3.1. Independent Atom Model
Structure refinement with spherical scattering factors from an independent atom model was performed with the SHELXL software [
12]. Selected bond lengths are given in
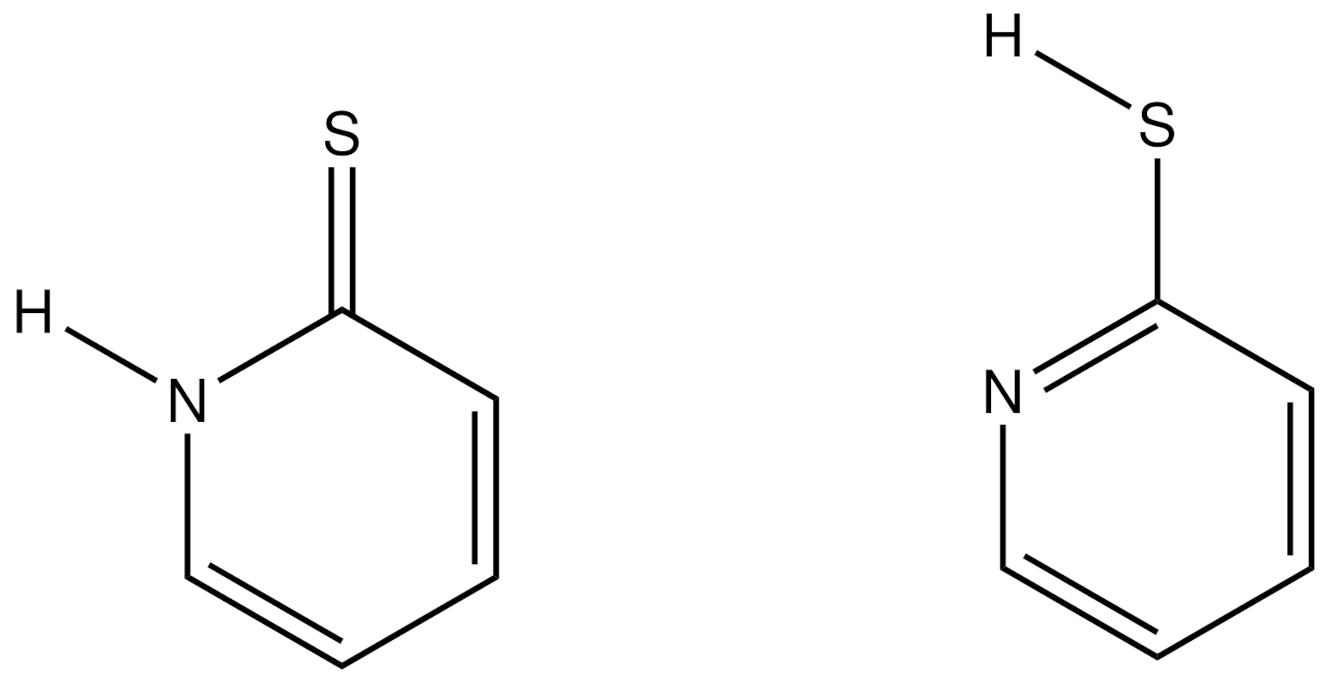
Table 2. The C–C bond lengths show a significant alternation. Bonds C2–C3 and C4–C5 are significantly shorter than C1–C2 and C3–C4. The C–S bond of 1.69981(19) Å confirms the double-bond character. All hydrogen atoms were located in difference Fourier maps and refined freely with isotropic displacement parameters. They clearly show the protonation of the nitrogen atom. Overall, this is consistent with the expectation for the thiopyridone form (
Scheme 1) and with the quantum-theoretical geometry of it, as calculated in 2002 in vacuo [
1]. A comparison with the room-temperature X-ray results of [
4] shows mainly the increased precision of the current data (
Table 2).
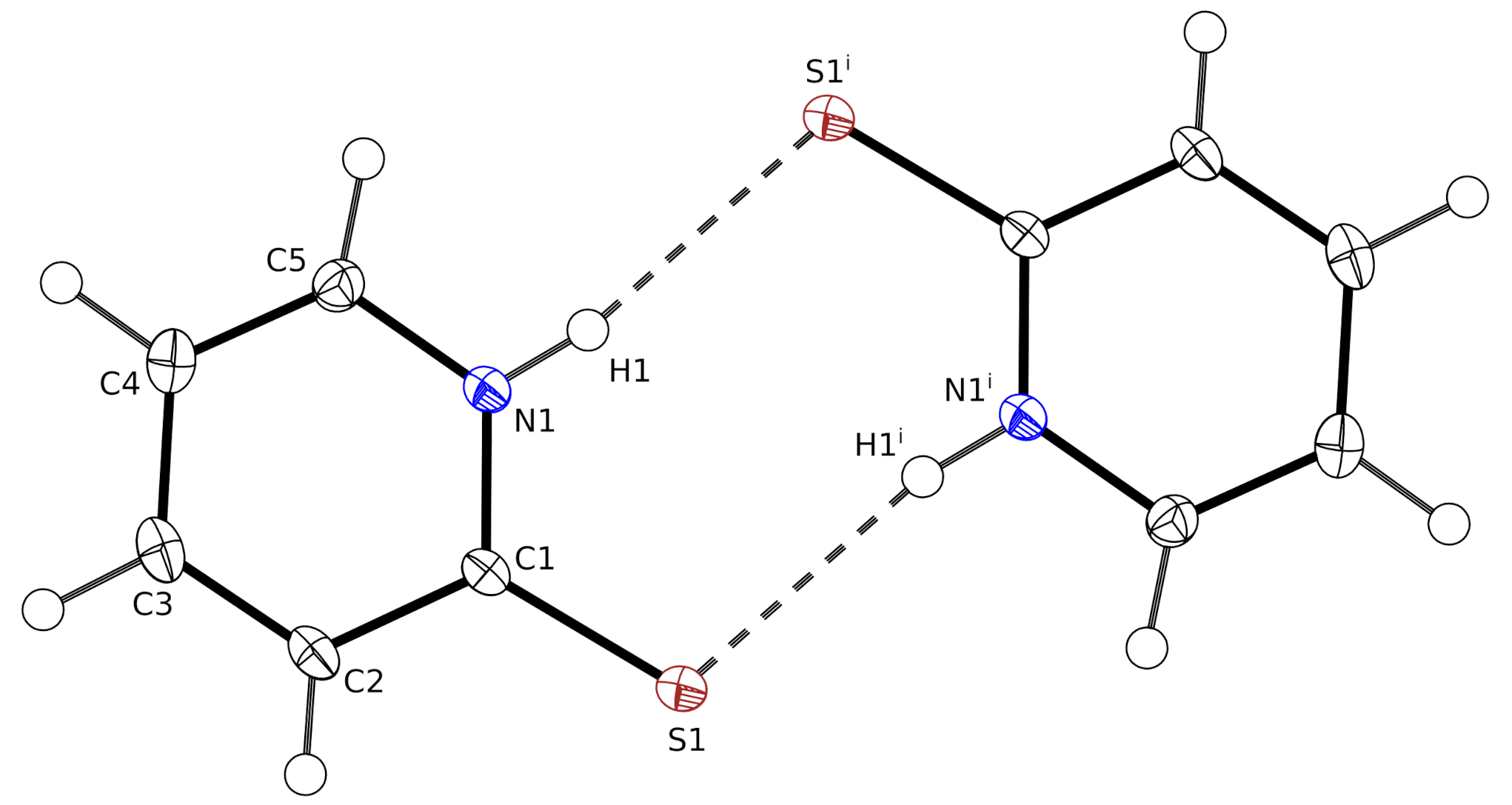
In the crystal, the molecules form centrosymmetric hydrogen-bonded dimers with the N–H group as donor and the sulfur as acceptor (
Figure 1). The graph set for this dimer is
. The corresponding inversion center is located at
, Wyckoff position
c.
The inversion at
, Wyckoff position
a, generates an antiparallel molecule with a perpendicular distance of 3.3949(1) Å. Based on the literature [
16], we consider such a short distance as an indication for a
stacking interaction. In this arrangement, the ring centers are not located exactly on top of each other. The distance between the ring centers is 3.6714(2) Å, resulting in a ring slippage of 1.40 Å.
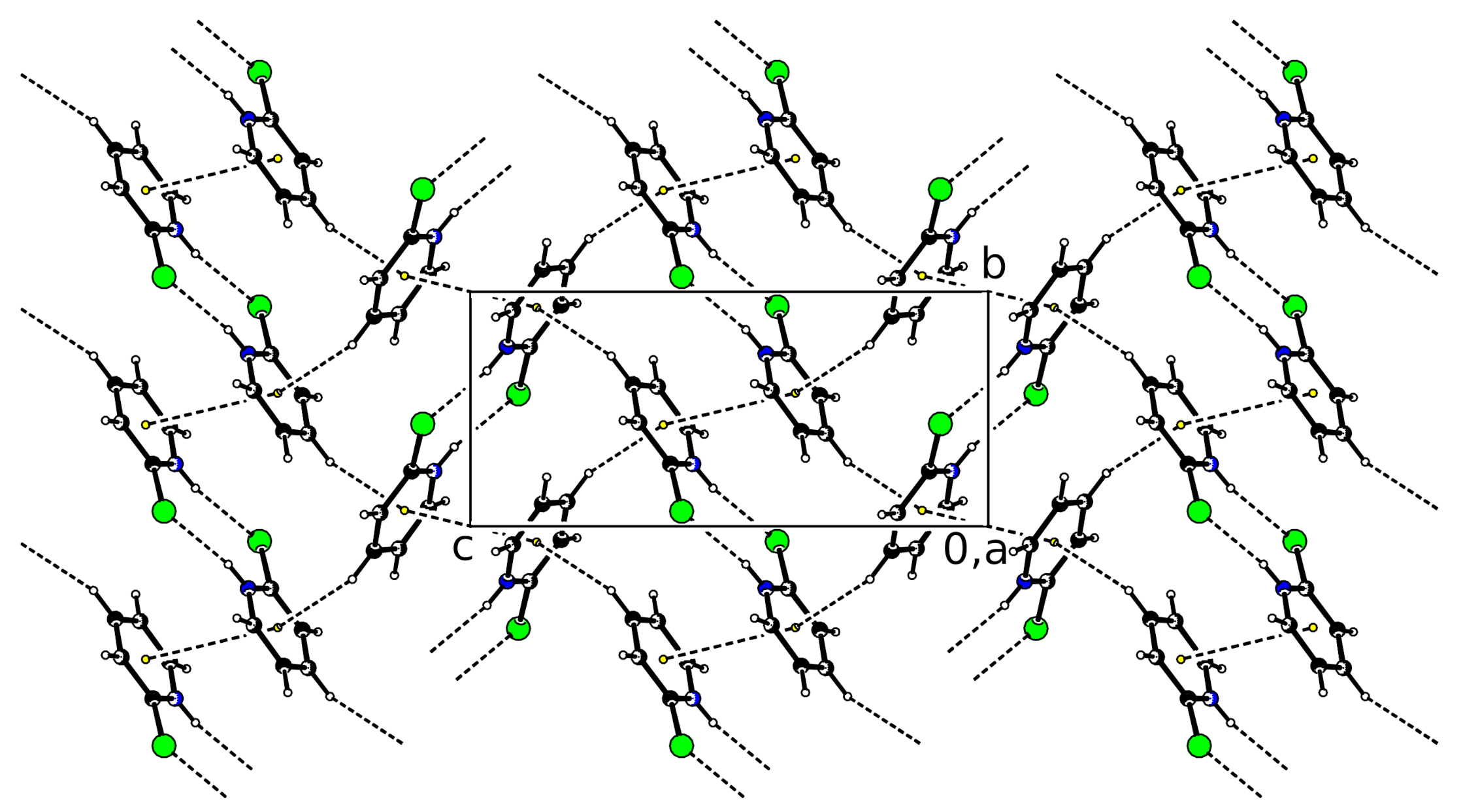
While the
stacking interaction concerns one side of the molecule, the opposite side is involved as acceptor of a C–H
interaction. C–H
interactions mainly have a dispersive nature. Despite their weakness, they can be influential [
17] as well as in small-molecule structures [
18] as in proteins [
19]. An overall impression of the intermolecular interactions in
(I) is depicted in
Figure 2. The weak interactions result in a layer formation in the
plane.
Symmetry considerations lead to the conclusion that the molecule is located in a general position without symmetry but the planarity of the molecule gives rise to an approximate mirror symmetry ( symmetry) with an r.m.s. deviation from ideal symmetry of only 0.017 Å. The formation of the hydrogen-bonded dimer disturbs this approximate symmetry only slightly. The r.m.s. deviation from ideal symmetry of the dimer is only 0.063 Å. The crystal field for the molecule, however, is non-symmetric, with stacking on one side, and a C–H interaction on the other side.
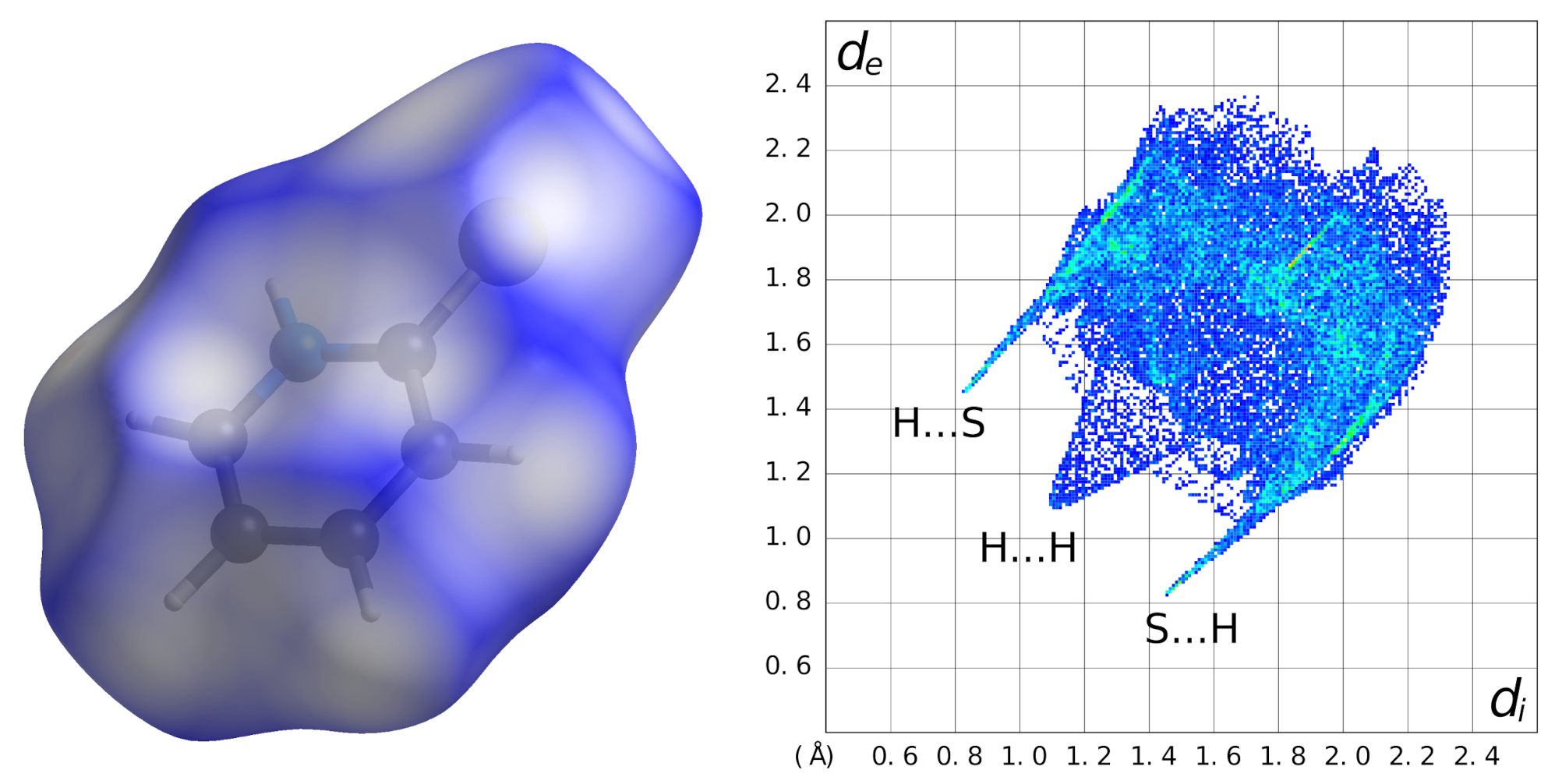
So far, the discussion of intermolecular interactions in
(I) has been based on pure geometrical calculations performed with the PLATON software [
20]. An alternative approach is the calculation and analysis of the Hirshfeld surface which is based on electron densities [
21]. The ratio between the electron density of the promolecule and the electron density of the procrystal is calculated, and an isosurface is drawn where this ratio is 0.5. It is a straightforward way to recognize all kinds of intermolecular interactions: the value of
becomes negative if the intermolecular distance is shorter than the van der Waals separation. For
(I), the Hirshfeld surface is shown in
Figure 3 (left). Hydrogen atoms occupy 63.1%, carbon 15.3%, nitrogen 2.8% and sulfur 18.8% of this surface. In the fingerprint plot (
Figure 3, right), H⋯S/S⋯H and H⋯H contacts are most prominent.
Following a procedure by Jelsch et al. [
22], it is possible to use the fingerprint plot (
Figure 3) to calculate an enrichment ratio
E for each intermolecular contact. Enrichment ratios larger than unity indicate contacts which are preferred in the crystal packing. The system tries to avoid contacts with
. For
(I), the enrichment ratios are given in
Table 3. The H⋯S/H⋯S and C⋯C contacts appear to be important for the packing which corresponds to the N–H⋯S hydrogen bonds and the
stacking (
discussed above). The enrichment ratio for the H⋯C contacts is only slightly above 1. The positive influence of the C–H
interactions might thus be small. From this analysis, H⋯H contacts are unfavorable. In this context, we note that the shortest intermolecular H⋯H contact is between H5 and H5
(
) with 2.345(10) Å (SHELXL results) or 2.18 Å (normalized C–H distances). This is smaller than the van der Waals distance of 2.40 Å.
For an analysis of the anisotropic displacement parameters, the result of the current refinement was subjected to a rigid-body TLS analysis using the THMA11 software [
23]. The weighted
R value for the TLS model is 0.028. (
with
). This low
R value is a clear sign that the observed atomic displacement parameters can be well fitted with a rigid-body TLS model.
The THMA11 program additionally performs a MSDA analysis (“Hirshfeld rigid-bond test” [
24]). The MSDA
for the S1–C1 bond is 0.0008 Å
, a value larger than expected. If this situation is related to the problems of this bond in the X–N analysis by Ohms et al. [
4] (
vide supra) cannot be decided on the independent atom model.
3.2. Thermal Expansion
A temperature-dependent diffraction experiment was performed to obtain better insight into the thermal behavior of
(I). In order to improve the internal consistency, the movable detector was kept at a fixed position for the duration of the experiment. The resulting unit cell parameters are provided in
Table S1 (Supplementary Materials). It appears that the largest expansion with temperature is along the
c axis, and the smallest along the
b axis (see
Figure S1, Supplementary Materials).
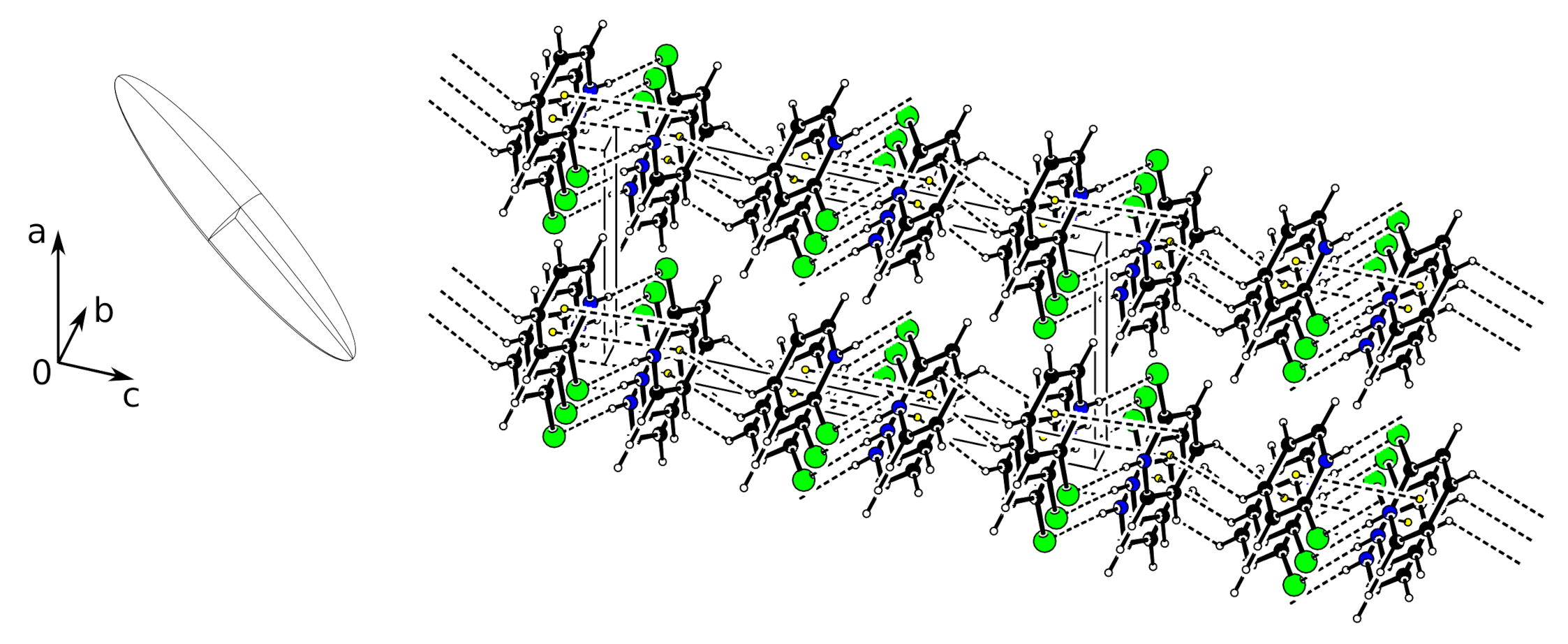
A more appropriate analysis of the temperature dependence of the unit cell is the calculation of the anisotropic thermal expansion tensor. This is a symmetric second-rank tensor [
25]. In the monoclinic case, the tensor has four independent values. For the calculation, we used the
STRAIN ANALYSIS routine of the PLATON software [
20] which uses the algorithm of Ohashi [
26]. According to the convention, the result is expressed in Cartesian space. (Definition for the current orthogonalization:
.) Based on the unit cell parameters at 100 and 260 K, we obtain the following unit strain tensor (
K
):
The corresponding eigenvalues are 122.1(6), 47.2(7) and 6.4(6) (
). Consequently, the representation of the tensor as an ellipsoid shows the large anisotropy (
Figure 4). The directions of the largest and the smallest expansion are both in the
plane. The largest expansion direction forms an angle of
with the
c axis, and
with the
a axis. This direction coincides approximately with the direction of the C–H
interaction (
vide supra).
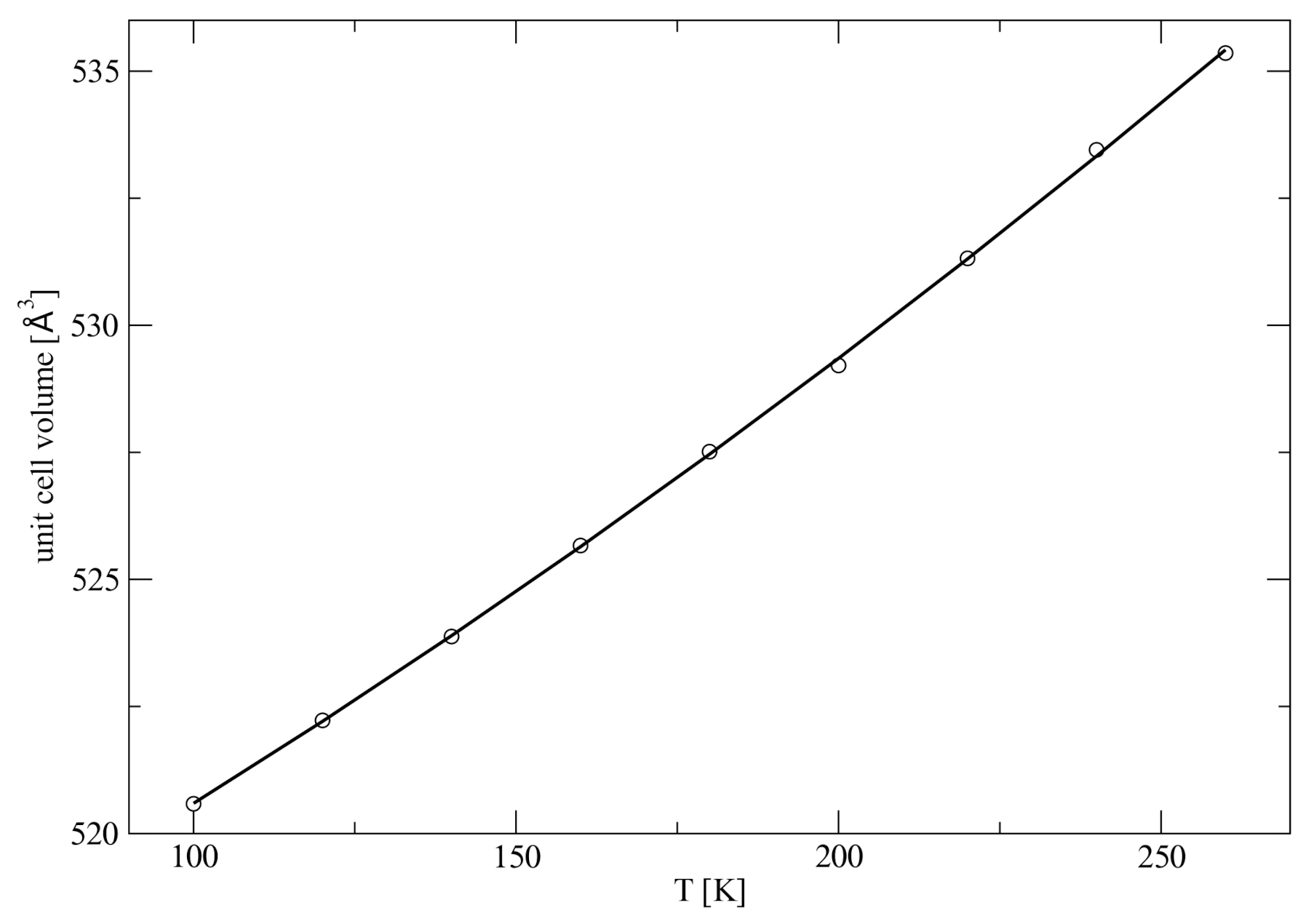
It should be noted that the expansion of the unit cell parameters (
Figure S1) cannot be properly fitted by a linear regression. Quadratic functions had to be used instead. Similarly, the volume expansion also needs a quadratic fit (
Figure 5).
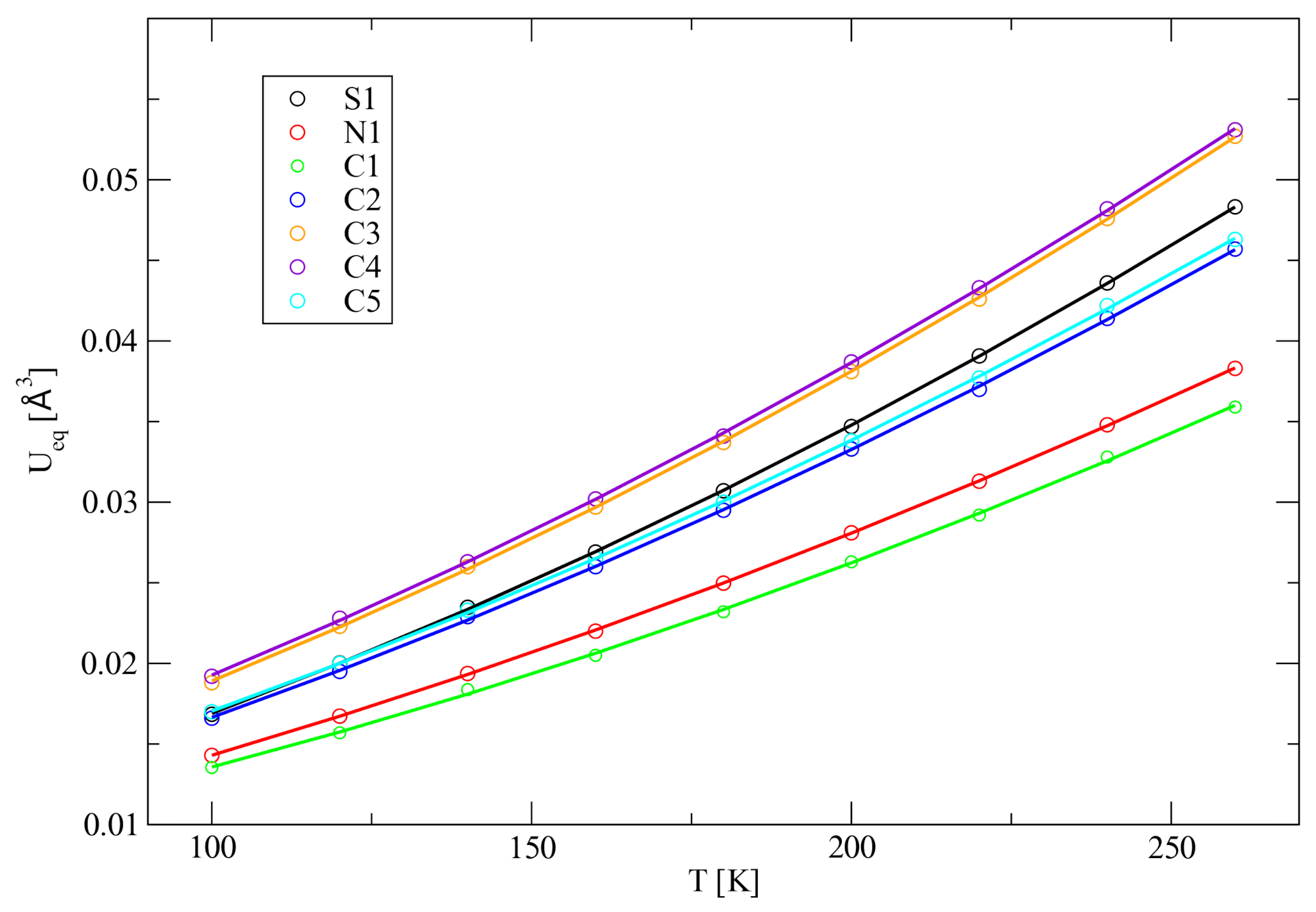
The measurements at nine different temperatures by single
scans not only allowed the determination of unit cell parameters but were also sufficient for structure refinements. Data completeness was 99.9–100.0% at
and 87.6–89.5% at the maximum resolution of
= 30.58–30.73
. From the refinement results, it becomes clear that the atomic displacement parameters do not increase linearly with temperature but a quadratic regression is needed to fit the data points (
Figure 6). Such non-linear behaviour can be associated with anharmonicity [
27]. A quadratic fit is also necessary to fit the temperature dependence of the eigenvalues of the T- and L-tensors, respectively, if the anisotropic displacement parameters are subjected to a TLS rigid-body fit (
Figures S2 and S3).
Table 4 presents the temperature dependence of the intermolecular C–H
interaction. We see an increase of 0.082(2) Å for the C3⋯Cg distance over the observed temperature range. This is larger than the increase in the
stacking interaction (0.0635(6) Å,
Table S3) and significantly larger than the increase in the N–H⋯S hydrogen bond (0.0090(14) Å,
Table S2). This observation is consistent with the shape and orientation of the thermal expansion tensor. We see here another example for the fact that the thermal expansion tensor is related to the strength of intermolecular bonds [
28]. The stronger the intermolecular bond, the smaller is the expansion.
3.3. Hirshfeld Atom Refinement
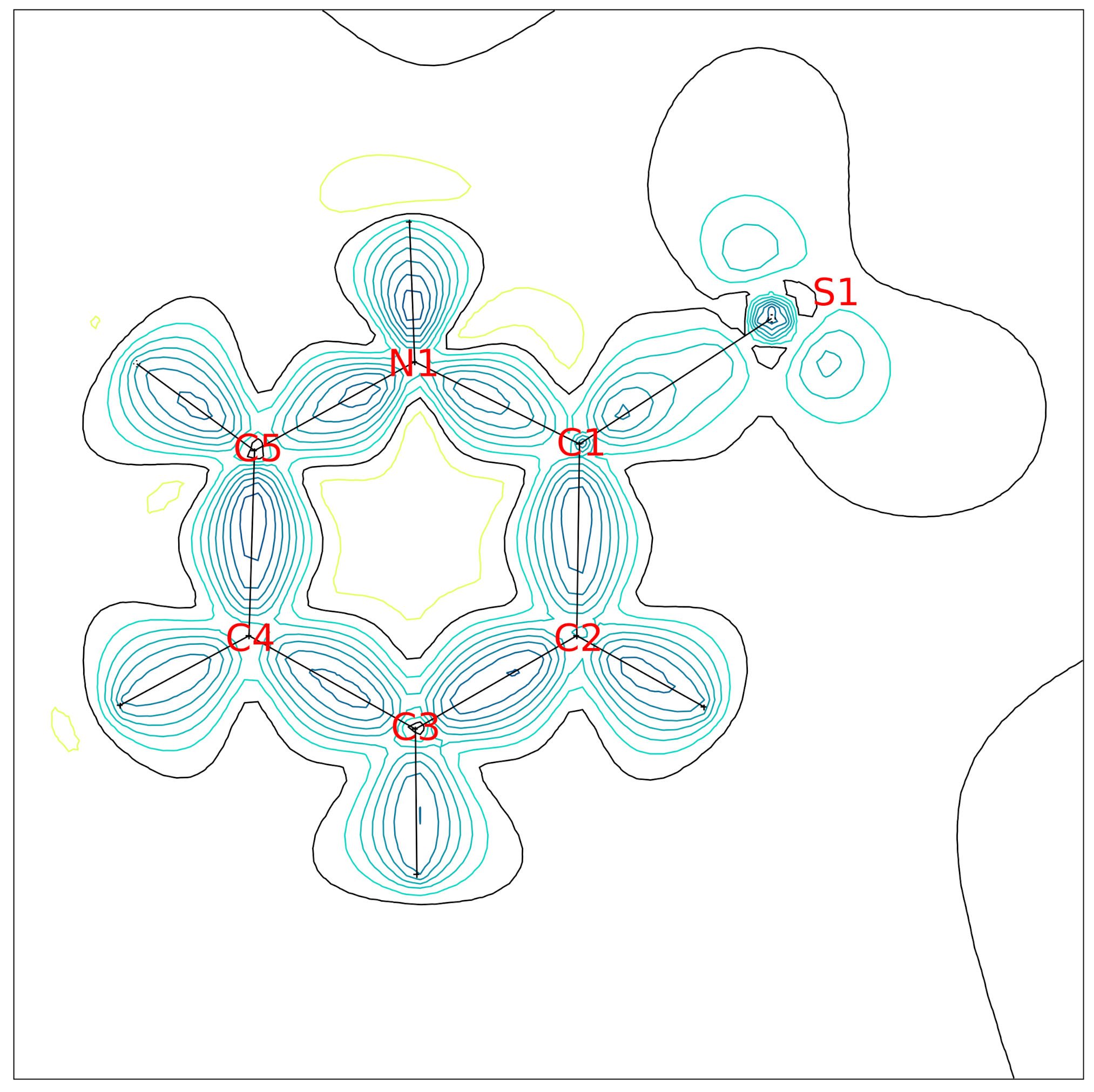
Analysis of the difference electron density of the spherical atom model (
Figure S4) strongly indicates that non-spherical contributions are present in the regions of the chemical bonds. It is a known shortcoming of the independent atom model. Additionally, we see a non-random distribution of residual electron density in the proximity of the sulfur atom. The fractal dimension plot [
29] of the difference density summarizes the deficits of the independent atom model (
Figure S5).
A rather recent but well-established approach is to replace the spherical atom model by non-spherical scattering factors. The latter can be retrieved from databases or obtained on the fly from quantum chemical wavefunction calculations. A possibility to partition the calculated electron density is based on Hirshfeld atoms, and the subsequent structure refinement is often called “Hirshfeld atom refinement (HAR)” [
30]. For the present case, we used the NoSpherA2 implementation [
10] as included in the Olex2 software [
13]. After the refinement with the non-spherical scattering factors, no significant residual electron density can be found on the sites of the bonds (
Figure 7). Clearly, the scattering factors have properly taken the bonding deformation density into account. Consistently, the
R-factor
improves from 1.98% (SHELXL) to 1.16% (NoSpherA2), and the fractal dimension plot (
Figure S6) has become more symmetrical.
Still, the residual electron density map of
(I) is not clean. What remains is non-random density at the sulfur atom. Because all electronic effects in the chemical bonds and the lone pairs should have been properly modeled by the non-spherical scattering factors, we speculate that the current harmonic model for the thermal motion is not sufficient. Anharmonic motion of sulfur atoms has been described before in the literature. For example, the authors of [
6] find “typical shashlik-like residual density patterns” if the anharmonic motion is only modeled with harmonic displacement parameters. This is in contrast with the present case of
(I), where there are four positive peaks at the sulfur in an approximately tetrahedral geometry. A possible explanation for the difference in the residual density pattern is a major difference in the crystal packing.
(I) is involved in hydrogen bonding while the sulfur in [
6] has no close intermolecular contacts. The different packing environment can certainly influence the atomic motion, resulting in a different residual density pattern when treated improperly (shashlik-like vs. tetrahedral). We also note that the packing in
(I) is more dense than the packing in [
6] (
Tables S4 and S5).
If non-spherical scattering factors are used, hydrogen atoms can be refined with anisotropic displacement parameters and the results reach the accuracy of neutron diffraction [
30]. In the present case of
(I), the introduction of anisotropic displacement parameters for the hydrogen atoms improves the structural model significantly (
Tables S6 and S7). A visualisation of the improvement is possible by comparing the calculated structure factors of the model with isotropic hydrogen atoms with those of anisotropic hydrogen atoms (
Figure S7). Several reflections exceed the
criterion.
Independent of the question about significance, we need to assess if the result is physically reasonable or not. The N–H distance is 1.035(3) Å, and the C–H distances are in the range 1.076(3)–1.084(3) Å. This corresponds very much to the expected values for neutron distances (1.020 and 1.085 Å, for N–H and C–H bonds [
31]). From a peanut plot (
Figure S8), the anisotropic parameters of the hydrogen atoms appear exaggerated. Still, the anisotropic hydrogen atoms from the NoSpherA2 result can be a starting point for the further steps of the analysis.
3.4. Multipole Refinement
The quality of the diffraction data allowed a multipole refinement. This approach is based on the Hansen–Coppens formalism [
32] to describe the electron density by a combination of spherical harmonics and the expansion parameters
and
. Essentially, it is a partitioning of the whole electron density into three parts: core density, spherical valence density and aspherical valence density:
For the multipole refinement, we used the XD software [
15]. When parameters up to the hexadecapole level are used, similar agreement factors are reached as in the Hirshfeld atom refinement with NoSpherA2 in Olex2 (
vide supra). Similarly, we again see non-random residual electron density close to the sulfur atom (
Figure S9).
In a next step, anharmonic motion parameters for the sulfur atom were introduced with a third-order Gram–Charlier expansion as implemented in the XD software. This led to a significant improvement according to the Hamilton
R-ratio test [
7,
33] (
Table S8). It was even possible to add the anharmonic motion parameters to all non-hydrogen atoms. The refined expansion coefficients are 1.9–28.6
for S, 0.3–6.3
for N, and 0.0–9.5
for C.
Following an idea from the literature [
34], the anisotropic displacement parameters of the hydrogen atoms were refined freely. In this step, the hydrogen positions were kept fixed at the coordinates from the Hirshfeld atom refinement. Quadrupole parameters for the hydrogen atoms were included. Similar to the literature results [
34], the resulting displacement parameters for hydrogen become smaller than from the Hirshfeld atom refinement and are physically more plausible (
Figure S10). The residual density of this last refinement model with anharmonic motion parameters for the non-hydrogen atoms and refined anisotropic parameters for the hydrogen atoms is featureless, as can be seen in the map (
Figure S11) and the fractal dimension plot (
Figure S12).
Figure 8 shows the deformation density in
(I). This is a very clean map and none of the unexplainable features from [
4] remain. In contrast to the X–N map of [
4], we do not detect a negative residual density on the sulfur position; and in our map, the free electron pairs of the sulfur are visible. There are several potential explanations for this improvement. First of all, the study of [
4] was an X-ray/neutron study (X–N study) while the current investigation is based on X-ray data only. X–N studies have several advantages and they are still very valuable today. A major disadvantage is that the diffraction experiments in X–N studies are usually performed on two different crystals. The crystal size and the crystal quality then differ and can result in scaling problems [
35]. We also believe that our lowering of the temperature to 100 K compared to the literature study at room temperature helped to improve the results. Additionally, the redundancy of the reflections in
(I) is rather high (
) compared to the literature study with a redundancy of
for X-rays and
for neutrons. A higher redundancy gives more reliable intensities and standard uncertainties. Finally, we detected anharmonic behaviour in
(I) and included Gram–Charlier parameters in the refinement. The resolution in [
4] was too low for such a treatment.
The deformation density is obtained by subtracting a promolecule based on spherical, independent atoms from the complete electron density. The choice of the promolecule brings some arbitrariness into the analysis and it is therefore now common practice to consider the total electron density [
36,
37]. A very popular procedure is the topological analysis according to Bader’s theory of
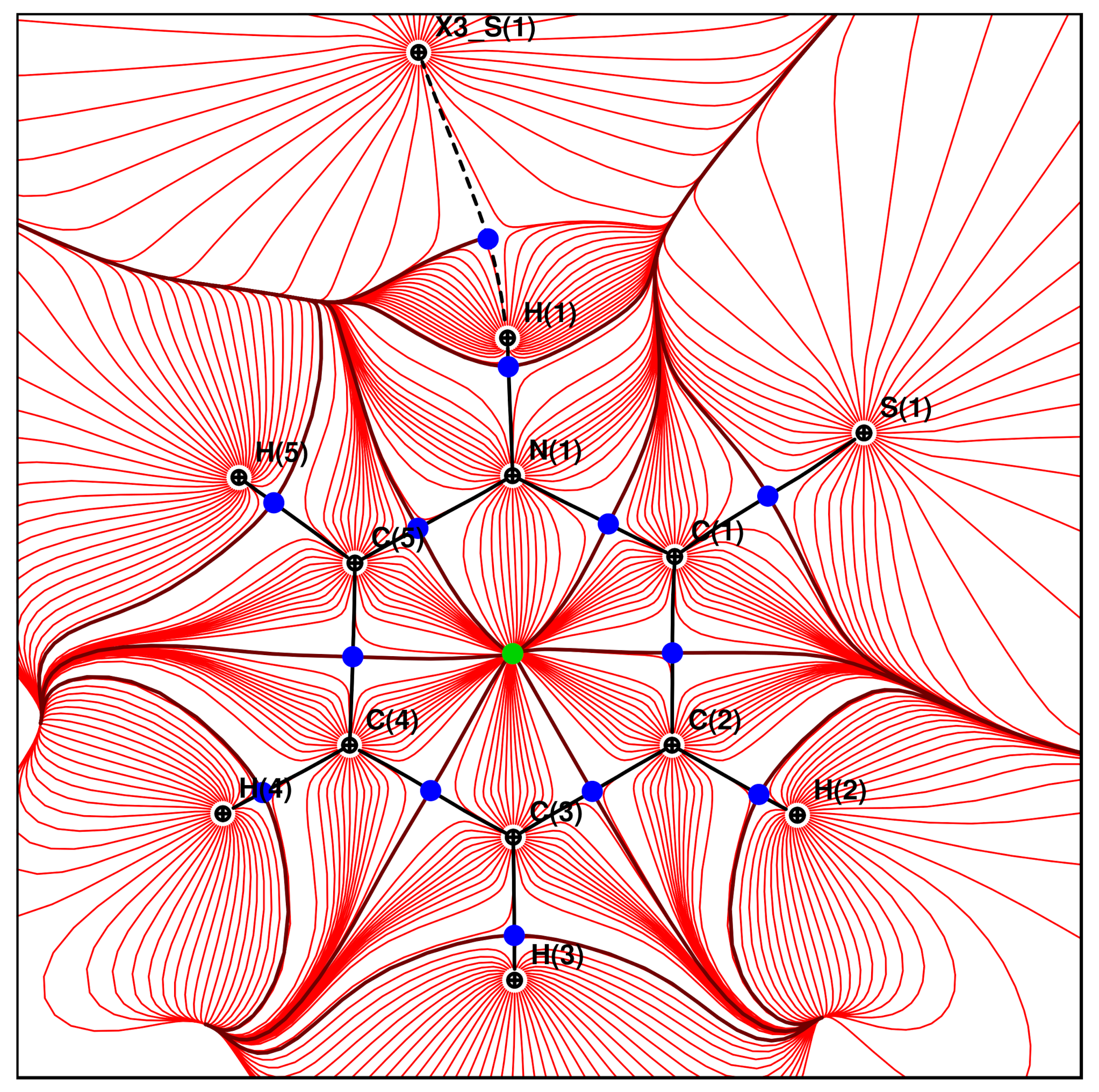
Atoms in Molecules [
38]. A trajectory plot of the electron density is shown in
Figure 9 and an analysis of the (3, −1) critical points is listed in
Table 5 and
Table 6. The bond lengths alternation discussed above is also seen in the Laplacian values and is thus consistent with the strengths of these covalent bonds. In addition, it was possible to detect the bond critical point for the intermolecular N–H⋯S hydrogen bond. Comparing the Laplacian value at this critical point with earlier studies [
39], it becomes clear that the N–H⋯S hydrogen bond is much weaker than typical O–H⋯O and most N–H⋯O hydrogen bonds. In another example, Laplacian values of 3.398 and 3.303 eÅ
were found at the critical points of O–H⋯N hydrogen bonds in an oxime [
40]. It is thus surprising that the weak N–H⋯S hydrogen bond in
(I) results in only little thermal expansion over a wide temperature range.
The search for bond critical points delivers an additional intermolecular interaction which has not come forward in the geometrical discussion above. It is between the C5–H5 group and the sulfur atom. Considering the thermal expansion of this contact (
Table S9), it has similar behaviour and strength as the described
stacking. This C–H⋯S interaction is in the crystallographic
a-direction and should thus connect the layers depicted in
Figure 4. Considering the enrichment ratios in
Table 3 which show that H⋯S contacts are favorable, the C5–H5⋯S1 contact might stabilize the crystal packing.
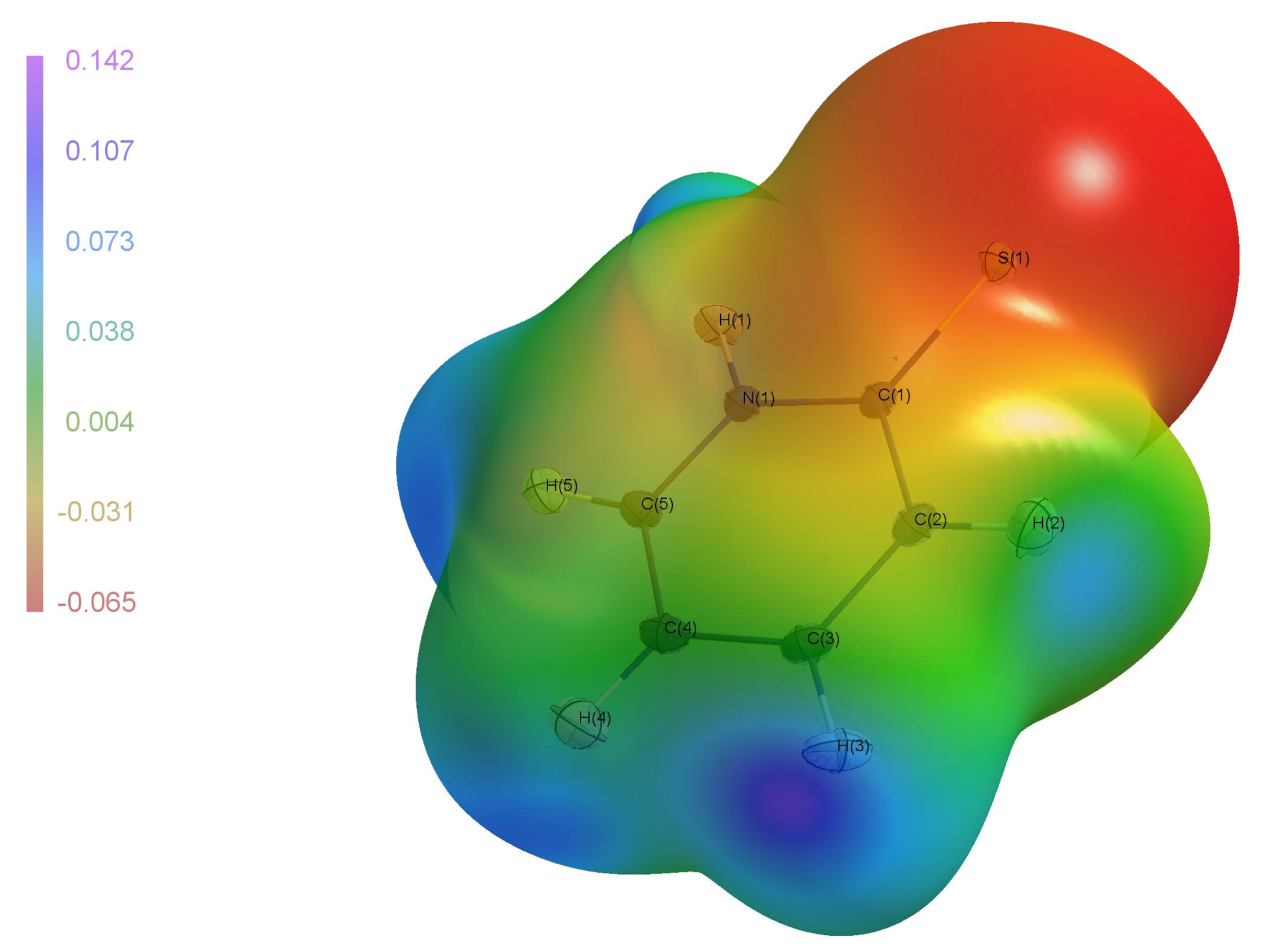
The electrostatic potential which was derived from the experimental electron density (
Figure 10) shows that negative regions are on the sulfur atom S1, while the positive regions are on the hydrogen atoms. By looking at the crystal packing with this picture in mind, we can state qualitatively that only the N–H⋯S hydrogen bond has an electrostatic character. Other attractive intermolecular forces in
(I) will have a dispersive nature.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}