
Mechanical Properties and Buckling of Kagome Graphene under Tension: A Molecular Dynamics Study



Abstract
:1. Introduction
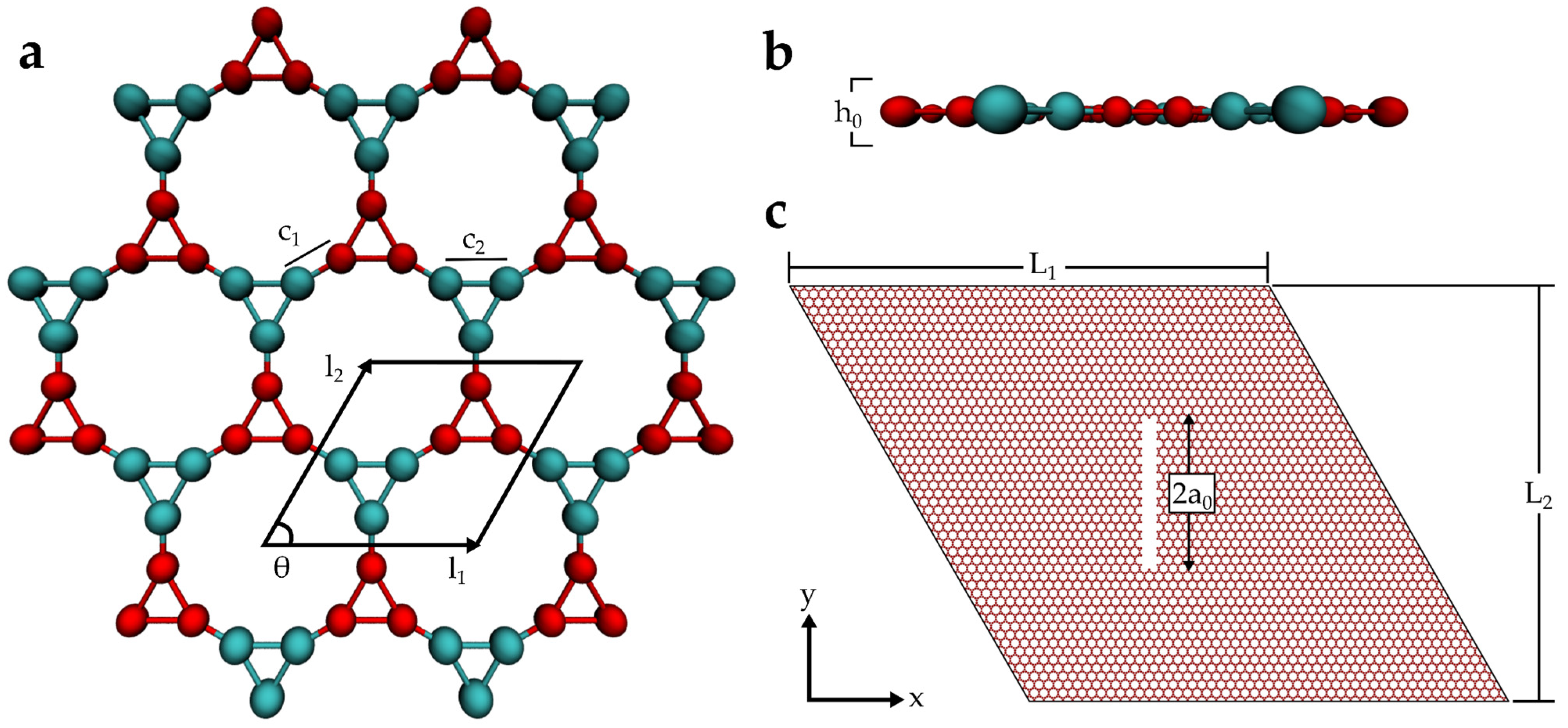
2. Materials and Methods
3. Results and Discussion
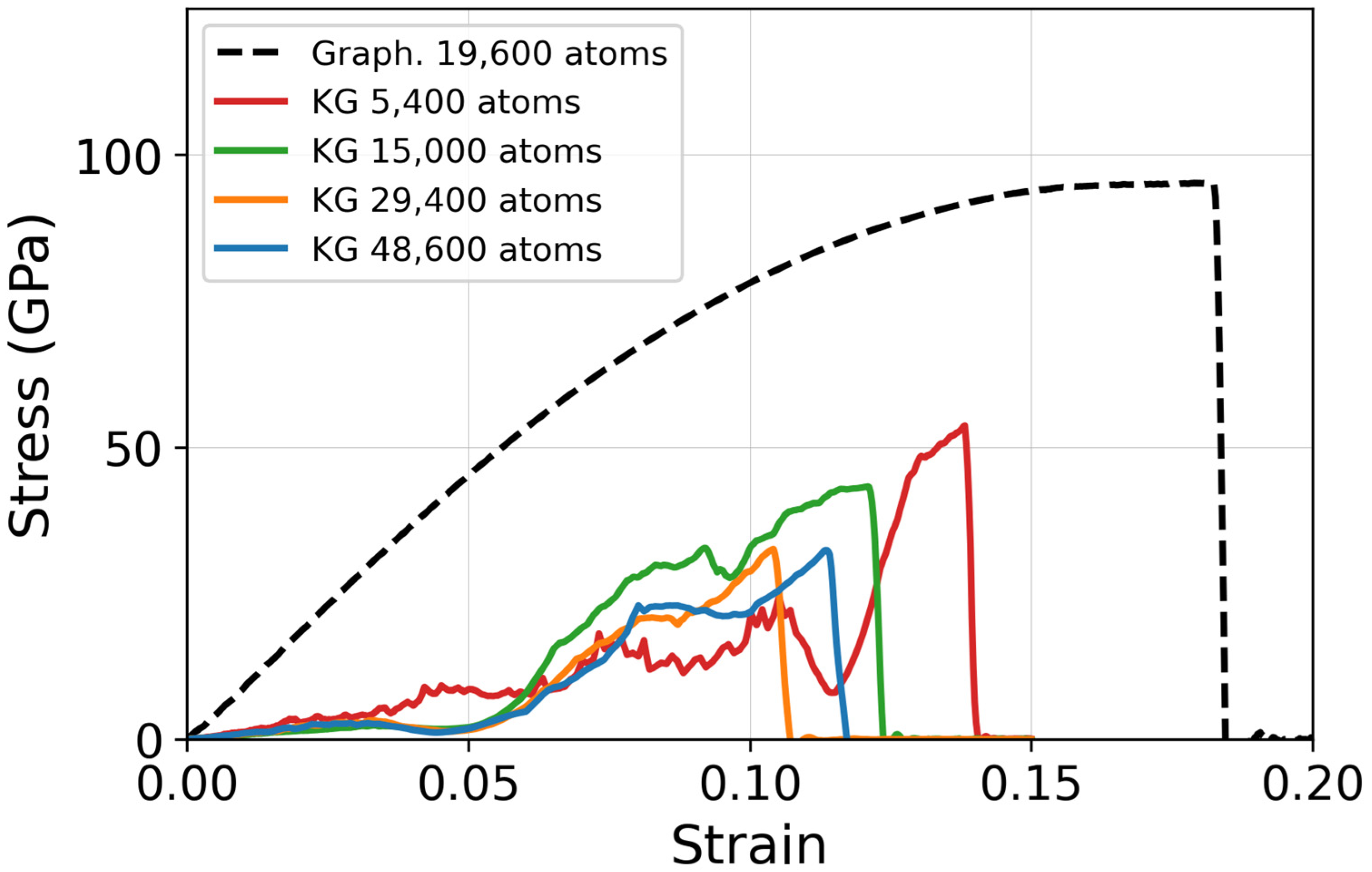
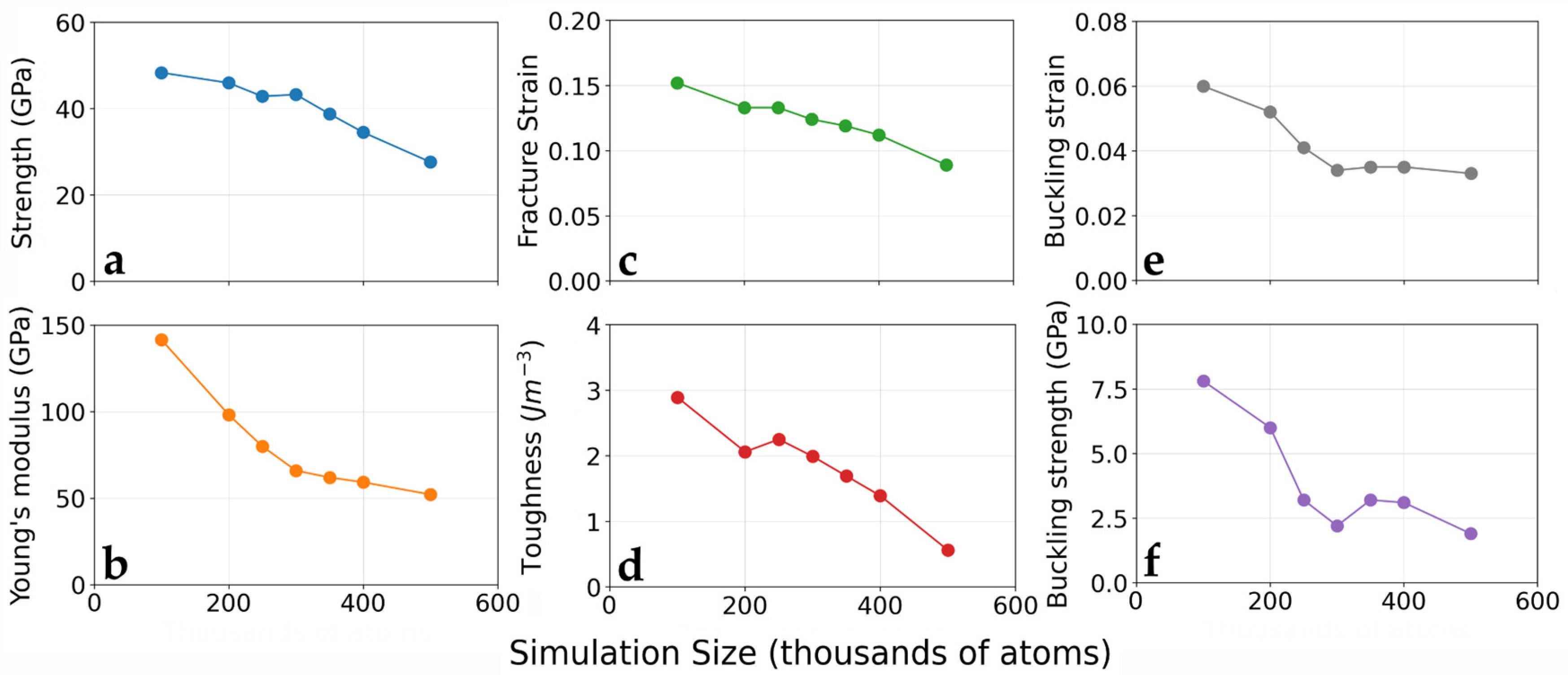
3.1. Simulation Size Effect
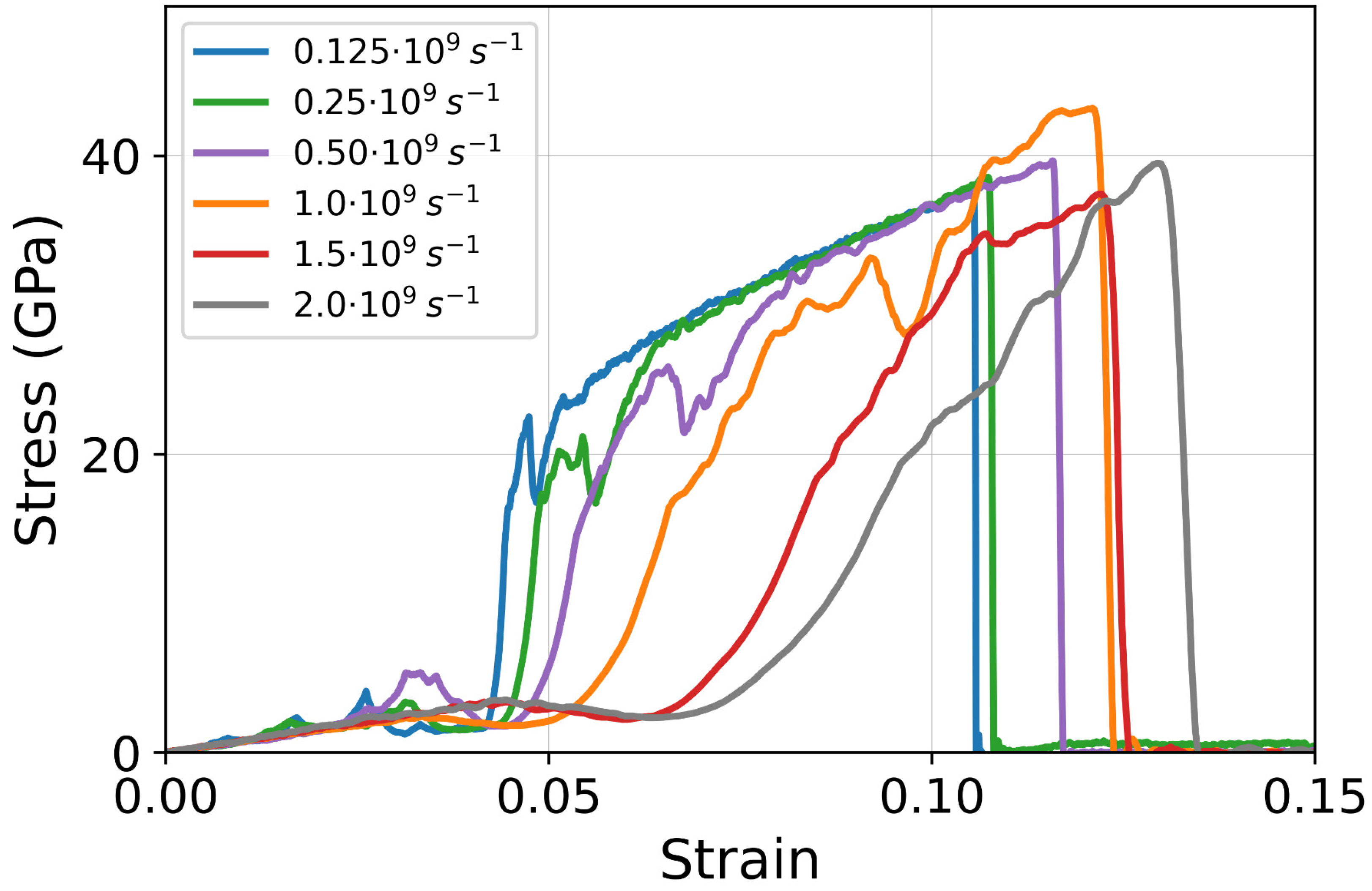
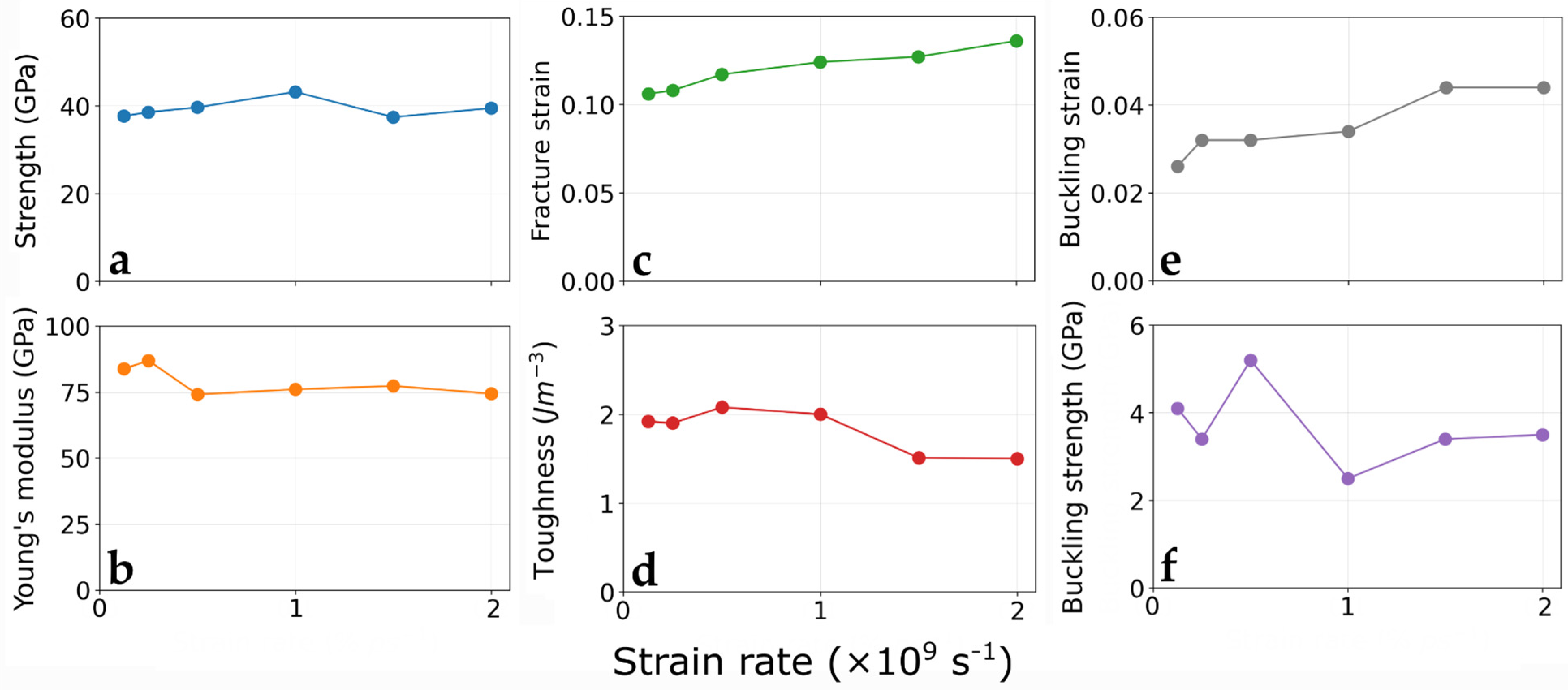
3.2. Strain Rate Effect
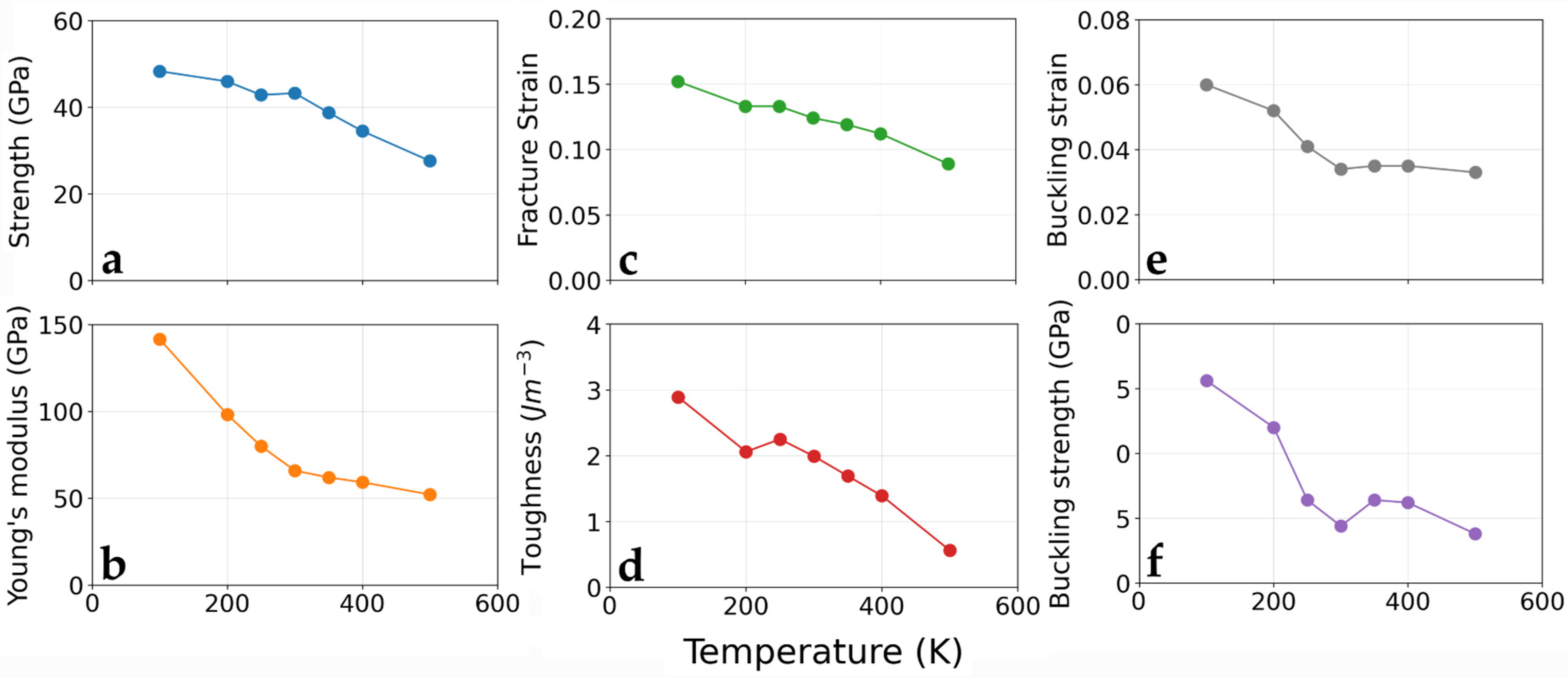
3.3. Temperature Effect
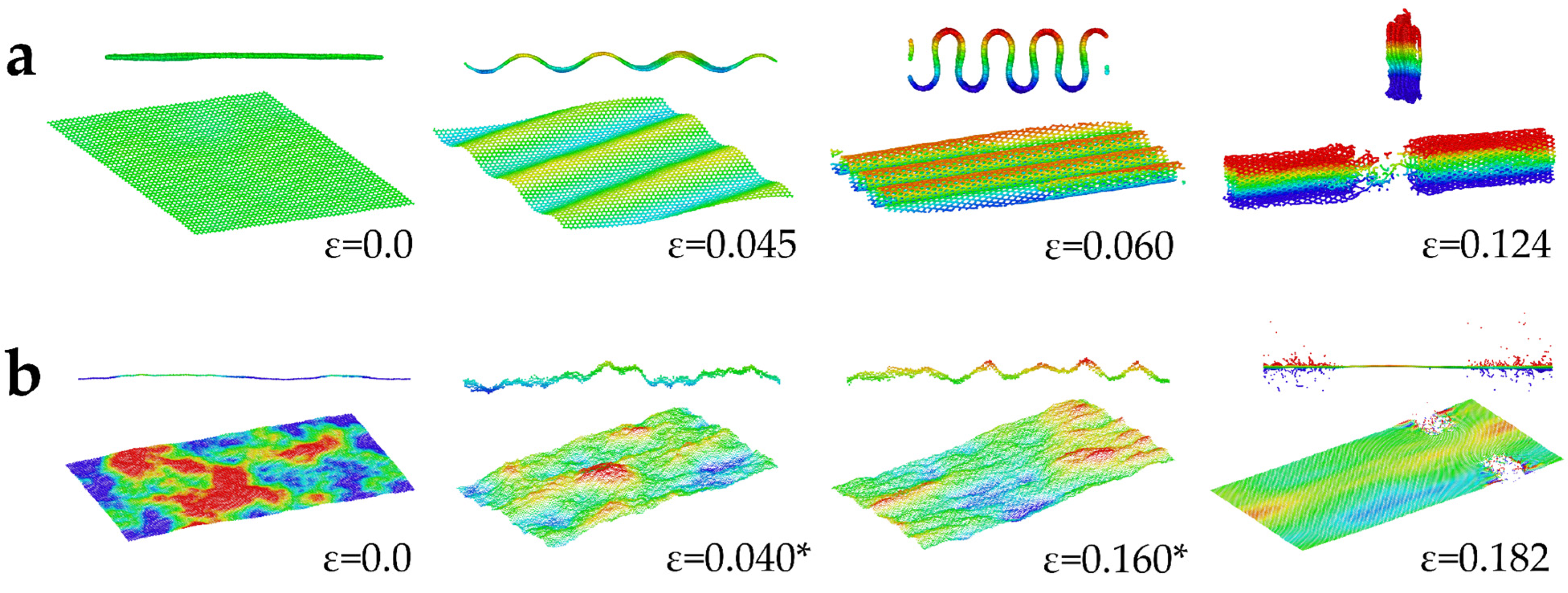
3.4. Fracture Mechanism
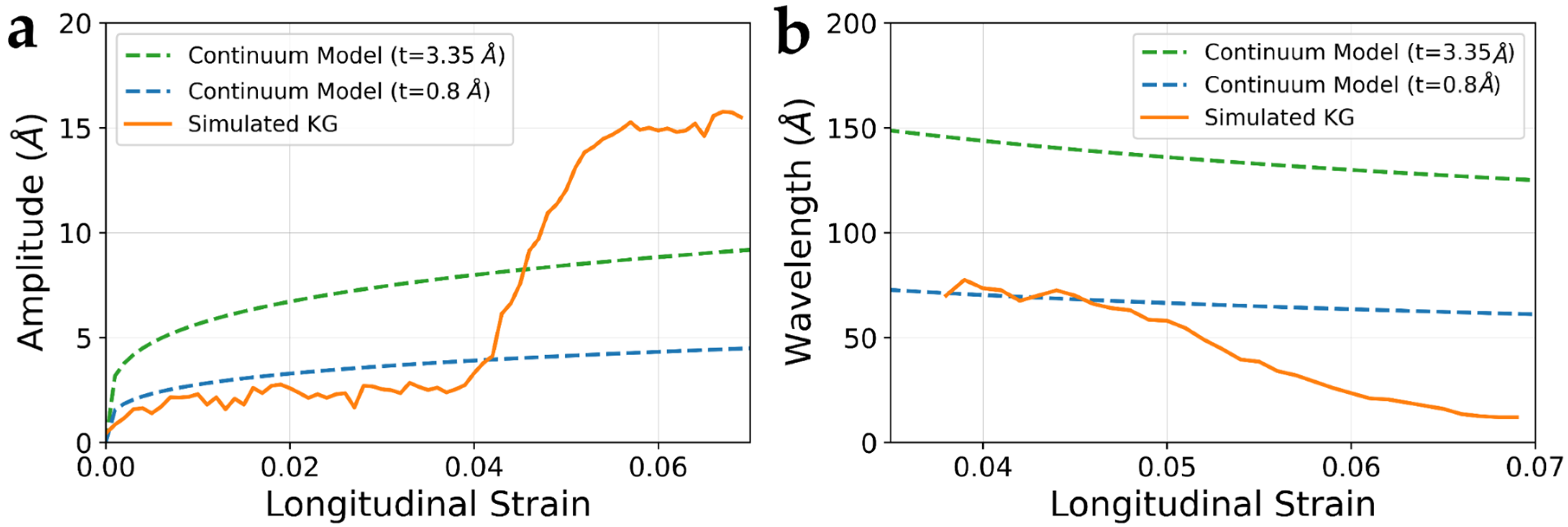
3.5. Wrinkling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Murali, S.; Cai, W.; Li, X.; Suk, J.W.; Potts, J.R.; Ruoff, R.S. Graphene and Graphene Oxide: Synthesis, Properties, and Applications. Adv. Mater. 2010, 22, 3906–3924. [Google Scholar] [CrossRef] [PubMed]
- Morozov, S.V.; Novoselov, K.S.; Katsnelson, M.I.; Schedin, F.; Elias, D.C.; Jaszczak, J.A.; Geim, A.K. Giant Intrinsic Carrier Mobilities in Graphene and Its Bilayer. Phys. Rev. Lett. 2008, 100, 016602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior Thermal Conductivity of Single-Layer Graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef]
- Sarikavak-Lisesivdin, B.; Lisesivdin, S.B.; Ozbay, E.; Jelezko, F. Structural Parameters and Electronic Properties of 2D Carbon Allotrope: Graphene with a Kagome Lattice Structure. Chem. Phys. Lett. 2020, 760, 138006. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, Y.Y.; Wang, H.; West, D.; Xie, Y.; Zhong, J.; Meunier, V.; Cohen, M.L.; Zhang, S.B. Carbon Kagome Lattice and Orbital-Frustration-Induced Metal-Insulator Transition for Optoelectronics. Phys. Rev. Lett. 2014, 113, 085501. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wei, N.; Fan, Z.; Jiang, J.-W.; Rabczuk, T. The Mechanical Properties of Three Types of Carbon Allotropes. Nanotechnology 2013, 24, 095702. [Google Scholar] [CrossRef]
- Jiang, Y.-X.; Yin, J.-X.; Denner, M.M.; Shumiya, N.; Ortiz, B.R.; Xu, G.; Guguchia, Z.; He, J.; Hossain, M.S.; Liu, X.; et al. Unconventional Chiral Charge Order in Kagome Superconductor KV3Sb5. Nat. Mater. 2021, 20, 1353–1357. [Google Scholar] [CrossRef]
- Jiang, N.; Ramanathan, A.; Bacsa, J.; La Pierre, H.S. Synthesis of a D1-Titanium Fluoride Kagome Lattice Antiferromagnet. Nat. Chem. 2020, 12, 691–696. [Google Scholar] [CrossRef]
- Pawlak, R.; Liu, X.; Ninova, S.; D’Astolfo, P.; Drechsel, C.; Liu, J.-C.; Häner, R.; Decurtins, S.; Aschauer, U.; Liu, S.-X.; et al. On-Surface Synthesis of Nitrogen-Doped Kagome Graphene. Angew. Chem. Int. Ed. 2021, 60, 8370–8375. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Cao, Q.; Geng, X.; Yang, Y.; Liu, S.; Peng, Q. The Mechanical Properties of Defective Graphyne. Crystals 2018, 8, 465. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Peng, Q.; Bai, Z.; Gao, F.; Igor, J. Proton Irradiation of Graphene: Insights from Atomistic Modeling. Nanoscale 2019, 11, 20754–20765. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Deng, B.; Zhu, H.; Lan, Y.; Shi, Y.; De, S.; Liu, L.; Chakraborty, P.; Gao, F.; Peng, Q. Magic Auxeticity Angle of Graphene. Carbon 2019, 149, 350–354. [Google Scholar] [CrossRef] [Green Version]
- Morresi, T.; Pedrielli, A.; a Beccara, S.; Gabbrielli, R.; Pugno, N.M.; Taioli, S. Structural, Electronic and Mechanical Properties of All-Sp2 Carbon Allotropes with Density Lower than Graphene. Carbon 2020, 159, 512–526. [Google Scholar] [CrossRef]
- Sun, H.; Mukherjee, S.; Daly, M.; Krishnan, A.; Karigerasi, M.H.; Singh, C.V. New Insights into the Structure-Nonlinear Mechanical Property Relations for Graphene Allotropes. Carbon 2016, 110, 443–457. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Stukowski, A. Visualization and Analysis of Atomistic Simulation Data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33. [Google Scholar] [CrossRef]
- O’Connor, T.C.; Andzelm, J.; Robbins, M.O. AIREBO-M: A Reactive Model for Hydrocarbons at Extreme Pressures. J. Chem. Phys. 2015, 142, 024903. [Google Scholar] [CrossRef]
- Peng, Q.; Wang, G.; Liu, G.-R.; De, S. Van Der Waals Density Functional Theory VdW-DFq for Semihard Materials. Crystals 2019, 9, 243. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Chen, Z.; De, S. A Density Functional Theory Study of the Mechanical Properties of Graphane with van Der Waals Corrections. Mech. Adv. Mater. Struct. 2015, 22, 717–721. [Google Scholar] [CrossRef]
- Kınacı, A.; Haskins, J.B.; Sevik, C.; Çağın, T. Thermal Conductivity of BN-C Nanostructures. Phys. Rev. B 2012, 86, 115410. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.W.; Ward, D.K.; Foster, M.E. An Analytical Bond-Order Potential for Carbon. J. Comp. Chem. 2015, 36, 1719–1735. [Google Scholar] [CrossRef]
- Brenner, D.W.; Shenderova, O.A.; Harrison, J.A.; Stuart, S.J.; Ni, B.; Sinnott, S.B. A Second-Generation Reactive Empirical Bond Order (REBO) Potential Energy Expression for Hydrocarbons. J. Phys. Condens. Matter 2002, 14, 783–802. [Google Scholar] [CrossRef]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A Reactive Potential for Hydrocarbons with Intermolecular Interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Ma, L.; Fan, F.; Zeng, Z.; Peng, C.; Loya, P.E.; Liu, Z.; Gong, Y.; Zhang, J.; Zhang, X.; et al. Fracture Toughness of Graphene. Nat. Commun. 2014, 5, 3782. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.; Sinclair, R.C.; Coveney, P.V. Uncertainty Quantification in Classical Molecular Dynamics. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2021, 379, 20200082. [Google Scholar] [CrossRef]
- Zhao, H.; Aluru, N.R. Temperature and Strain-Rate Dependent Fracture Strength of Graphene. J. Appl. Phys. 2010, 108, 064321. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Lan, L.; Tan, H. The Physics of Wrinkling in Graphene Membranes under Local Tension. Phys. Chem. Chem. Phys. 2013, 15, 2764–2773. [Google Scholar] [CrossRef]
- Zhao, C.; Liu, F.; Kong, Z.; Yan, T.; Ding, F. The Wrinkle Formation in Graphene on Transition Metal Substrate: A Molecular Dynamics Study. Int. J. Smart Nano Mater. 2021, 3, 277–287. [Google Scholar] [CrossRef]
- Lui, C.H.; Liu, L.; Mak, K.F.; Flynn, G.W.; Heinz, T.F. Ultraflat Graphene. Nature 2009, 462, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Rakshit, B.; Mahadevan, P. Absence of Rippling in Graphene under Biaxial Tensile Strain. Phys. Rev. B 2010, 82, 153407. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.; Miao, F.; Chen, Z.; Zhang, H.; Jang, W.; Dames, C.; Lau, C.N. Controlled Ripple Texturing of Suspended Graphene and Ultrathin Graphite Membranes. Nat. Nanotechnol. 2009, 4, 562–566. [Google Scholar] [CrossRef] [Green Version]
- Tapasztó, L.; Dumitrică, T.; Kim, S.J.; Nemes-Incze, P.; Hwang, C.; Biró, L.P. Breakdown of Continuum Mechanics for Nanometre-Wavelength Rippling of Graphene. Nat. Phys. 2012, 8, 739–742. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Wang, B.; Wu, J.; Yang, R.; Dunn, M.L. Bending Rigidity and Gaussian Bending Stiffness of Single-Layered Graphene. Nano Lett. 2013, 13, 26–30. [Google Scholar] [CrossRef] [Green Version]
- Shoaib, H.; Peng, Q.; Alsayoud, A.Q. Atomic Insights into Fracture Characteristics of Twisted Tri-Layer Graphene. Crystals 2021, 11, 1202. [Google Scholar] [CrossRef]
- Fasolino, A.; Los, J.H.; Katsnelson, M.I. Intrinsic Ripples in Graphene. Nat. Mater. 2007, 6, 858–861. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Xu, S.; Xie, Y.; Zhong, C.; Wu, C.; Zhang, S.B. Ferromagnetism and Wigner Crystallization in Kagome Graphene and Related Structures. Phys. Rev. B 2018, 98, 035135. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.H.; Gong, K.; Wang, Q. Controlling the Formation of Wrinkles in a Single Layer Graphene Sheet Subjected to In-Plane Shear. Carbon 2011, 49, 3107–3112. [Google Scholar] [CrossRef]
- Cerda, E.; Mahadevan, L. Geometry and Physics of Wrinkling. Phys. Rev. Lett. 2003, 90, 074302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerda, E.; Ravi-Chandar, K.; Mahadevan, L. Wrinkling of an Elastic Sheet under Tension. Nature 2002, 419, 579–580. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Berry, V. Wrinkled, Rippled and Crumpled Graphene: An Overview of Formation Mechanism, Electronic Properties, and Applications. Mater. Today 2016, 19, 197–212. [Google Scholar] [CrossRef]
- Grafold-Zheng, Y.; Wei, N.; Fan, Z.; Xu, L.; Huang, Z. Mechanical Properties of Grafold: A Demonstration of Strengthened Graphene. Nanotechnology 2011, 22, 479501. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Calculated Value (This Study) | Calculated Value [8] |
|---|---|---|
| θ | 30.0° | 30.0° |
| l1 | 5.45 Å | -- |
| l2 | 5.45 Å | -- |
| h0 | 3.35 Å | 3.47 Å |
| c1 | 1.32 Å | 1.30 Å |
| c2 | 1.62 Å | 1.59 Å |
| L1 | 15.6–46.8 nm | 13 nm |
| L2 | 13.5–40.5 nm | 13 nm |
| a0 | 1.0–6.0 nm | -- |
| Property | Graphene | KG (This Study) | KG [13] 1 |
|---|---|---|---|
| Simulated atoms | 40,000 | 48,600 | 4050 |
| Young’s Modulus (GPa) | 937 ± 4 | 96 ± 23 | 121.5 ± 1.6 |
| Ultimate Strength (GPa) | 95.2 ± 0.1 | 43.1 ± 1.6 | 8.8 |
| Strain at Failure | 0.186 ± 0.003 3 | ~0.047 ± 0.0043 2 | 0.234 3 |
| 0.124 ± 0.0014 3 | |||
| Toughness (J m−3) | 12.2 ± 0.3 | 1.92 ± 0.13 | NA |
| 1.0 nm | 2.0 nm | 4.0 nm | 6.0 nm | |
|---|---|---|---|---|
| 1.69 | 0.952 | 0.541 | 0.373 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wavrunek, T.; Peng, Q.; Abu-Zahra, N. Mechanical Properties and Buckling of Kagome Graphene under Tension: A Molecular Dynamics Study. Crystals 2022, 12, 292. https://doi.org/10.3390/cryst12020292
Wavrunek T, Peng Q, Abu-Zahra N. Mechanical Properties and Buckling of Kagome Graphene under Tension: A Molecular Dynamics Study. Crystals. 2022; 12(2):292. https://doi.org/10.3390/cryst12020292
Chicago/Turabian StyleWavrunek, Trevor, Qing Peng, and Nidal Abu-Zahra. 2022. "Mechanical Properties and Buckling of Kagome Graphene under Tension: A Molecular Dynamics Study" Crystals 12, no. 2: 292. https://doi.org/10.3390/cryst12020292