
Molecular Structures of the Pyridine-2-olates PhE(pyO)3 (E = Si, Ge, Sn)—[4+3]-Coordination at Si, Ge vs. Heptacoordination at Sn

Abstract
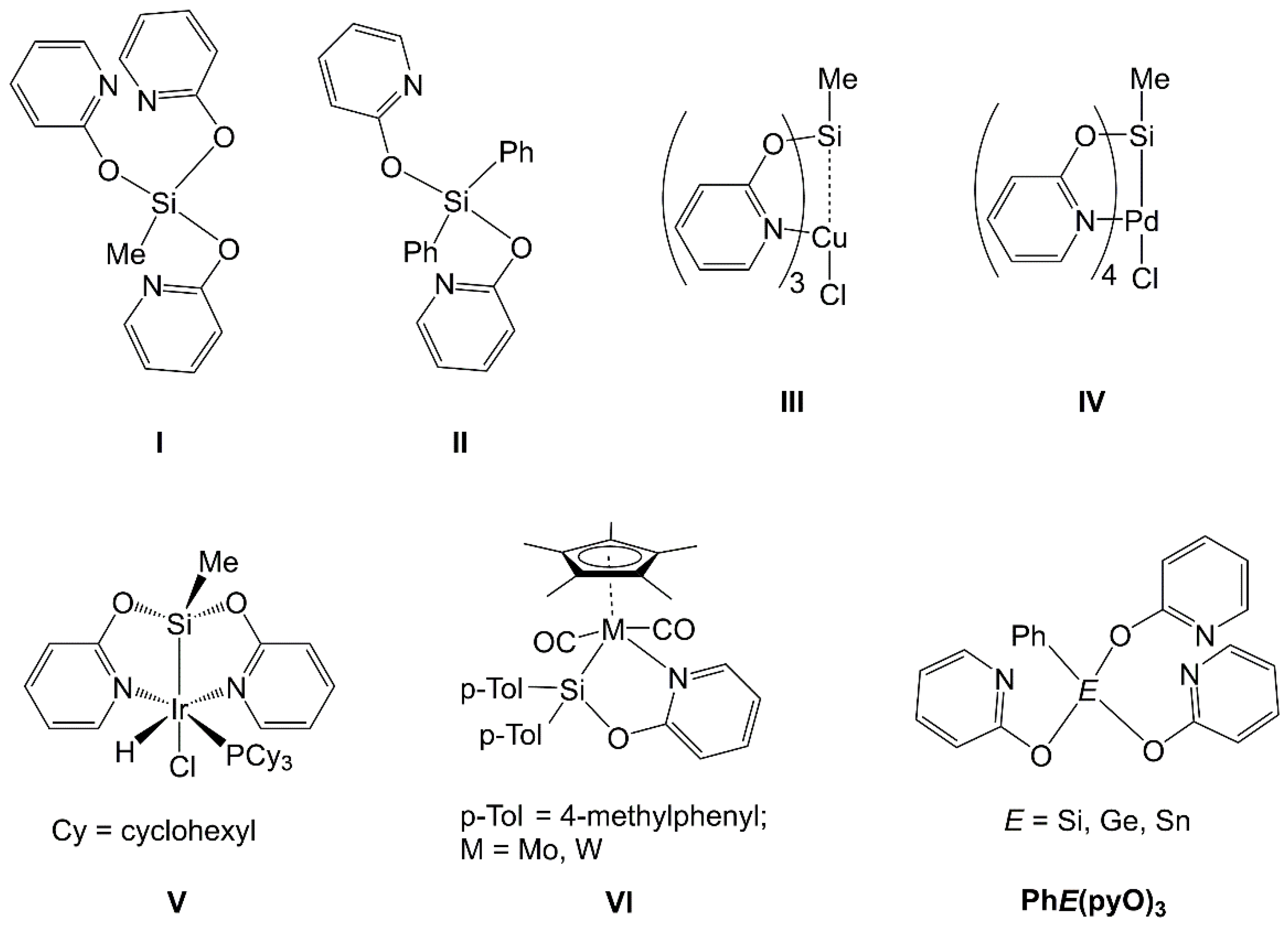
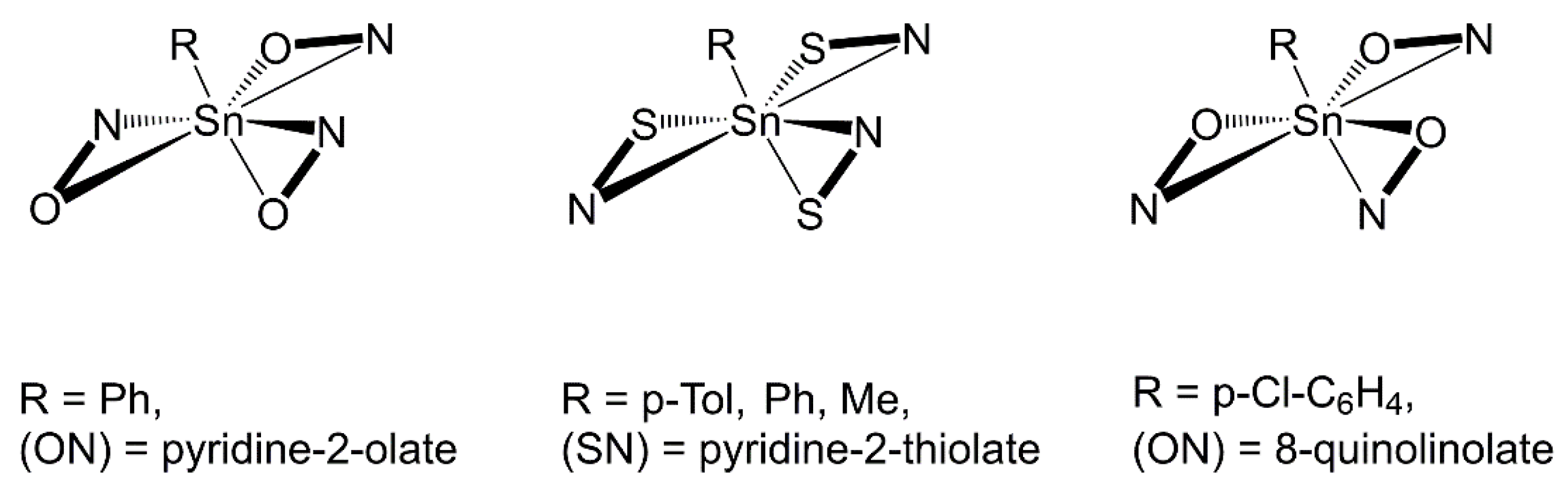
:1. Introduction
2. Materials and Methods
2.1. General Considerations
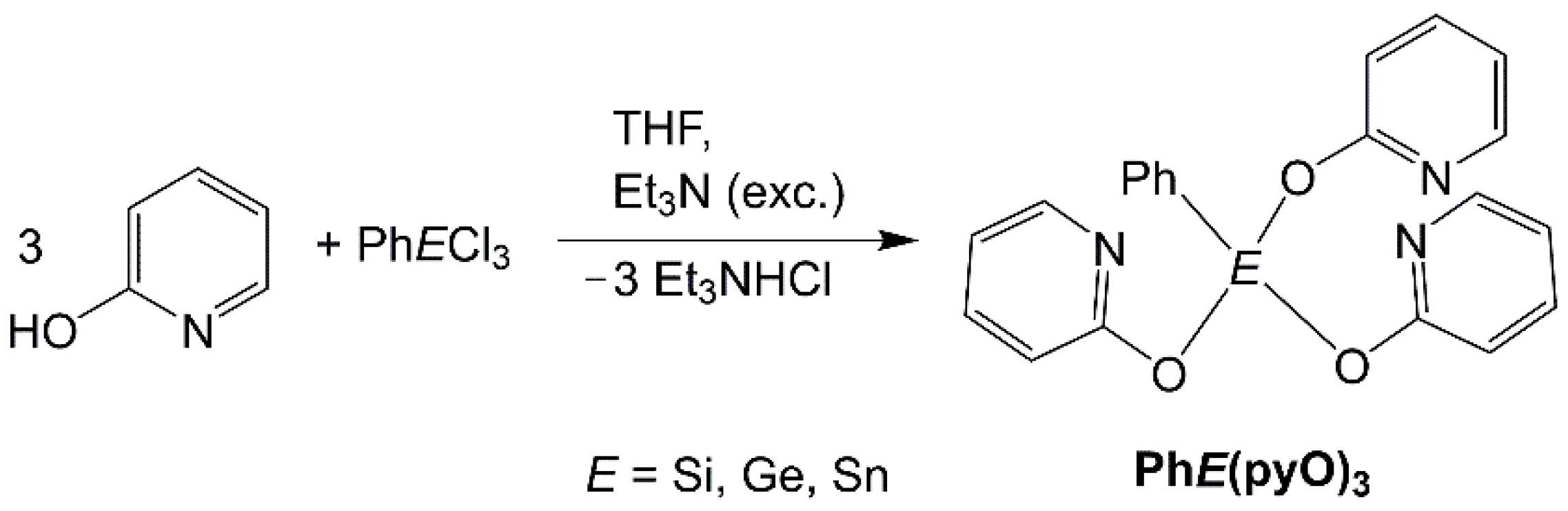
2.2. Syntheses and Characterization
3. Results and Discussion
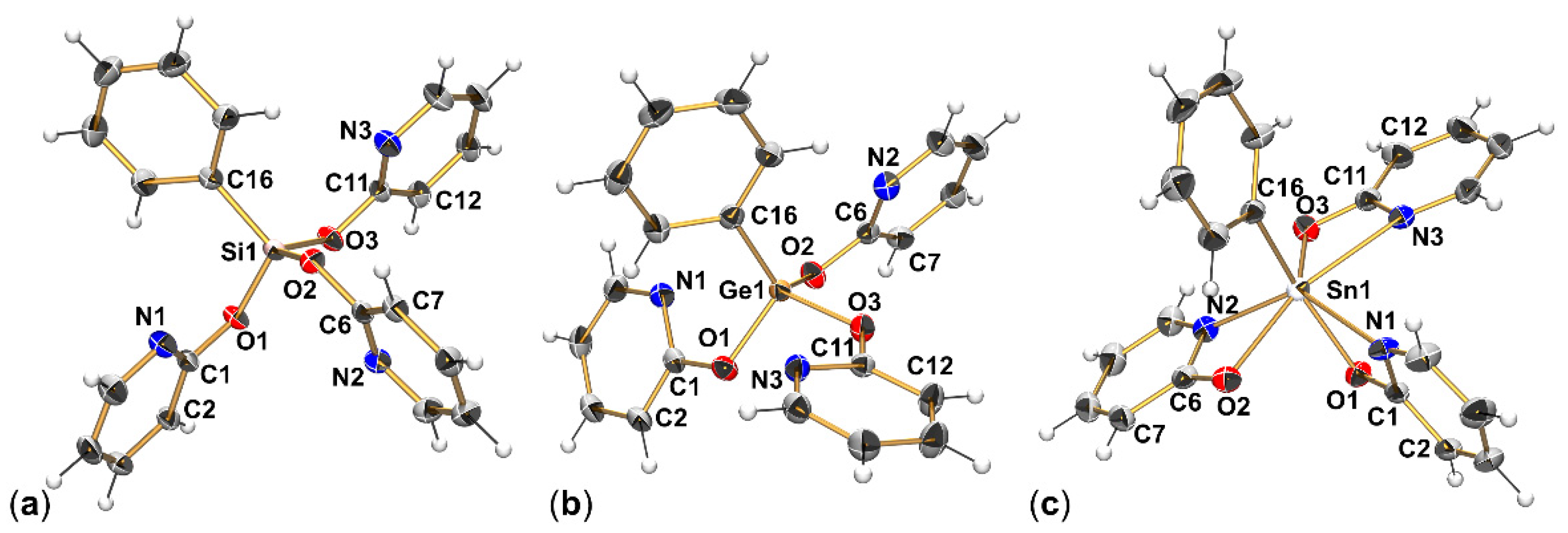
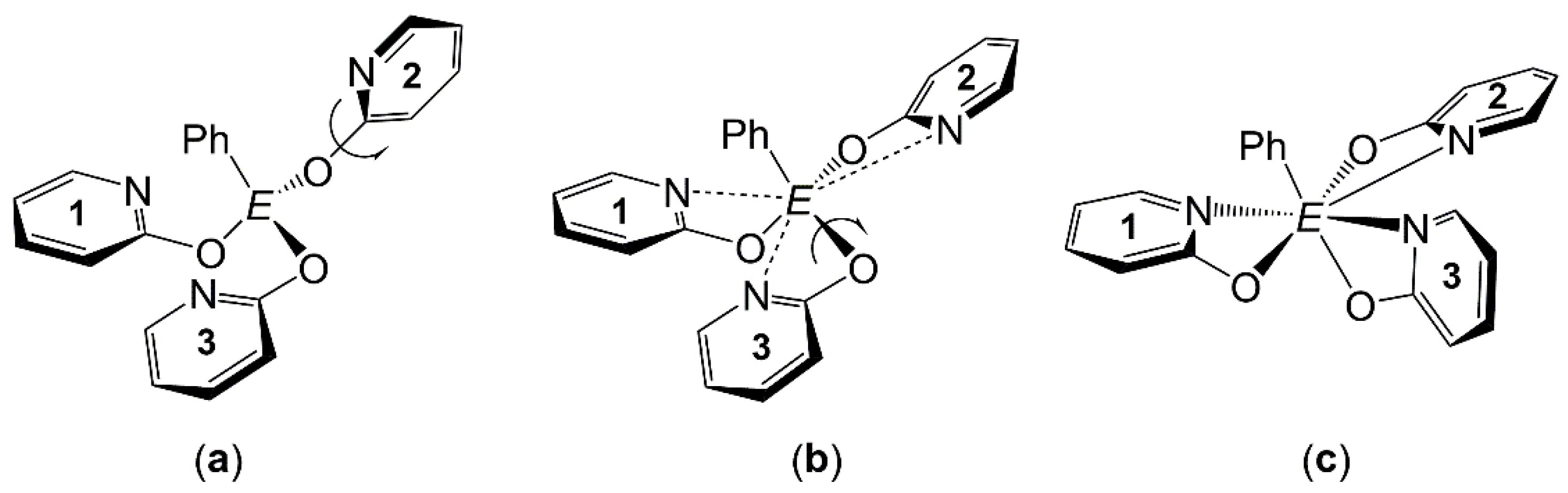
3.1. Crystallographic Analysis of the Molecular Structures of PhE(pyO)3 (E = Si, Ge, Sn)
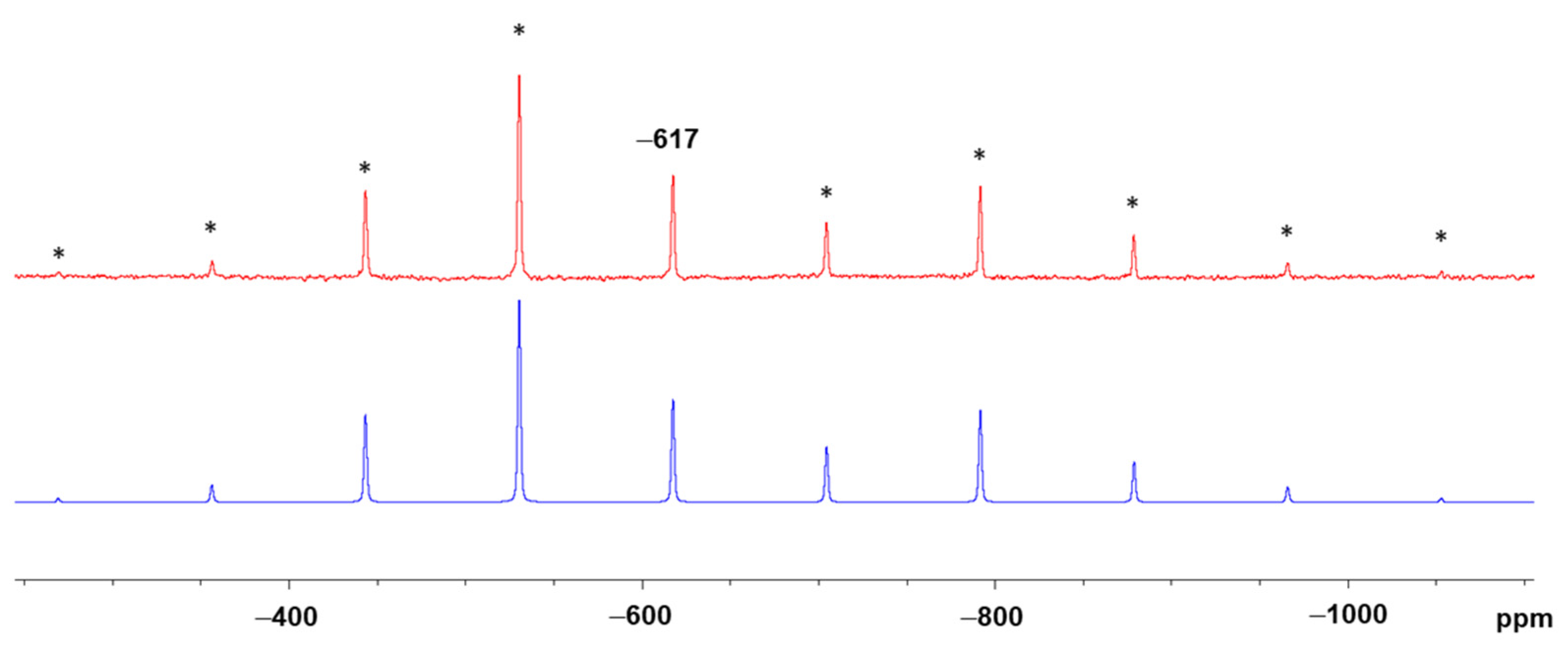
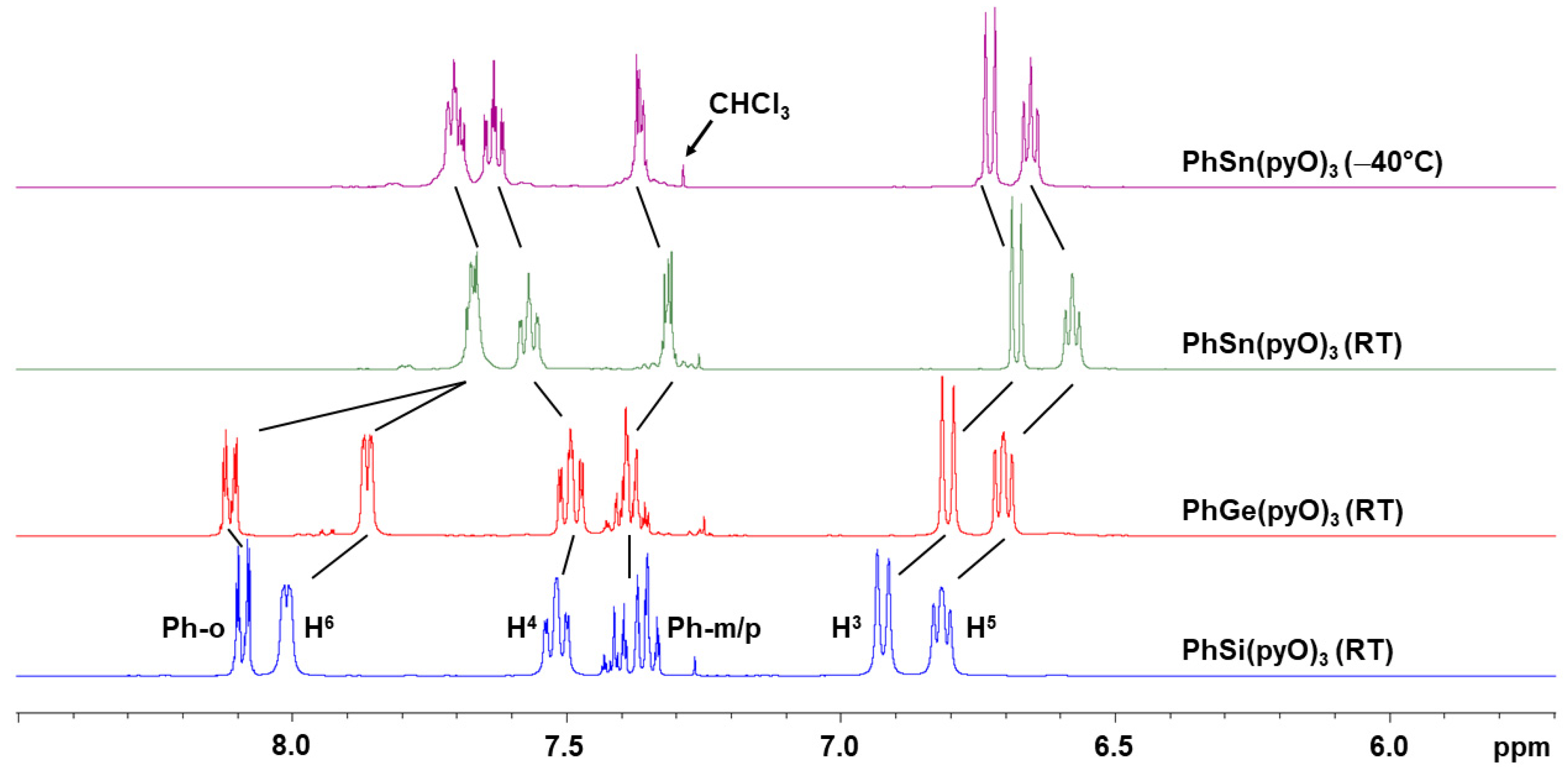
3.2. NMR Spectroscopic Analyses of PhE(pyO)3 (E = Si, Ge, Sn)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | PhSi(pyO)3 · THF 1 | PhSi(pyO)3 · CHCl3 2 | PhGe(pyO)3 | PhSn(pyO)3 3 |
|---|---|---|---|---|
| Formula | C25H25N3O4Si | C22H18Cl3N3S3Si | C21H17GeN3O3 | C21H17N3O3Sn |
| Mr | 459.57 | 506.83 | 431.96 | 478.07 |
| T (K) | 200(2) | 180(2) | 180(2) | 150(2) |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | monoclinic | monoclinic | triclinic | monoclinic |
| Space group | P21/c | P21/c | P | P21 |
| a (Å) | 12.1634(6) | 12.2304(4) | 9.7609(3) | 8.4129(3) |
| b (Å) | 17.0246(10) | 16.8989(4) | 9.7775(3) | 10.9605(2) |
| c (Å) | 11.4268(7) | 11.3681(3) | 9.8668(3) | 10.8580(3) |
| α (°) | 90 | 90 | 82.957(2) | 90 |
| β (°) | 101.450(4) | 101.160(2) | 83.434(2) | 98.385(2) |
| γ (°) | 90 | 90 | 86.564(2) | 90 |
| V (Å3) | 2319.1(2) | 2395.13(11) | 927.42(5) | 990.51(5) |
| Z | 4 | 4 | 2 | 2 |
| ρcalc (g·cm−1) | 1.32 | 1.46 | 1.55 | 1.60 |
| μMoKα (mm−1) | 0.1 | 0.5 | 1.7 | 1.3 |
| F(000) | 968 | 1040 | 440 | 476 |
| θmax (°), Rint | 28.0, 0.0243 | 28.0, 0.0454 | 28.0, 0.0428 | 29.0, 0.0270 |
| Completeness | 99.8% | 99.9% | 100% | 99.9% |
| Reflns collected | 15,889 | 26,422 | 24,367 | 56,321 |
| Reflns unique | 5587 | 5563 | 4480 | 5258 |
| Restraints | 0 | 73 | 0 | 1 |
| Parameters | 253 | 353 | 254 | 254 |
| GoF | 1.072 | 1.053 | 1.077 | 1.256 |
| R1, wR2 [I > 2σ(I)] | 0.0425, 0.1078 | 0.0420, 0.0983 | 0.0310, 0.0687 | 0.0184, 0.0416 |
| R1, wR2 (all data) | 0.0639, 0.1203 | 0.0570, 0.1059 | 0.0368, 0.0708 | 0.0203, 0.0433 |
| Largest peak/hole (e·Å−3) | 0.23, −0.39 | 0.30, −0.34 | 0.30, −0.50 | 0.93, −0.96 |
| Bond 1 | PhSi(pyO)3 · CHCl3 | PhGe(pyO)3 | PhSn(pyO)3 |
|---|---|---|---|
| O1–C1 | 1.362(2) | 1.350(2) | 1.314(4) |
| O2–C6 | 1.364(2) | 1.350(2) | 1.288(4) |
| O3–C11 | 1.360(2) | 1.350(2) | 1.315(4) |
| N1–C1 | 1.322(2) | 1.327(3) | 1.347(4) |
| N2–C6 | 1.321(2) | 1.324(3) | 1.348(4) |
| N3–C11 | 1.319(2) | 1.324(3) | 1.337(4) |
| C1–C2 | 1.386(2) | 1.391(3) | 1.408(4) |
| C5–C6 | 1.387(2) | 1.389(3) | 1.421(4) |
| C11–C12 | 1.392(2) | 1.391(3) | 1.403(4) |
| C2–C3 | 1.379(2) | 1.379(3) | 1.375(5) |
| C7–C8 | 1.379(2) | 1.376(3) | 1.368(5) |
| C12–C13 | 1.379(2) | 1.374(3) | 1.380(6) |
| C3–C4 | 1.385(3) | 1.387(3) | 1.399(5) |
| C8–C9 | 1.386(2) | 1.388(3) | 1.394(6) |
| C13–C14 | 1.384(3) | 1.389(3) | 1.391(6) |
| C4–C5 | 1.372(3) | 1.376(3) | 1.370(5) |
| C9–C10 | 1.371(3) | 1.375(3) | 1.379(5) |
| C14–C15 | 1.374(3) | 1.372(3) | 1.378(6) |
| N1–C5 | 1.345(2) | 1.343(3) | 1.341(4) |
| N2–C10 | 1.349(2) | 1.349(3) | 1.347(4) |
| N3–C15 | 1.347(2) | 1.347(3) | 1.353(4) |
| Compound | E | Solvent | E···N(pyS) Coordination? 1 | δ13C(C2) | δ13C(C6) |
|---|---|---|---|---|---|
| PhClSi(pyS)2 [40] | Si | CD2Cl2 | 1 | 167.5 | 140.8 |
| MeClSi(pyS)2 [40] | Si | CD2Cl2 | 1 | 168.1 | 140.4 |
| Si(pyS)4 [41] | Si | CDCl3 | 0.5 | 163.4 | 144.7 |
| Sn(pyS)4 [41] | Sn | CDCl3 | 0.5 | 162.9 | 145.7 |
| Ph2Si(pyS)2 [3] | Si | CDCl3 | 0 | 158.1 | 147.5 |
| Me2Si(pyS)2 [3] | Si | CDCl3 | 0 | 158.7 | 148.8 |
References
- Kazimierczuk, K.; Dołęga, A. Department of A. Synthesis and structural characterization of new cyclic siloxane with functionalized organic substituents. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 1140–1143. [Google Scholar] [CrossRef]
- Ehrlich, L.; Gericke, R.; Brendler, E.; Wagler, J. (2-Pyridyloxy)silanes as Ligands in Transition Metal Coordination Chemistry. Inorganics 2018, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Seidel, A.; Weigel, M.; Ehrlich, L.; Gericke, R.; Brendler, E.; Wagler, J. Molecular Structures of the Silicon Pyridine-2-(thi)olates Me3Si(pyX), Me2Si(pyX)2 and Ph2Si(pyX)2 (py = 2-Pyridyl, X = O, S), and Their Intra- and Intermolecular Ligand Exchange in Solution. Crystals 2022, 12, 1054. [Google Scholar] [CrossRef]
- Sun, J.; Ou, C.; Wang, C.; Uchiyama, M.; Deng, L. Silane-Functionalized N-Heterocyclic Carbene−Cobalt Complexes Containing a Five-Coordinate Silicon with a Covalent Co−Si Bond. Organometallics 2015, 34, 1546–1551. [Google Scholar] [CrossRef]
- Julián, A.; Garcés, K.; Lalrempuia, R.; Jaseer, E.A.; García-Orduña, P.; Fernández-Alvarez, F.J.; Lahoz, F.J.; Oro, L.A. Reactivity of Ir–NSiN Complexes: Ir-Catalyzed Dehydrogenative Silylation of Carboxylic Acids. ChemCatChem 2018, 10, 1027–1034. [Google Scholar] [CrossRef]
- Kanno, Y.; Komuro, T.; Tobita, H. Direct Conversion of a Si−C(aryl) Bond to Si−Heteroatom Bonds in the Reactions of η3-α-Silabenzyl Molybdenum and Tungsten Complexes with 2-Substituted Pyridines. Organometallics 2015, 34, 3699–3705. [Google Scholar] [CrossRef]
- This refers to a search in the Cambridge Structure Database using ConQuest version 2022.2.0.
- Hadjikakou, K.; Jurkschat, K.; Schürmann, M. Novel organotin(IV) compounds derived from bis(organostannyl)methanes: Synthesis and crystal structures of bis[diphenyl(pyridin-2-onato)stannyl]methane and bis[bromophenyl(pyrimidine-2-thionato)stannyl]methane · C7H8. J. Organomet. Chem. 2006, 691, 1637–1642. [Google Scholar] [CrossRef]
- Ma, C.; Li, Q.; Zhang, R. A novel self-assembling of mixed tri- and dibutyltin macrocyclic complex with solvothermal synthesis method. Inorg. Chim. Acta 2009, 362, 2937–2940. [Google Scholar] [CrossRef]
- Haribabu, J.; Tamura, Y.; Yokoi, K.; Balachandran, C.; Umezawa, M.; Tsuchiya, K.; Yamada, Y.; Karvembu, R.; Aoki, S. Synthesis and Anticancer Properties of Bis- and Mono(cationic peptide) Hybrids of Cyclometalated Iridium(III) Complexes: Effect of the Number of Peptide Units on Anticancer Activity. Eur. J. Inorg.Chem. 2021, 2021, 1796–1814. [Google Scholar] [CrossRef]
- Zhou, J.; Yu, G.; Yang, J.; Shi, B.; Ye, B.; Wang, M.; Huang, F.; Stang, P.J. Polymeric Nanoparticles Integrated from Discrete Organoplatinum(II) Metallacycle by Stepwise Post-assembly Polymerization for Synergistic Cancer Therapy. Chem. Mater. 2020, 32, 4564–4573. [Google Scholar] [CrossRef]
- Yusof, E.N.M.; Ravoof, T.B.S.A.; Page, A.J. Cytotoxicity of Tin(IV)-based compounds: A review. Polyhedron 2021, 198, 115069. [Google Scholar] [CrossRef]
- Gericke, R.; Wagler, J. Ruthenium complexes of phosphino derivatives of carboxylic amides: Synthesis and characterization of tridentate P,E2 and tetradentate P,E3 (E = N,O) ligands and their reactivity towards [RuCl2(PPh3)3]. Polyhedron 2017, 125, 57–67. [Google Scholar] [CrossRef]
- Herzfeld, J.; Berger, A.E. Sideband intensities in NMR spectra of samples spinning at the magic angles. J. Chem. Phys. 1980, 73, 6021–6030. [Google Scholar] [CrossRef]
- Mason, J. Conventions for the reporting of nuclear magnetic shielding (or shift) tensors suggest by participants in the NATO AEW in NMR Shielding Constants at the University of Maryland, College Park, July 1992. Solid State Nucl. Magn. Reson. 1993, 2, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Program for the Solution of Crystal Structures; SHELXS-97; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Program for the Refinement of Crystal Structures; SHELXL-2014/7; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, L.J. ORTEP-3 for windows—A version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- POV-RAY (Version 3.7), Trademark of Persistence of Vision Raytracer Pty. Ltd.,Williamstown, Victoria (Australia). Copyright Hallam Oaks Pty. Ltd., 1994–2004. Available online: http://www.povray.org/download/ (accessed on 28 June 2021).
- Herzog, U.; Rheinwald, G. Novel Chalcogenides of Silicon with Bicyclo[2.2.2]octane Skeletons, MeSi(SiMe2E)3MR (E = S, Se, Te; M = Si, Ge, Sn; R = Me, Ph). Organometallics 2001, 20, 5369–5374. [Google Scholar] [CrossRef]
- Schaeffer, C.D., Jr.; Zuckerman, J.J. Pulsed fourier transform NMR of substituted aryltrimethyltin derivatives: II. (119Sn-13C) coupling constants and 13C chemical shifts of meta- and para-derivatives. J. Organomet. Chem. 1973, 55, 97–110. [Google Scholar] [CrossRef]
- Mantina, M.; Chamberlin, A.C.; Valero, R.; Cramer, C.J.; Truhlar, D.G. Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113, 5806–5812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuniewski, A.; Rosiak, D.; Chojnaki, J.; Becker, B. Coordination polymers and molecular structures among complexes of mercury(II) halides with selected 1-benzoylthioureas. Polyhedron 2015, 90, 47–57. [Google Scholar] [CrossRef]
- Kraft, B.M.; Brennessel, W.W. Chelation and Stereodynamic Equilibria in Neutral Hypercoordinate Organosilicon Complexes of 1-Hydroxy-2-pyridinone. Organometallics 2014, 33, 158–171. [Google Scholar] [CrossRef] [Green Version]
- Schürmann, M.; Huber, F. Tris(2-pyridinethiolato)(p-tolyl)tin(IV), [Sn(C5H4NS)3(C7H7)]. Acta Crystallogr. C 1994, 50, 206–209. [Google Scholar] [CrossRef]
- Huber, F.; Schmiedgen, R.; Schürmann, M.; Barbieri, R.; Ruisi, G.; Silvestri, A. Mono-organotin(IV) and Tin(IV) Derivatives of 2-Mercaptopyridine and 2-Mercaptopyrimidine: X-ray Structures of Methyl-tris(2-pyridinethiolato)tin(IV) and Phenyl-tris(2-pyridinethiolato)tin(IV)·1.5CHCl3. Appl. Organomet. Chem. 1997, 11, 869–888. [Google Scholar] [CrossRef]
- Schürmann, M.; Schmiedgen, R.; Huber, F.; Silvestri, A.; Ruisi, G.; Barbieri Paulsen, A.; Barbieri, R. Mono-aryltin(IV) and mono-benzyltin(IV) complexes with pyridine-2-carboxylic acid and 8-hydroxyquinoline. X-ray structure of p-chloro-phenyl-tris(8-quinolinato)tin(IV)·2CHCl3. J. Organomet. Chem. 1999, 584, 103–117. [Google Scholar] [CrossRef]
- Schöne, D.; Gerlach, D.; Wiltzsch, C.; Brendler, E.; Heine, T.; Kroke, E.; Wagler, J. A Distorted Trigonal Antiprismatic Cationic Silicon Complex with Ureato Ligands: Syntheses, Crystal Structures and Solid State 29Si NMR Properties. Eur. J. Inorg. Chem. 2010, 2010, 461–467. [Google Scholar] [CrossRef]
- Bohme, U. CSD Private Communication2020, CCDC 2025957.
- Yang, J.; Verkade, J.G. Non-catalyzed addition reactions of Cl3SiSiCl3 with 1,2-diketones, 1,2-quinones and with a 1,4-quinone. J. Organomet. Chem. 2002, 651, 15–21. [Google Scholar] [CrossRef]
- Feng, Y.-L.; Zhang, F.-X.; Kuang, D.-Z.; Yang, C.-L. Two Novel Dibutyltin Complexes with Trimers and Hexanuclear Based on the Bis(5-Cl/Me-salicylaldehyde) Carbohydrazide: Syntheses, Structures, Fluorescent Properties and Herbicidal Activity. Chin. J. Struct. Chem. 2020, 39, 682–692. [Google Scholar] [CrossRef]
- Ma, C.; Tian, G.; Zhang, R. New triorganotin(IV) complexes of polyfunctional S,N,O-ligands: Supramolecular structures based on π⋅⋅⋅π and/or C–H⋅⋅⋅π interactions. J. Organomet. Chem. 2006, 691, 2014–2022. [Google Scholar] [CrossRef]
- Boyer, J.; Brelière, C.; Carré, F.; Corriu, R.J.P.; Kpoton, A.; Poirier, M.; Royo, G.; Young, J.C. Five-co-ordinated Silicon Compounds: Geometry of Formation by Intramolecular Co-ordination. Crystal Structure of 2-( Dimethylaminomethyl)phenyl-I -naphthylsilane. J. Chem. Soc. Dalton. Trans. 1989, 1, 43–51. [Google Scholar] [CrossRef]
- Engelhardt, G.; Radeglia, R.; Jancke, H.; Lippmaa, E.; Mägi, M. Zur Interpretation 29Si-NMR-chemischer Verschiebungen. Org. Magn. Reson. 1973, 5, 561–566. [Google Scholar] [CrossRef]
- Gericke, R.; Wagler, J. Ruthenium Complexes of Stibino Derivatives of Carboxylic Amides: Synthesis and Characterization of Bidentate Sb,E, Tridentate Sb,E2, and Tetradentate Sb,E3 (E = N and O) Ligands and Their Reactivity Toward [RuCl2(PPh3)3]. Inorg. Chem. 2020, 59, 6359–6375. [Google Scholar] [CrossRef]
- Gericke, R.; Wagler, J. (2-Pyridyloxy)arsines as ligands in transition metal chemistry: A stepwise As(III) → As(II) → As(I) reduction. Dalton Trans. 2020, 49, 10042–10051. [Google Scholar] [CrossRef] [PubMed]
- Baus, J.A.; Burschka, C.; Bertermann, R.; Fonseca Guerra, C.; Bickelhaupt, F.M.; Tacke, R. Neutral Six-Coordinate and Cationic Five-Coordinate Silicon(IV) Complexes with Two Bidentate Monoanionic N,S-Pyridine-2-thiolato(−) Ligands. Inorg. Chem. 2013, 52, 10664–10676. [Google Scholar] [CrossRef]
- Wächtler, E.; Gericke, R.; Kutter, S.; Brendler, E.; Wagler, J. Molecular structures of pyridinethiolato complexes of Sn(II), Sn(IV), Ge(IV), and Si(IV). Main Group Met. Chem. 2013, 36, 181–191. [Google Scholar] [CrossRef]
- Wahlicht, S.; Brendler, E.; Heine, T.; Zhechkov, L.; Wagler, J. 7-Azaindol-1-yl(organo)silanes and Their PdCl2 Complexes: Pd-Capped Tetrahedral Silicon Coordination Spheres and Paddlewheels with a Pd-Si Axis. Organometallics 2014, 33, 2479–2488. [Google Scholar] [CrossRef]
- Autschbach, J.; Sutter, K.; Truflandier, L.A.; Brendler, E.; Wagler, J. Atomic Contributions from Spin-Orbit Coupling to 29Si NMR Chemical Shifts in Metallasilatrane Complexes. Chem. Eur. J. 2012, 18, 12803–12813. [Google Scholar] [CrossRef]
- Wagler, J.; Brendler, E. Metallasilatranes: Palladium(II) and Platinum(II) as Lone-Pair Donors to Silicon(IV). Angew. Chem. Int. Ed. 2010, 49, 624–627. [Google Scholar] [CrossRef]
- Gualco, P.; Lin, T.-P.; Sircoglou, M.; Mercy, M.; Ladeira, S.; Bouhadir, G.; Pérez, L.M.; Amgoune, A.; Maron, L.; Gabbaï, F.P.; et al. Gold–Silane and Gold–Stannane Complexes: Saturated Molecules as σ-Acceptor Ligands. Angew. Chem. Int. Ed. 2009, 48, 9892–9895. [Google Scholar] [CrossRef] [PubMed]
- Kameo, H.; Kawamoto, T.; Bourissou, D.; Sakaki, S.; Nakazawa, H. Evaluation of the σ-Donation from Group 11 Metals (Cu, Ag, Au) to Silane, Germane, and Stannane Based on the Experimental/Theoretical Systematic Approach. Organometallics 2015, 34, 1440–1448. [Google Scholar] [CrossRef]
- Grobe, J.; Lütke-Brochtrup, K.; Krebs, B.; Läge, M.; Niemeyer, H.-H.; Würthwein, E.-U. Alternativ-Liganden XXXVIII. Neue Versuche zur Synthese von Pd(0)- und Pt(0)-Komplexen des Tripod-Phosphanliganden FSi(CH2CH2PMe2)3. Z. Naturforsch. 2007, 62b, 55–65. [Google Scholar] [CrossRef]
- Grobe, J.; Krummen, N.; Wehmschulte, R.; Krebs, B.; Läge, M. Alternativ-Liganden. XXXI Nickelcarbonylkomplexe mit Tripod-Liganden des Typs XM’(OCH2PMe2)n(CH2CH2PR2)3-n (M’ = Si, Ge; n = 0-3). Z. Anorg. Allg. Chem. 1994, 620, 1645–1658. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. C 2015, 71, 9–18. [Google Scholar] [CrossRef]








| Bond | PhSi(pyO)3 · CHCl3 | PhGe(pyO)3 | PhSn(pyO)3 |
|---|---|---|---|
| E1–O1 | 1.640(1) | 1.790(2) | 2.120(2) |
| E1–O2 | 1.636(1) | 1.777(2) | 2.257(2) |
| E1–O3 | 1.642(1) | 1.785(2) | 2.133(2) |
| E1–N1 | 2.968(2) | 2.789(2) | 2.281(3) |
| E1–N2 | 3.002(2) | 2.923(2) | 2.212(3) |
| E1–N3 | 3.000(2) | 2.908(2) | 2.406(3) |
| E1–C16 | 1.842(2) | 1.919(2) | 2.138(2) |
| O1-E1-O2 | 115.85(6) | 101.36(6) | 84.64(9) |
| O1-E1-O3 | 96.91(6) | 103.17(6) | 93.00(9) |
| O2-E1-O3 | 112.54(6) | 98.20(6) | 138.85(9) |
| O1-C1-N1 | 117.50(13) | 116.77(17) | 113.0(3) |
| O2-C6-N2 | 117.30(13) | 117.65(17) | 113.9(3) |
| O3-C11-N3 | 117.45(13) | 117.89(17) | 113.5(3) |
| O1-C1-C2 | 117.55(13) | 119.37(18) | 126.2(3) |
| O2-C6-C7 | 117.96(13) | 118.23(18) | 126.7(3) |
| O3-C11-C12 | 117.47(13) | 118.03(17) | 124.3(3) |
| Compound | E···N(pyS) Coordination? 1 | δ 13C(C2) | δ 13C(C6) |
|---|---|---|---|
| PhSi(pyO)3 | 0 | 160.3 | 147.3 |
| PhGe(pyO)3 | intermediate | 163.3 | 145.9 |
| PhSn(pyO)3 | 1 | 167.0 | 141.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuß, S.; Brendler, E.; Wagler, J. Molecular Structures of the Pyridine-2-olates PhE(pyO)3 (E = Si, Ge, Sn)—[4+3]-Coordination at Si, Ge vs. Heptacoordination at Sn. Crystals 2022, 12, 1802. https://doi.org/10.3390/cryst12121802
Kuß S, Brendler E, Wagler J. Molecular Structures of the Pyridine-2-olates PhE(pyO)3 (E = Si, Ge, Sn)—[4+3]-Coordination at Si, Ge vs. Heptacoordination at Sn. Crystals. 2022; 12(12):1802. https://doi.org/10.3390/cryst12121802
Chicago/Turabian StyleKuß, Sarah, Erica Brendler, and Jörg Wagler. 2022. "Molecular Structures of the Pyridine-2-olates PhE(pyO)3 (E = Si, Ge, Sn)—[4+3]-Coordination at Si, Ge vs. Heptacoordination at Sn" Crystals 12, no. 12: 1802. https://doi.org/10.3390/cryst12121802