | D019 | hP16 | B2 | cF96 | |
| a0 (Å) | 6.56 | 2.84 | 3.51 | 3.86 | 2.82 | 5.77 | 5.10 | 2.96 | 11.31 |
| 6.60 [35] | 2.88 [36] | 3.57 [37] | 3.85 [38] | 2.83 [39] | 5.78 [40] | 5.10 [41] | 3.01 [42] | 11.28 [43] | |
| b0 (Å) | 7.31 | - | - | - | - | - | - | - | - |
| 7.35 [35] | - | - | - | - | - | - | - | - | |
| c0 (Å) | 4.77 | - | - | 8.59 | 4.05 | 4.64 | 8.32 | - | - |
| 4.80 [35] | - | - | 8.58 [38] | 4.07 [39] | 4.65 [40] | 8.30 [41] | - | - | |
| Ef (eV) | −0.43 | −0.68 | −0.51 | −0.36 | −0.41 | −0.29 | −0.38 | −0.36 | −0.31 |
| −0.42 ab | −0.69 ab | −0.47 ab | −0.38 [44] | −0.42 [44] | −0.26 [44] | −0.36 [45] | −0.35 [46] | −0.28 [45] | |
| B0 (GPa) | 92.6 | 152.9 | 156.4 | 71.5 | 136.8 | 112.1 | 130.8 | 123.4 | 83.4 |
| 113.1 ab | 160.4 ab | 183.0 ab | 103.0 [47] | 112.1 [47] | 111.9 [47] | 163.4 [48] | 142.0 [45] | 119.8 [49] |
| Pearson | Source | a0 (Å) | Ec (eV) | Ef (eV) | B0 (GPa) | |
|---|---|---|---|---|---|---|
| AlNi2Ti | cF16 | lmp | 5.82 | 4.81 | −0.53 | 148.8 |
| MP [50] | 5.89 | 6.42 | −0.62 | 162.0 | ||
| Al16Ni7Ti6 | cF116 | lmp | 12.09 | 4.44 | −0.50 | 78.7 |
| MP [51] | 11.80 | 5.66 | −0.56 | 130.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Li, J.; Lai, W.; Liu, J.; Liu, B. Interatomic Potential to Predict the Favored Glass-Formation Compositions and Local Atomic Arrangements of Ternary Al-Ni-Ti Metallic Glasses. Crystals 2022, 12, 1065. https://doi.org/10.3390/cryst12081065
Yang Q, Li J, Lai W, Liu J, Liu B. Interatomic Potential to Predict the Favored Glass-Formation Compositions and Local Atomic Arrangements of Ternary Al-Ni-Ti Metallic Glasses. Crystals. 2022; 12(8):1065. https://doi.org/10.3390/cryst12081065
Chicago/Turabian StyleYang, Qilin, Jiahao Li, Wensheng Lai, Jianbo Liu, and Baixin Liu. 2022. "Interatomic Potential to Predict the Favored Glass-Formation Compositions and Local Atomic Arrangements of Ternary Al-Ni-Ti Metallic Glasses" Crystals 12, no. 8: 1065. https://doi.org/10.3390/cryst12081065