
Multi-Component Diffusion in the Vicinity of a Growing Crystal


Abstract
:1. Introduction
2. Materials and Method
2.1. Method
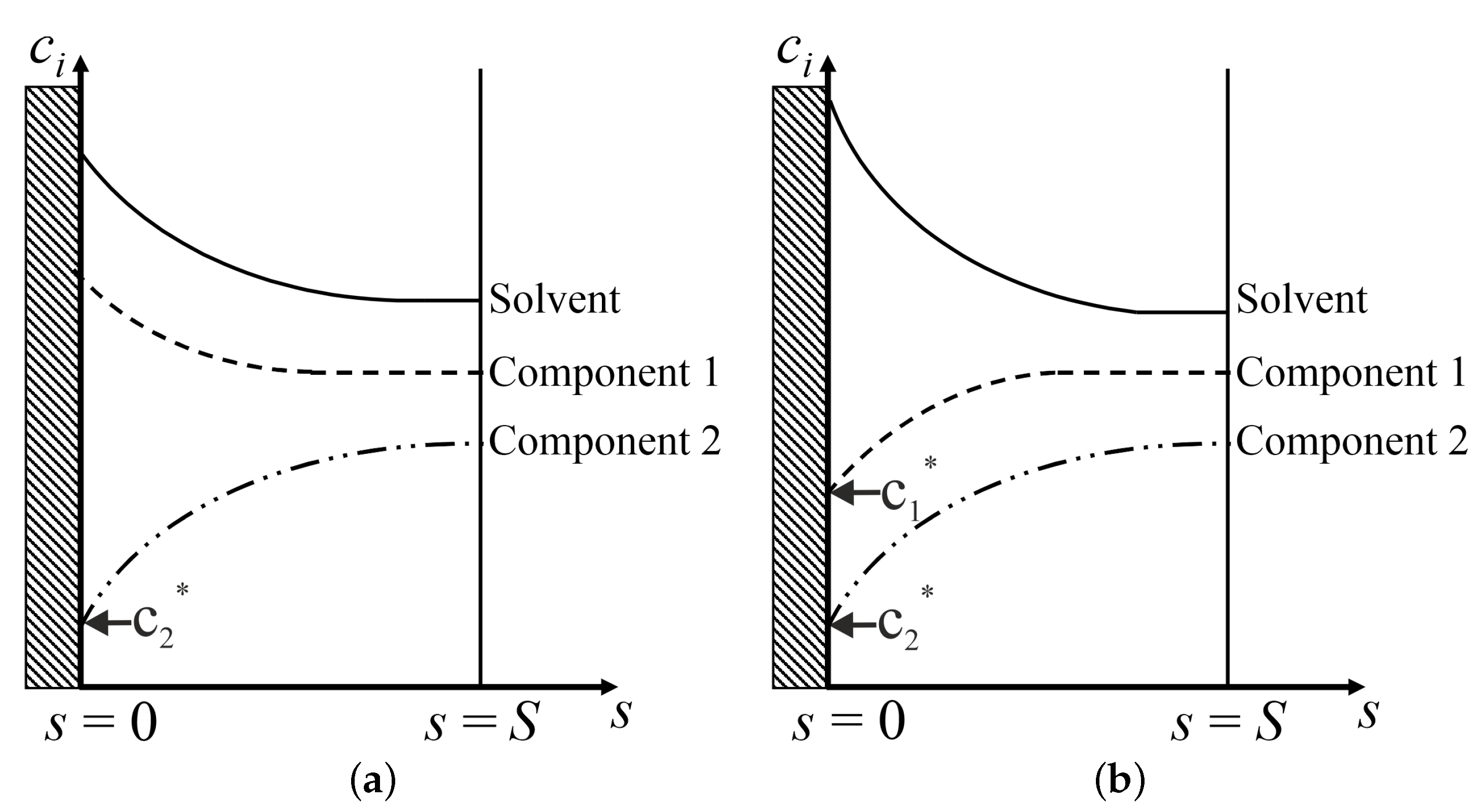
2.2. Model Set-Up
3. Results
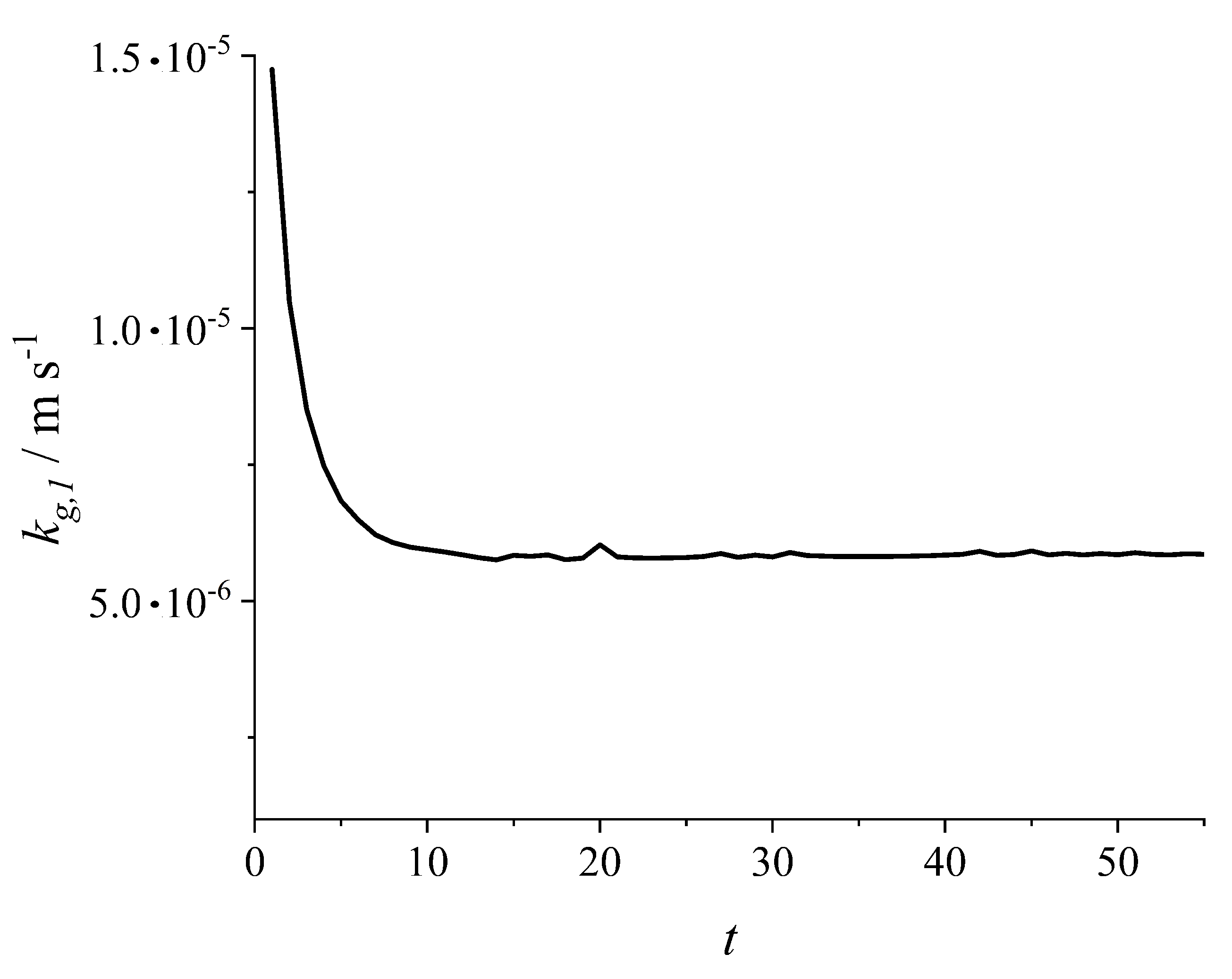
3.1. Solvate Crystallization of a Single Component
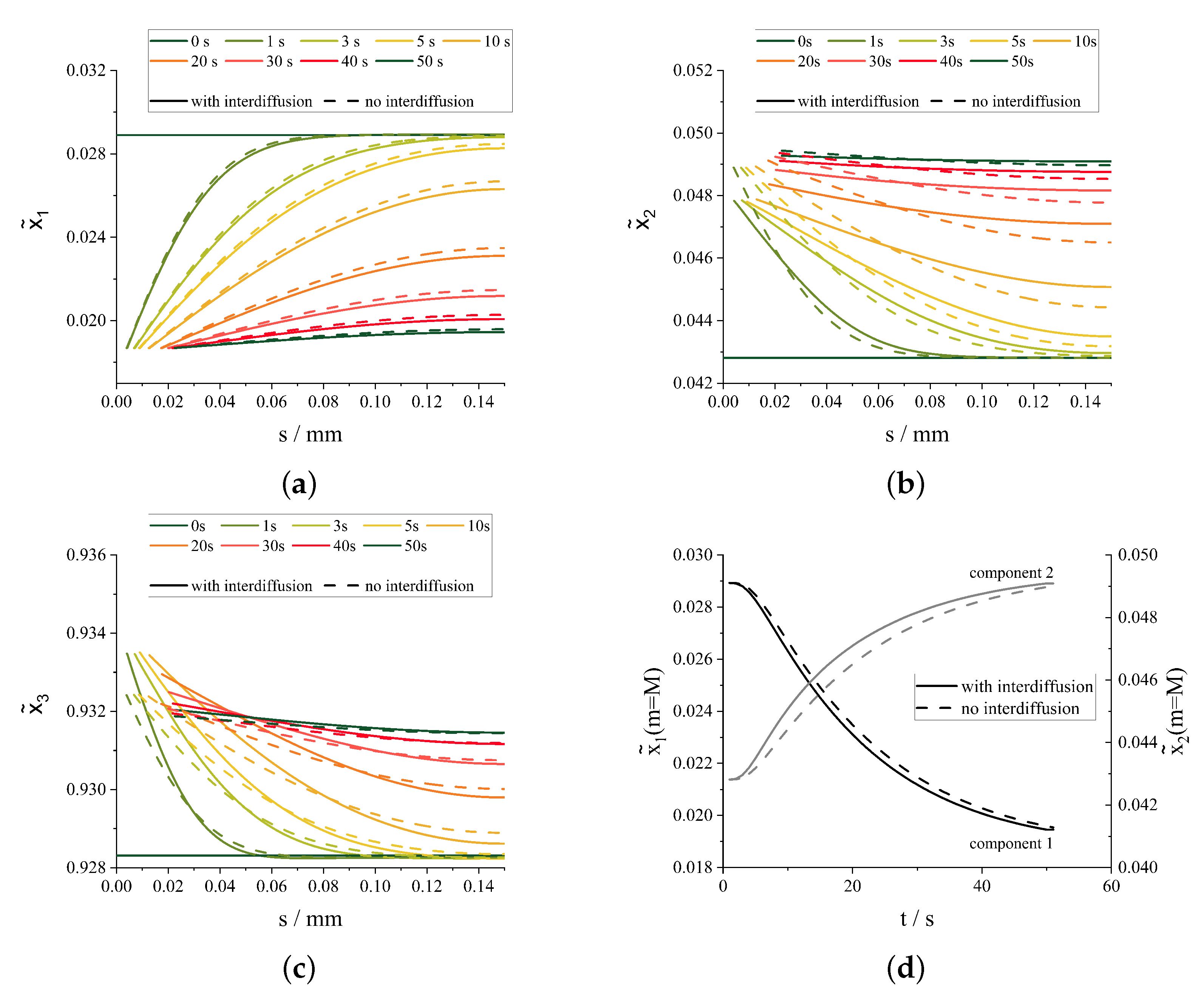
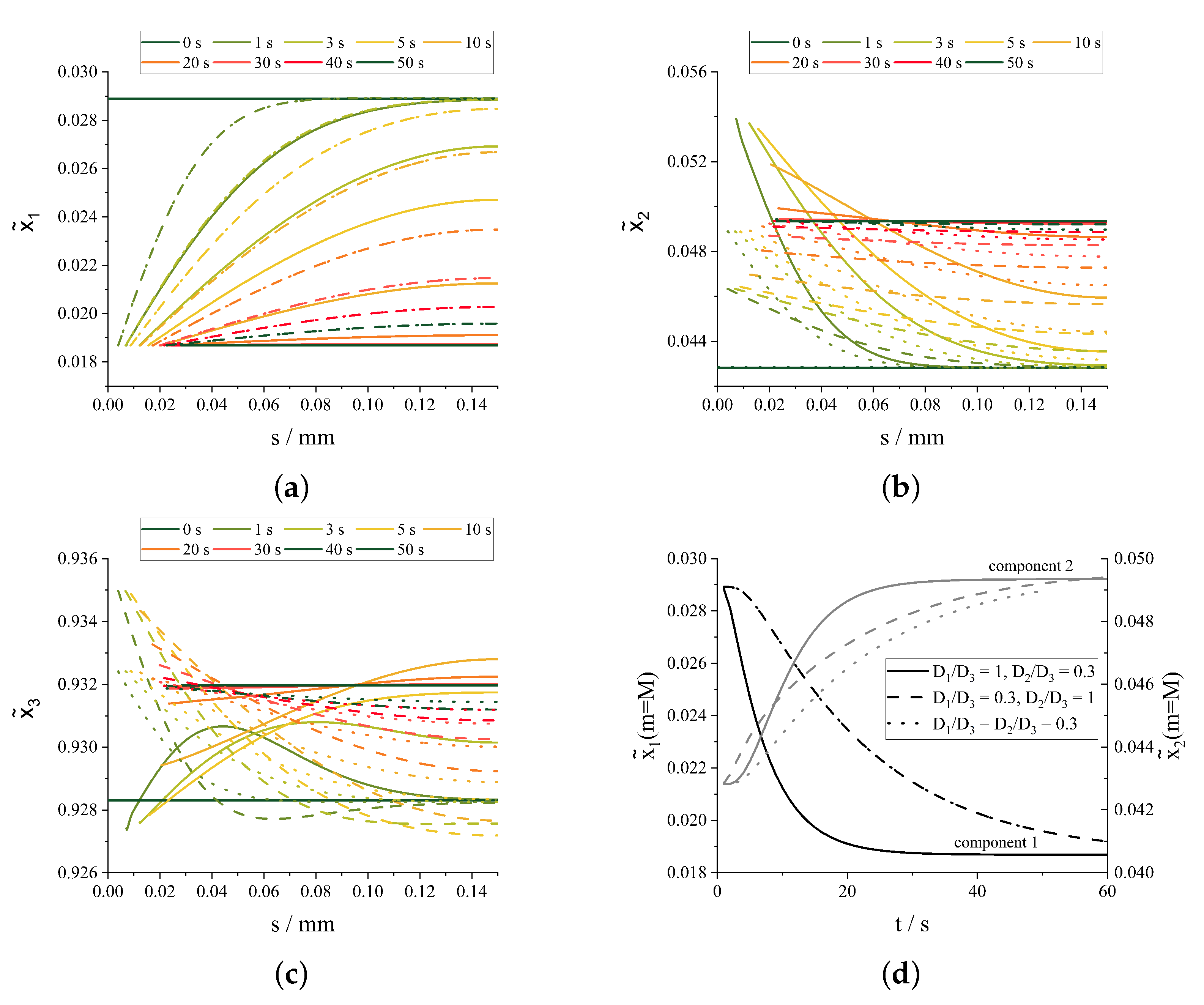
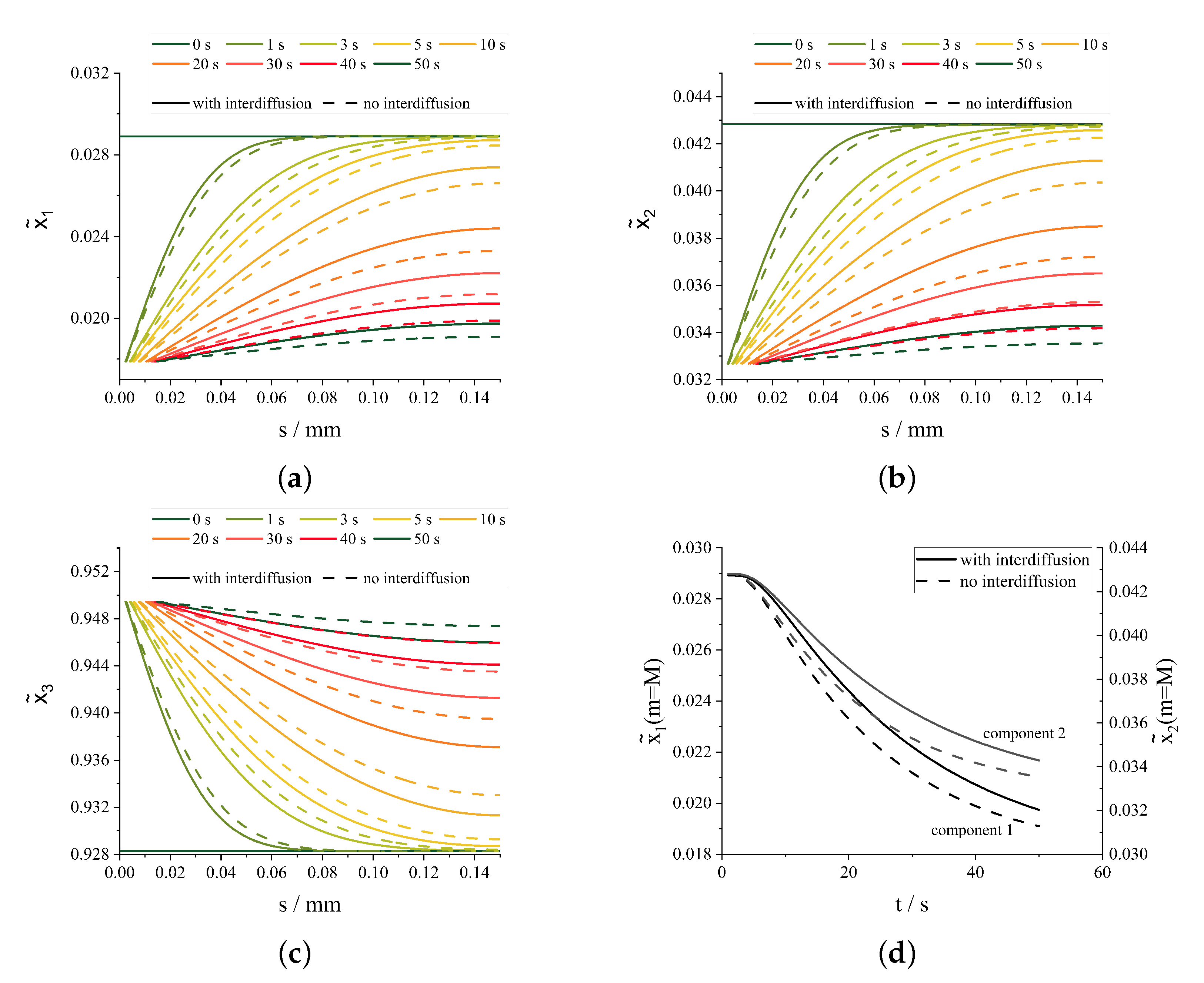
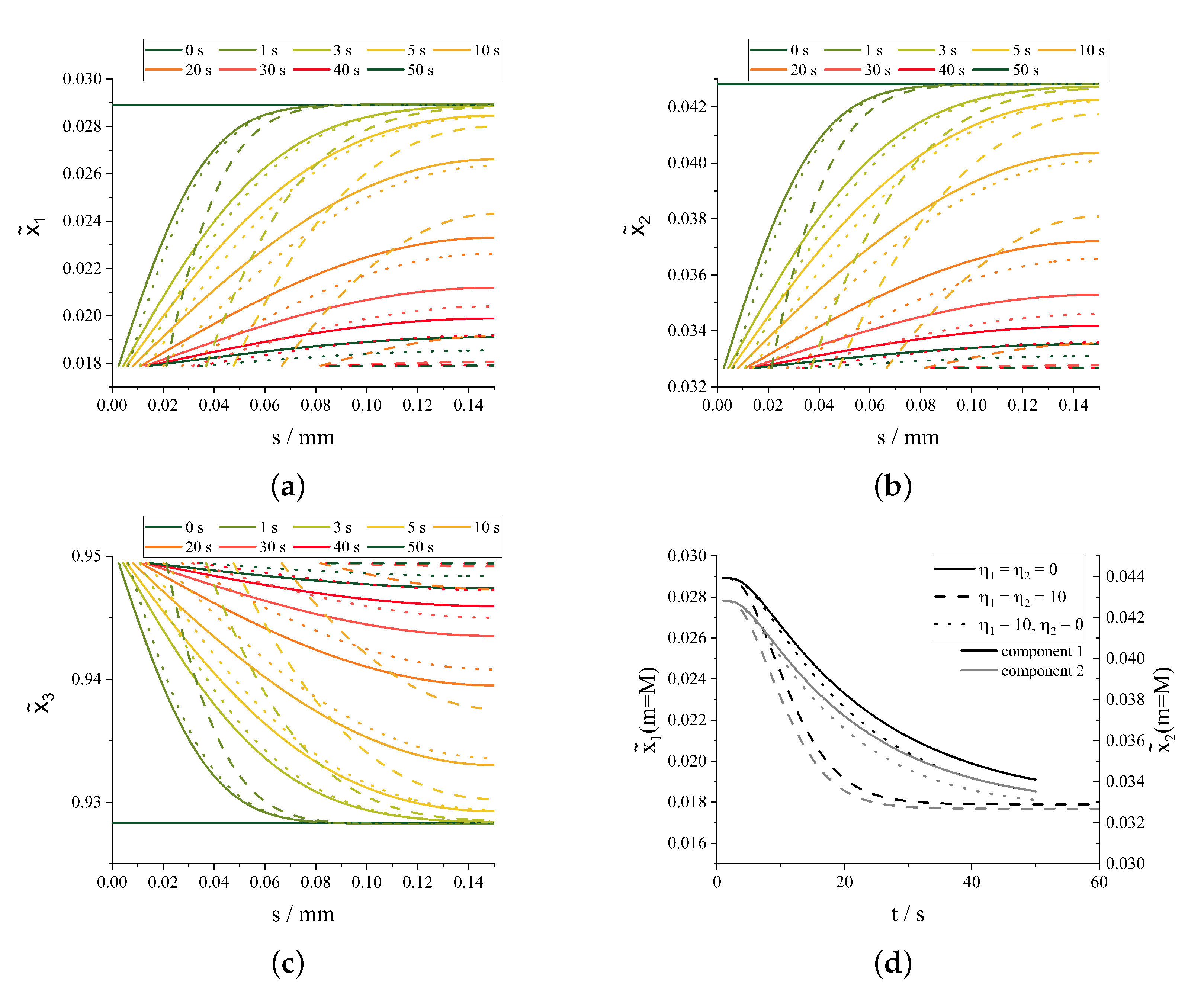
3.2. Simultaneous Crystallization of Multiple Components
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guignon, B.; Duquenoy, A.; Dumoulin, E.D. Fluid bed encapsulation of particles: Principles and practice. Dry. Technol. 2002, 20, 419–447. [Google Scholar] [CrossRef]
- Degrève, J.; Baeyens, J.; van de Velden, M.; de Laet, S. Spray-agglomeration of NPK-fertilizer in a rotating drum granulator. Powder Technol. 2006, 163, 188–195. [Google Scholar] [CrossRef]
- Suresh, P.; Sreedhar, I.; Vaidhiswaran, R.; Venugopal, A. A comprehensive review on process and engineering aspects of pharmaceutical wet granulation. Chem. Eng. J. 2017, 328, 785–815. [Google Scholar] [CrossRef]
- Römbach, E.; Ulrich, J. Self-controlled coating process for drugs. Cryst. Growth Des. 2007, 7, 1618–1622. [Google Scholar] [CrossRef]
- Shin, G.S.; Choi, W.G.; Na, S.; Ryu, S.O.; Moon, T. Rapid crystallization in ambient air for planar heterojunction perovskite solar cells. Electron. Mater. Lett. 2017, 13, 72–76. [Google Scholar] [CrossRef]
- Sakai, N.; Pathak, S.; Chen, H.W.; Haghighirad, A.A.; Stranks, S.D.; Miyasaka, T.; Snaith, H.J. The mechanism of toluene-assisted crystallization of organic–inorganic perovskites for highly efficient solar cells. J. Mater. Chem. A 2016, 4, 4464–4471. [Google Scholar] [CrossRef]
- Nijdam, J.; Trouillet, V.; Kachel, S.; Scharfer, P.; Schabel, W.; Kind, M. Coat formation of surface-active proteins on aqueous surfaces during drying. Colloids Surf. B Biointerfaces 2014, 123, 53–60. [Google Scholar] [CrossRef]
- Seo, K.S.; Han, H.K. Multilayer-coated tablet of clopidogrel and rosuvastatin: Preparation and in vitro/in vivo characterization. Pharmaceutics 2019, 11, 313. [Google Scholar] [CrossRef] [Green Version]
- Song, K.S.; Lim, J.; Yun, S.; Kim, D.; Kim, Y. Composite fouling characteristics of CaCO3 and CaSO4 in plate heat exchangers at various operating and geometric conditions. Int. J. Heat Mass Transf. 2019, 136, 555–562. [Google Scholar] [CrossRef]
- Lv, Y.; Lu, K.; Ren, Y. Composite crystallization fouling characteristics of normal solubility salt in double-pipe heat exchanger. Int. J. Heat Mass Transf. 2020, 156, 119883. [Google Scholar] [CrossRef]
- Yuan, J.J.; Stepanski, M.; Ulrich, J. Fremdstoffeinflüsse auf Kristallwachstumsgeschwindigkeiten bei der Kristallisation aus Lösungen. Chem. Ing. Tech. 1990, 62, 645–646. [Google Scholar] [CrossRef]
- Rauls, M.; Bartosch, K.; Kind, M.; Lacmann, R.; Mersmann, A. The influence of impurities on crystallization kinetics–a case study on ammonium sulfate. J. Cryst. Growth 2000, 213, 116–128. [Google Scholar] [CrossRef]
- Vergara, A.; Paduano, L.; Vitagliano, V.; Sartorio, R. Multicomponent diffusion in solutions where crystals grow. Mater. Chem. Phys. 2000, 66, 126–131. [Google Scholar] [CrossRef]
- Kubota, N. Effect of impurities on the growth kinetics of crystals. Cryst. Res. Technol. J. Exp. Ind. Crystallogr. 2001, 36, 749–769. [Google Scholar] [CrossRef]
- Louhi-Kultanen, M.; Kallas, J.; Partanen, J.; Sha, Z.; Oinas, P.; Palosaari, S. The influence of multicomponent diffusion on crystal growth in electrolyte solutions. Chem. Eng. Sci. 2001, 56, 3505–3515. [Google Scholar] [CrossRef]
- Zago, G.P.; Penha, F.M.; Seckler, M.M. Product characteristics in simultaneous crystallization of NaCl and CaSO4 from aqueous solution with seeding. Desalination 2020, 474, 114180. [Google Scholar] [CrossRef]
- Penha, F.M.; Andrade, F.R.D.; Lanzotti, A.S.; Moreira Junior, P.F.; Zago, G.P.; Seckler, M.M. In Situ Observation of Epitaxial Growth during Evaporative Simultaneous Crystallization from Aqueous Electrolytes in Droplets. Crystals 2021, 11, 1122. [Google Scholar] [CrossRef]
- Chiarella, R.A.; Davey, R.J.; Peterson, M.L. Making co-crystals the utility of ternary phase diagrams. Cryst. Growth Des. 2007, 7, 1223–1226. [Google Scholar] [CrossRef]
- Urbanus, J.; Roelands, C.M.; Verdoes, D.; Jansens, P.J.; ter Horst, J.H. Co-crystallization as a separation technology: Controlling product concentrations by co-crystals. Cryst. Growth Des. 2010, 10, 1171–1179. [Google Scholar] [CrossRef]
- Mersmann, A. Crystallization Technology Handbook; CRC Press: Boca Raton, FL, USA, 2001. [Google Scholar]
- Sun, C.C. Cocrystallization for successful drug delivery. Expert Opin. Drug Deliv. 2013, 10, 201–213. [Google Scholar] [CrossRef]
- Pawar, N.; Saha, A.; Nandan, N.; Parambil, J.V. Solution cocrystallization: A scalable approach for cocrystal production. Crystals 2021, 11, 303. [Google Scholar] [CrossRef]
- Burton, J.A.; Prim, R.C.; Slichter, W.P. The distribution of solute in crystals grown from the melt. Part I. Theoretical. J. Chem. Phys. 1953, 21, 1987–1991. [Google Scholar] [CrossRef]
- Garside, J.; Mersmann, A.; Nývlt, J. Measurement of Crystal Growth and Nucleation Rates; IChemE: Rugby, UK, 2002. [Google Scholar]
- Eder, C.; Choscz, C.; Müller, V.; Briesen, H. Jamin-interferometer-setup for the determination of concentration and temperature dependent face-specific crystal growth rates from a single experiment. J. Cryst. Growth 2015, 426, 255–264. [Google Scholar] [CrossRef]
- Lin, H.; Rosenberger, F.; Alexander, J.I.; Nadarajah, A. Convective-diffusive transport in protein crystal growth. J. Cryst. Growth 1995, 151, 153–162. [Google Scholar] [CrossRef]
- Gupta, A.; Shim, S.; Issah, L.; McKenzie, C.; Stone, H.A. Diffusion of multiple electrolytes cannot be treated independently: Model predictions with experimental validation. Soft Matter 2019, 15, 9965–9973. [Google Scholar] [CrossRef]
- Caspari, W.A. CCCXXIV.—The system sodium carbonate–sodium sulphate–water. J. Chem. Soc. Trans. 1924, 125, 2381–2387. [Google Scholar] [CrossRef]
- von Plessen, H. Sodium Sulfates. In Ullmann’s Encyclopedia of Industrial Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2000. [Google Scholar] [CrossRef]
- Thieme, C. Sodium Carbonates. In Ullmann’s Encyclopedia of Industrial Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2000. [Google Scholar] [CrossRef]
- Taylor, R.; Krishna, R. Multicomponent Mass Transfer; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1993; Volume 2. [Google Scholar]
- Rehfeldt, S.; Stichlmair, J. Measurement and calculation of multicomponent diffusion coefficients in liquids. Fluid Phase Equilibria 2007, 256, 99–104. [Google Scholar] [CrossRef]
- Krishna, R.; van Baten, J.M. The darken relation for multicomponent diffusion in liquid mixtures of linear alkanes: An investigation using molecular dynamics (MD) simulations. Ind. Eng. Chem. Res. 2005, 44, 6939–6947. [Google Scholar] [CrossRef]
- Kim, S.U.; Srinivasan, V. A Method for Estimating Transport Properties of Concentrated Electrolytes from Self-Diffusion Data. J. Electrochem. Soc. 2016, 163, A2977. [Google Scholar] [CrossRef]
- Nielsen, J.M.; Adamson, A.W.; Cobble, J.W. The self-diffusion coefficients of the ions in aqueous sodium chloride and sodium sulfate at 25. J. Am. Chem. Soc. 1952, 74, 446–451. [Google Scholar] [CrossRef]
- Liu, X.; Schnell, S.K.; Simon, J.M.; Krüger, P.; Bedeaux, D.; Kjelstrup, S.; Bardow, A.; Vlugt, T.J.H. Diffusion coefficients from molecular dynamics simulations in binary and ternary mixtures. Int. J. Thermophys. 2013, 34, 1169–1196. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Tanner, J.E. Spin diffusion measurements: Spin echoes in the presence of a time–dependent field gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef] [Green Version]
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input and Examples for PHREEQC Version 3: A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; U.S. Geological Survey: Denver, CO, USA, 2013.
- Marion, G.M.; Mironenko, M.V.; Roberts, M.W. FREZCHEM: A geochemical model for cold aqueous solutions. Comput. Geosci. 2010, 36, 10–15. [Google Scholar]
- Hirt, C.W.; Nichols, B.D. Volume of fluid (VOF) method for the dynamics of free boundaries. J. Comput. Phys. 1981, 39, 201–225. [Google Scholar] [CrossRef]
- Helfenritter, C.; Kind, M. Determination of crystal growth rates in multi-component solutions. Cryst. Growth Des. 2022. in review process. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Value | Source |
|---|---|---|
| (saturated at 25 °C) | 0.162 | [28] |
| (saturated at 25 °C) | 0.179 | [28] |
| (saturated at 20 °C) | 0.11 | [28] |
| (saturated at 20 °C) | 0.15 | [28] |
| (both solids) | 1300 (1470) kg m−3 | assumption (actual values [29,30]) |
| 1300 (1346) kg m−3 | assumption (calc., PhreeqC at 25 °C and ) | |
| 0.142 kg mol−1 | ||
| 0.106 kg mol−1 | ||
| 0.018 kg mol−1 | ||
| m | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helfenritter, C.; Kind, M. Multi-Component Diffusion in the Vicinity of a Growing Crystal. Crystals 2022, 12, 872. https://doi.org/10.3390/cryst12060872
Helfenritter C, Kind M. Multi-Component Diffusion in the Vicinity of a Growing Crystal. Crystals. 2022; 12(6):872. https://doi.org/10.3390/cryst12060872
Chicago/Turabian StyleHelfenritter, Christoph, and Matthias Kind. 2022. "Multi-Component Diffusion in the Vicinity of a Growing Crystal" Crystals 12, no. 6: 872. https://doi.org/10.3390/cryst12060872